Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
To get the human SOD1 sequence, I went to UniProt. The ID for this protein is P00441 and the sequence is the following:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Now, if the mutation is A4V, that means that in position 4 there’s a change from alanine to valine. The mutated sequence is then the following:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
That mutated sequence was introduced in the PepMLM-650M Colab, changing the peptide length to 12 and the number of binders to 4:
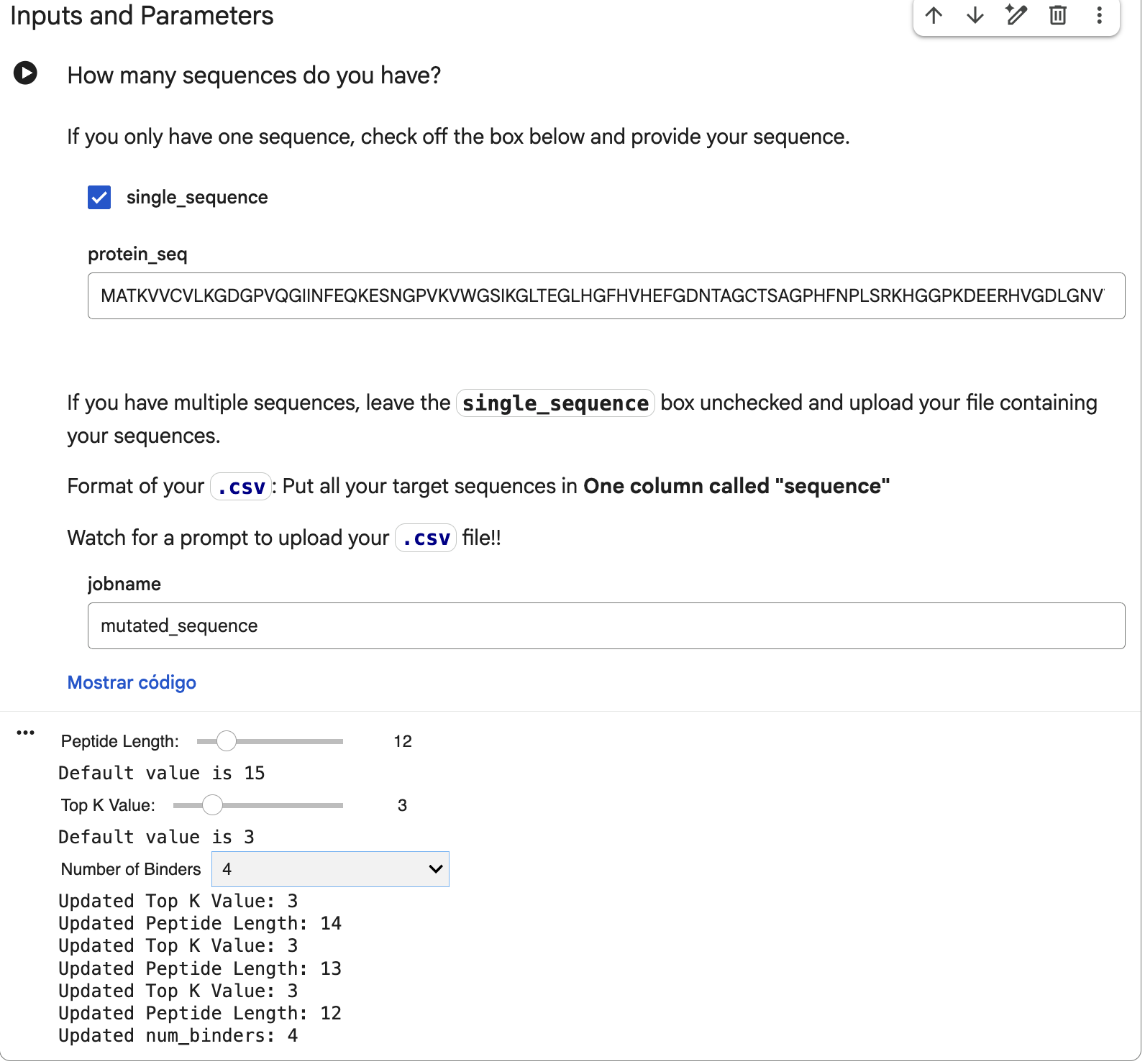
And after generating the peptides, here’s the results:
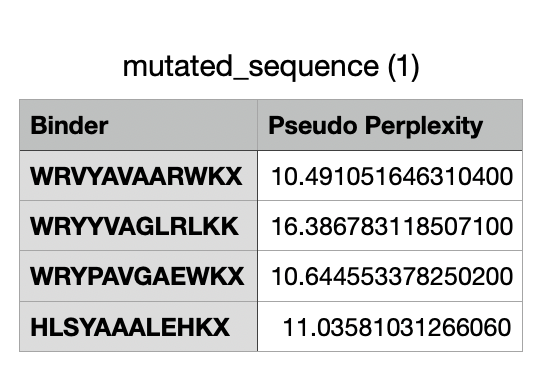
For the four generated binders, perplexity values vary. But, in general, a lower perplexity means a more confident model of the protein.
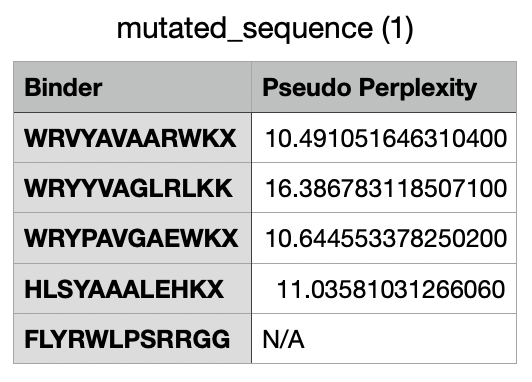
And this would be the list of all the binders, including the known binder last.
Part 2: Evaluate Binders with AlphaFold3
Before entering the mutated SOD1 sequence with each binder, I need to modify the sequence for each one because AlphaFold3 does not read B, J, O, U, X, Z characters, which are unknown in the sequence. I googled if there’s a way to solve this, and found in a Reddit post that AlphaFold suggests changing these to “A” (alanine), so these would be the new binder sequences:
- WRVYAVAARWKA
- WRYYVAGLRLKK
- WRYPAVGAEWKA
- HLSYAAALEHKA
- FLYRWLPSRRGG (known binder)
Now, in AlphaFold, I introduced the mutant SOD1 sequence and then added the binder sequence like this:
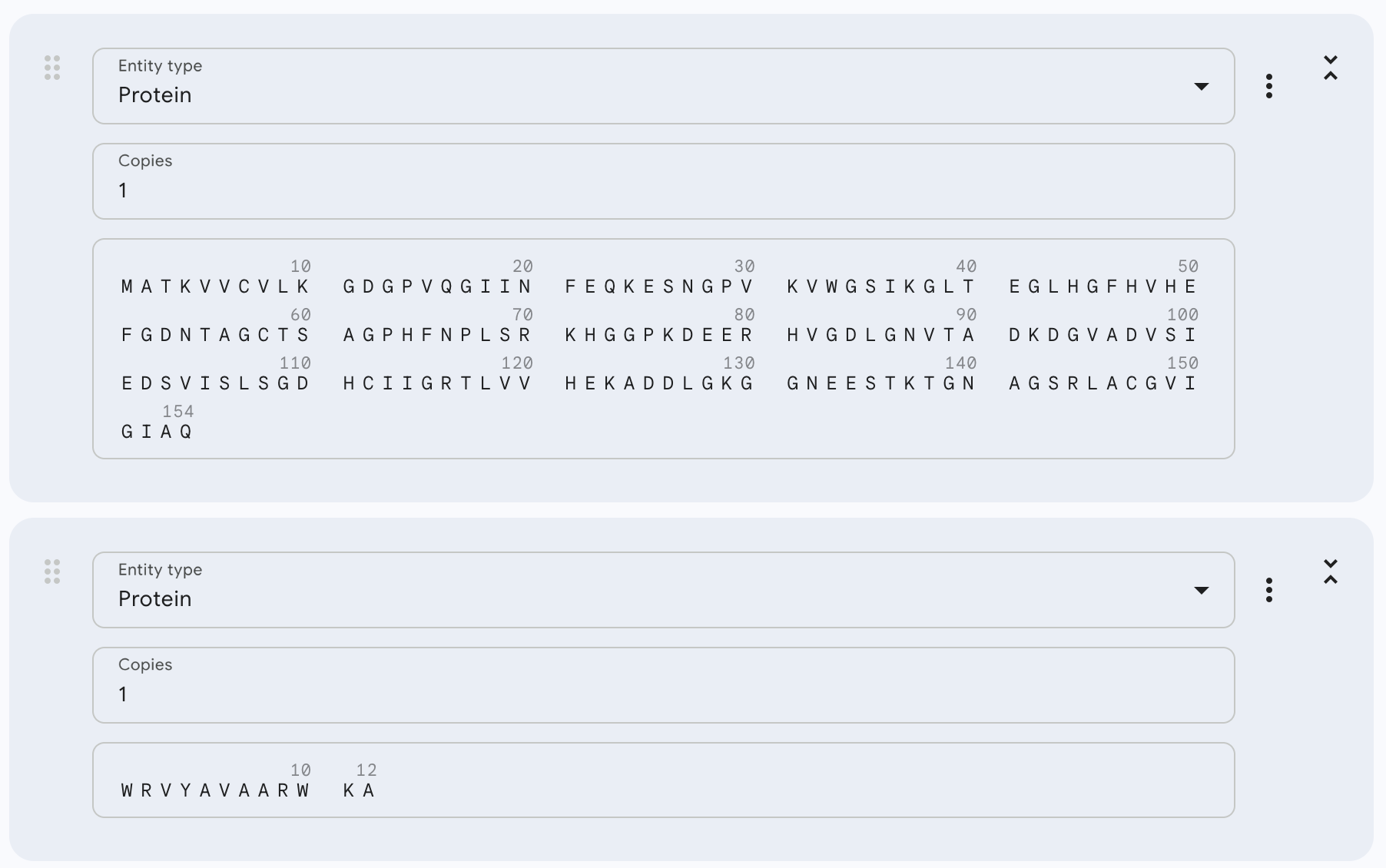
After running the sequence with each binder, the AlphaFold Server creates an entry with an ipTM score, a pTM score, the 3D model of the protein with its binder, and a graph.
Here are the ipTM scores for all peptides:
a (WRVYAVAARWKA) = 0.34 b (WRYYVAGLRLKK) = 0.3 c (WRYPAVGAEWKA) = 0.36 d (HLSYAAALEHKA) = 0.28 e(FLYRWLPSRRGG - known binder) = 0.35
Here are the models generated by AlphaFold for each:
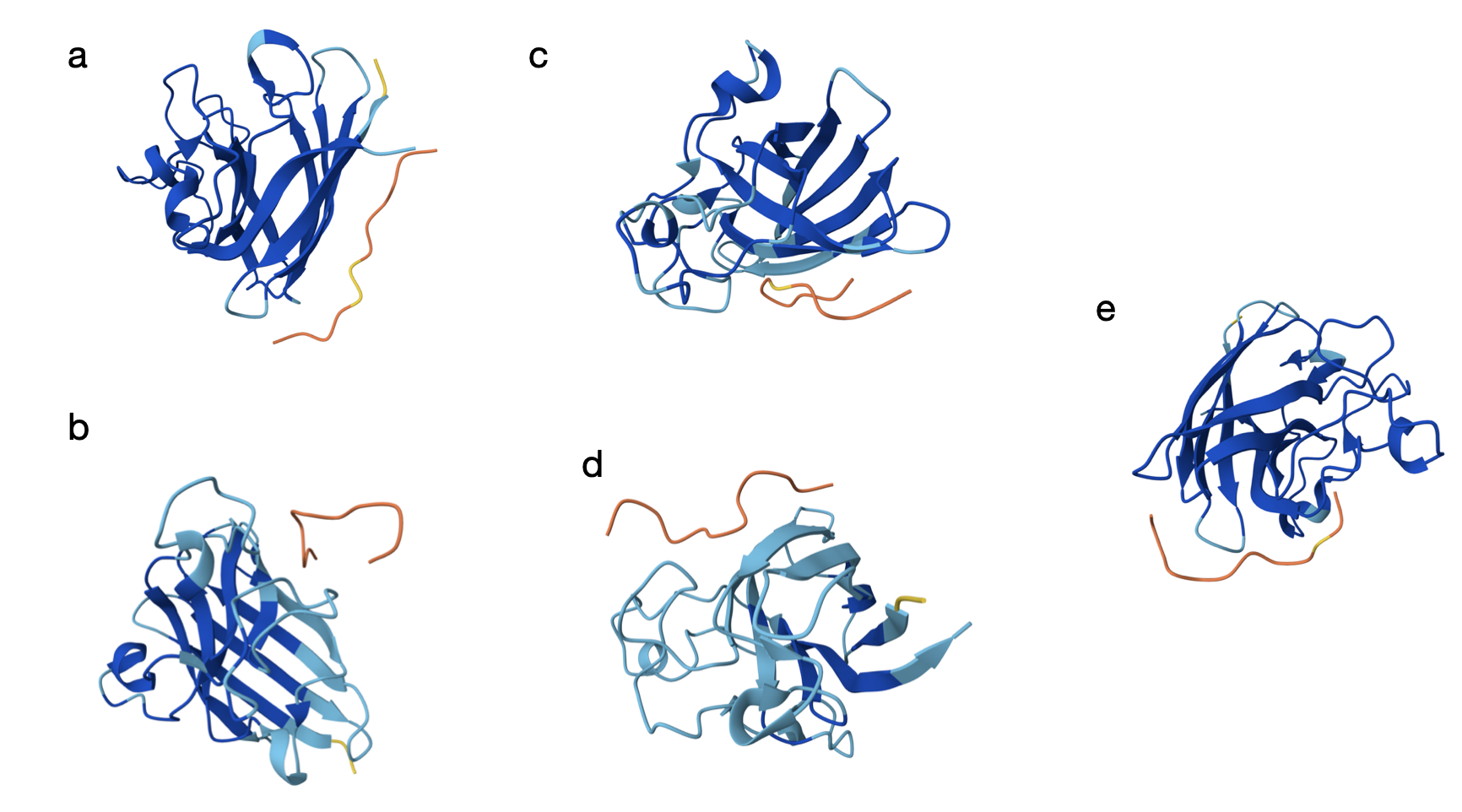
From the models, the orange and yellow segments (the peptides) appear to bind on the outer surface of the SOD1 monomer rather than in the core. In most panels (a–c and e), it seems the peptide is located along the edge of the β-barrel, but not inserting into the barrel. The binding site does not look centered directly at the extreme N-terminus where the A4V mutation lies, but it is still in the same general surface region. As shown in the models, they seem mostly surface-bound with only partial shallow contacts (which is a bit discouraging).
The ipTM scores are all fairly modest and clustered in a narrow range (0.28–0.36), which suggests weak-to-moderate confidence in the predicted complexes. The best PepMLM-generated peptide is c (WRYPAVGAEWKA) with 0.36, which slightly exceeds the known binder (FLYRWLPSRRGG, 0.35) used as a control for this particular test. Peptides a (0.34) and b (0.30) are similar but slightly lower, while d (0.28) is the weakest. At least one generated peptide (c) performs a bit better than the known binder based on ipTM.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
In PeptiVerse, I introduced the mutated human SOD1 sequence with each one of the five binders. Here are the results for each of them:
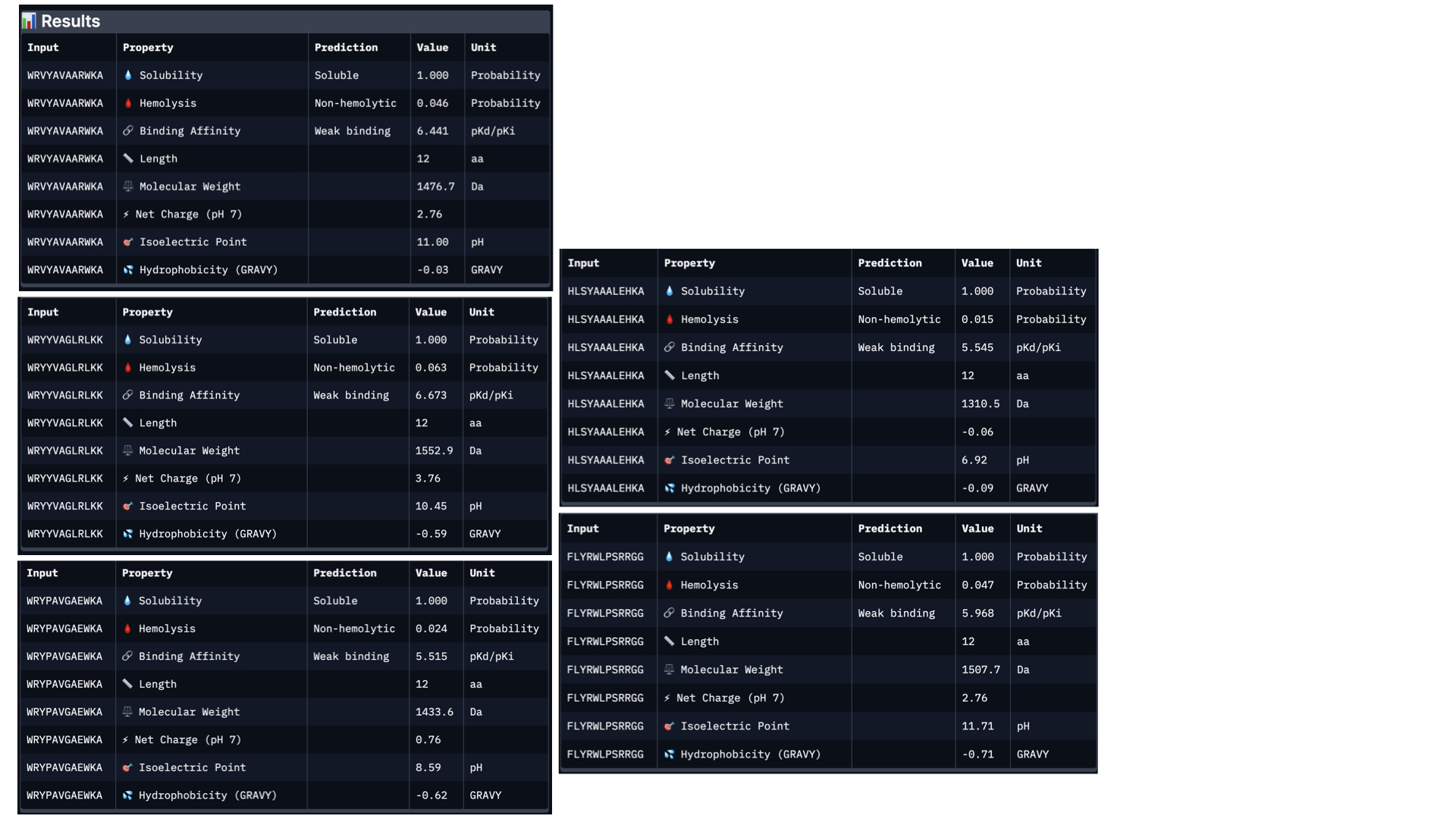
Analysing all peptides, it seems that all of them are predicted to be soluble, which is therapeutically good. Additionally, the hemolysis probability indicates how likely is the peptide able to destroy red blood cells; the lower the value, the better. All peptides show a relatively low hemolysis probability, being peptide #d the safest (sequence HLSYAAALEHKA). In terms of binding ability, a higher value indicates a stronger predicted binging; being peptide #b (sequence WRYYVAGLRLKK) the strongest. Finally, lower hydrophobicity levels are better because the peptides are more soluble. Just like with solubility, all have acceptable hydrophobicity levels.
In conclusion, the best peptide could be peptide #a (WRVYAVAARWKA), showing good solubility, low hydrophobicity, relatively strong predicted binding affinity, and low hemolysis risk. Overall, this peptide represents the most balanced option for further development, taking into account the results from PeptiVerse alone.
When comparing the two prediction methods (AlphaFold and PeptiVerse), there is no clear link between them. Peptide b had the strongest predicted binding affinity in PeptiVerse but a lower score in AlphaFold3, while peptide c scored highest in AlphaFold3 but did not have the strongest predicted affinity. This proves the two tools are measuring different things.
On the positive side, none of the peptides raised safety concerns, as all were predicted to be soluble and had low risk of harming red blood cells.
Taking everything into account, peptide a (WRVYAVAARWKA) offers the best overall balance. It performs reasonably well in both prediction methods and has good safety properties.
Part 4: Generate Optimized Peptides with moPPIt
These are the settings for the moPPit Colab I adjusted:
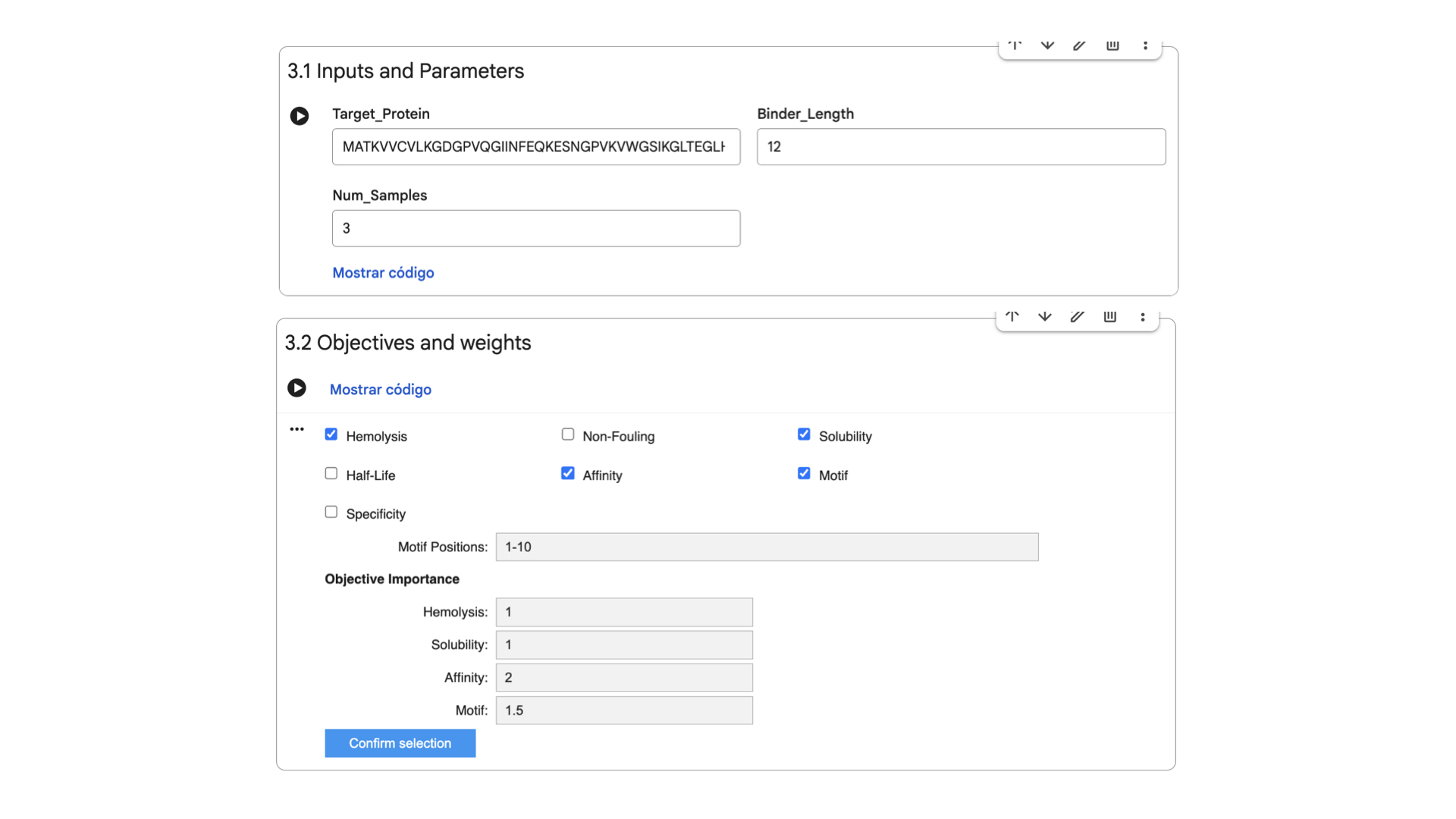
Hemolysis and Solubility have the same weight to maintain therapeutic properties (as discussed in Part 3). Affinity has the highest weight to achieve strong binding, while Motif maintains a medium weight to ensure binding near the mutation.
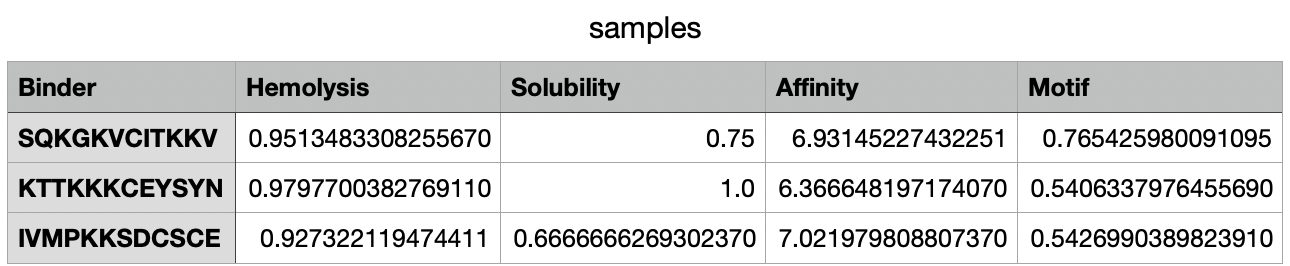
After 38 minutes running, the .csv with the results was created:
Part C: Final Project: L-Protein Mutants
For this part, I decided to follow the step by step for L-Protein Engineering, which is Option 1: Mutagenesis.
After running the ESM-2 Colab, I downloaded the L-protein mutants dataset as a csv and uploaded it to the Colab; and then obtained the file top_30_protein_mutations_scores.csv.
These are the top 30 protein mutations that come from the ESM-2 model. The model produces an LLR score for each mutation type at each residue position. If the LLR score is higher than 2, the mutation is strongly supported, between 1 and 2 is tolerated, between 0 and 1 is weakly tolerated, and below 0 is most probably damaging.
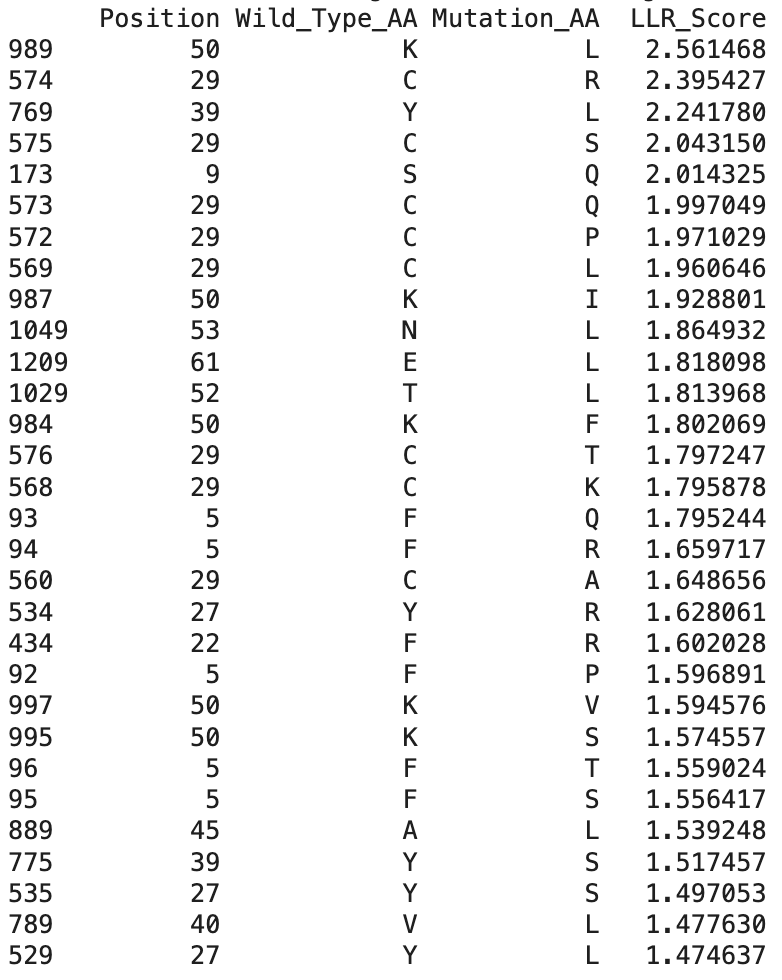
And here are the identified mutations from the experimental results:
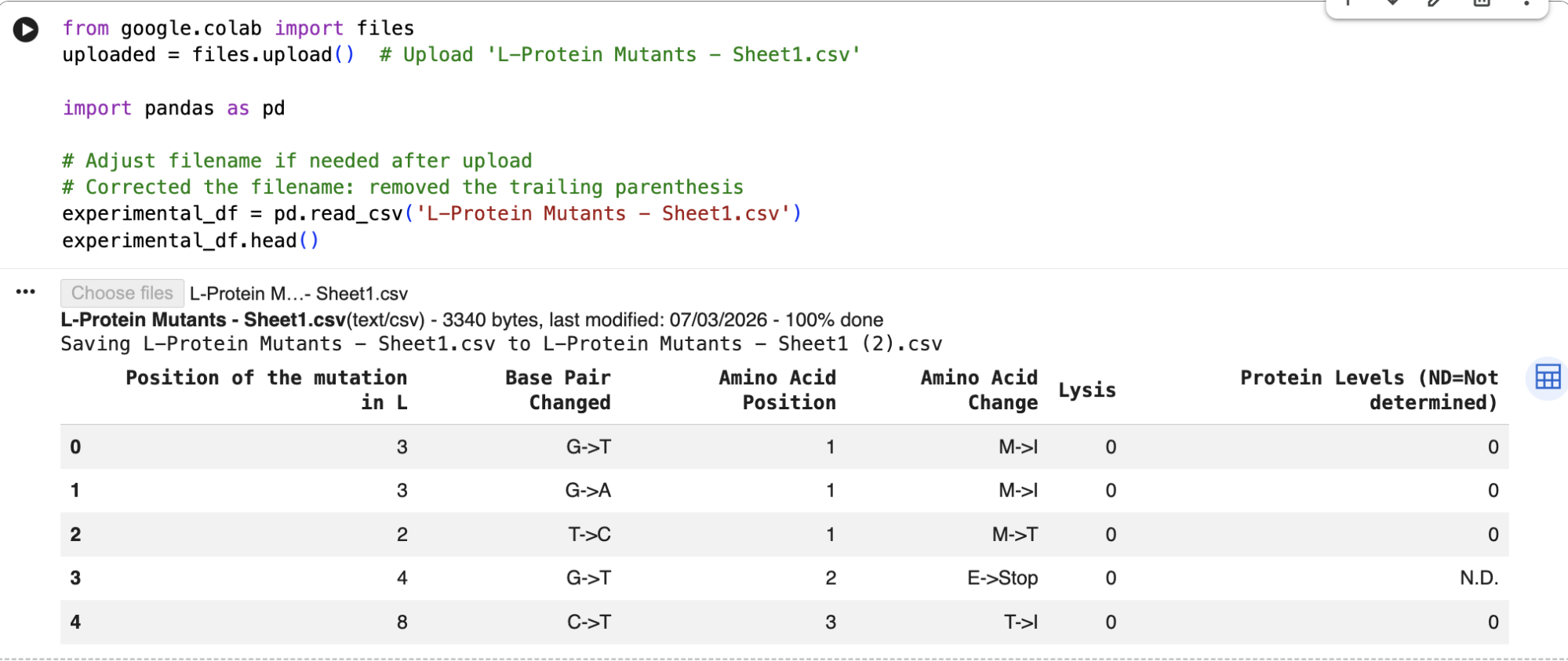
When comparing ESM-2 model predictions with the data from the real experiments, I can see that several top LLR mutations occur at the same positions experimentally, which means that the mutations are tolerated:
- Position 29 (C)
- Position 50 (K)
- Position 39 (Y)
The highest scoring mutation in the ESM-2 model predictions was K50L, with a LLR score of 2.56. This is a position in the transmembrane region, and is a change from lysine to leucine.
Also, some experimentally functional mutations from the L-Protein mutants dataset correspond to positive LLR scores from the ESM-2 model predictions. This means that the evolutionary model partially successfully identifies mutations that maintain protein stability and function.
Now, moving onto picking the mutations, I wanted to pick the mutations that are not the most obvious, so: that come from experimentally validated data, that are not the most repeated mutations in the table, and that can affect different biochemical properties.
The first thing to check is that the mutation shows lysis activity (so lysis = 1 on the table), and then check the region (soluble/transmembrane) and reflect on the type of amino acid. Here are the mutations I picked:
1. P13L
The experimental dataset shows Lysis = 1 and Protein = 1, the mutation is in the soluble region, and changing proline to leucine may increase folding flexibility (Yu et al., 2015) in the soluble domain of the protein.
2. R19H
The experimental dataset shows Lysis = 1, the mutation is in the soluble region, and changing arginine to histidine may modify electrostatic interactions (according to Muller et al., 2019), that could be interesting to see.
3. R20W
The experimental dataset shows Lysis = 1, the mutation is in the soluble region, and changing arginine to tryptophan may alter protein to protein interactions because of its hydrophobic characteristics, maybe stabilizing the protein further (Swift & Stewart, 1991).
4. A45P
The experimental dataset shows Lysis = 1, the mutation is in the transmembrane region, and changing alanine to proline could help in forming or changing the pore structure (Lee et al., 2003)
5. R18G
The experimental dataset shows Lysis = 1, the mutation is in the soluble region, and changing arginine to glycine could help in flexibility and folding of the protein (Gekko et al., 1994).
After choosing these 5 mutations, I will predict the 3D structure of the mutated L-protein and comparing it to the wild type.
Here’s the original wild type sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
And here’s the sequence with the 5 chosen mutations:
METRFPQQSQQTLASTNGHWPFKHEDYPCRRQQRSSTLYVLIFLPIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
I then went to ESM Fold and downloaded the PDB file to the predicted protein structure for the wild type sequence, and the predicted protein structure for my mutant sequence:
I uploaded those PDB files to PyMOL to visualize both structures better:
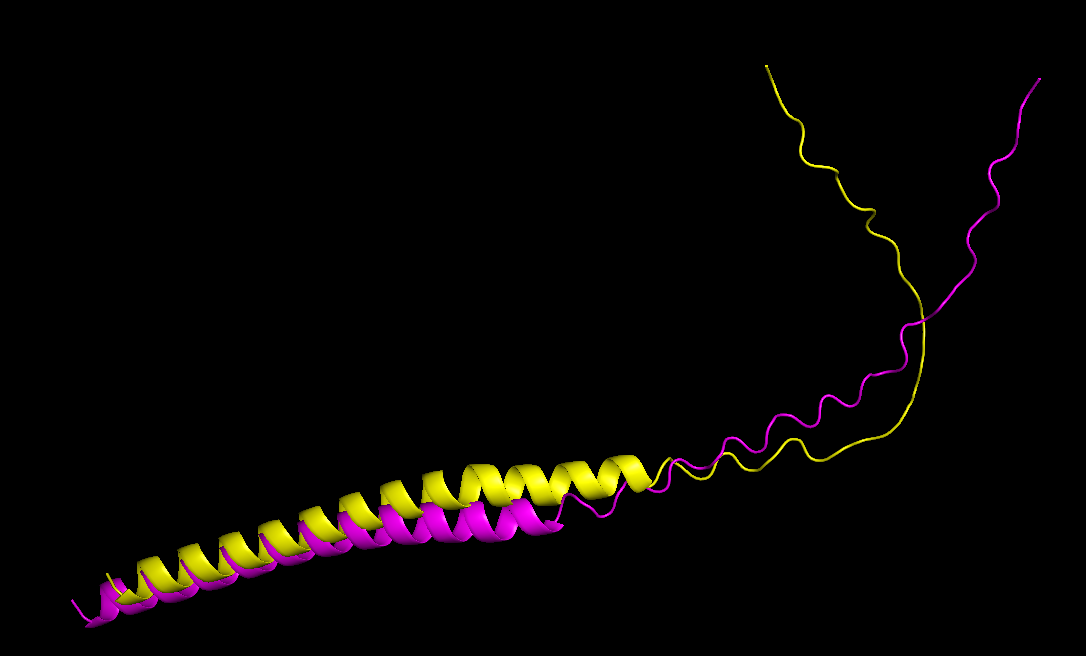
The mutant sequence is yellow, and the wild type is pink.
Comparing both structures, the mutant model maintained a similar overall fold, only with a more folded coil, which could mean that the mutations are unlikely to disrupt protein structure.
References:
Muller, L., Jackson, S. N., & Woods, A. S. (2019). Histidine, the less interactive cousin of arginine. European Journal of Mass Spectrometry, 25(2), 212-218.
Yu, H., Zhao, Y., Guo, C., Gan, Y., & Huang, H. (2015). The role of proline substitutions within flexible regions on thermostability of luciferase. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1854(1), 65-72.
Swift, S., & Stewart, G. S. (1991). The molecular biology of tryptophan synthase: A model for protein-protein interaction. Biotechnology and Genetic Engineering Reviews, 9(1), 229-294.
Lee, D. J. S., Keramidas, A., Moorhouse, A. J., Schofield, P. R., & Barry, P. H. (2003). The contribution of proline 250 (P-2′) to pore diameter and ion selectivity in the human glycine receptor channel. Neuroscience letters, 351(3), 196-200.
Gekko, K., Kunori, Y., Takeuchi, H., Ichihara, S., & Kodama, M. (1994). Point mutations at glycine-121 of Escherichia coli dihydrofolate reductase: important roles of a flexible loop in the stability and function. The Journal of Biochemistry, 116(1), 34-41.