Week 2 Homework Section 2: DNA Read, Write and Edit
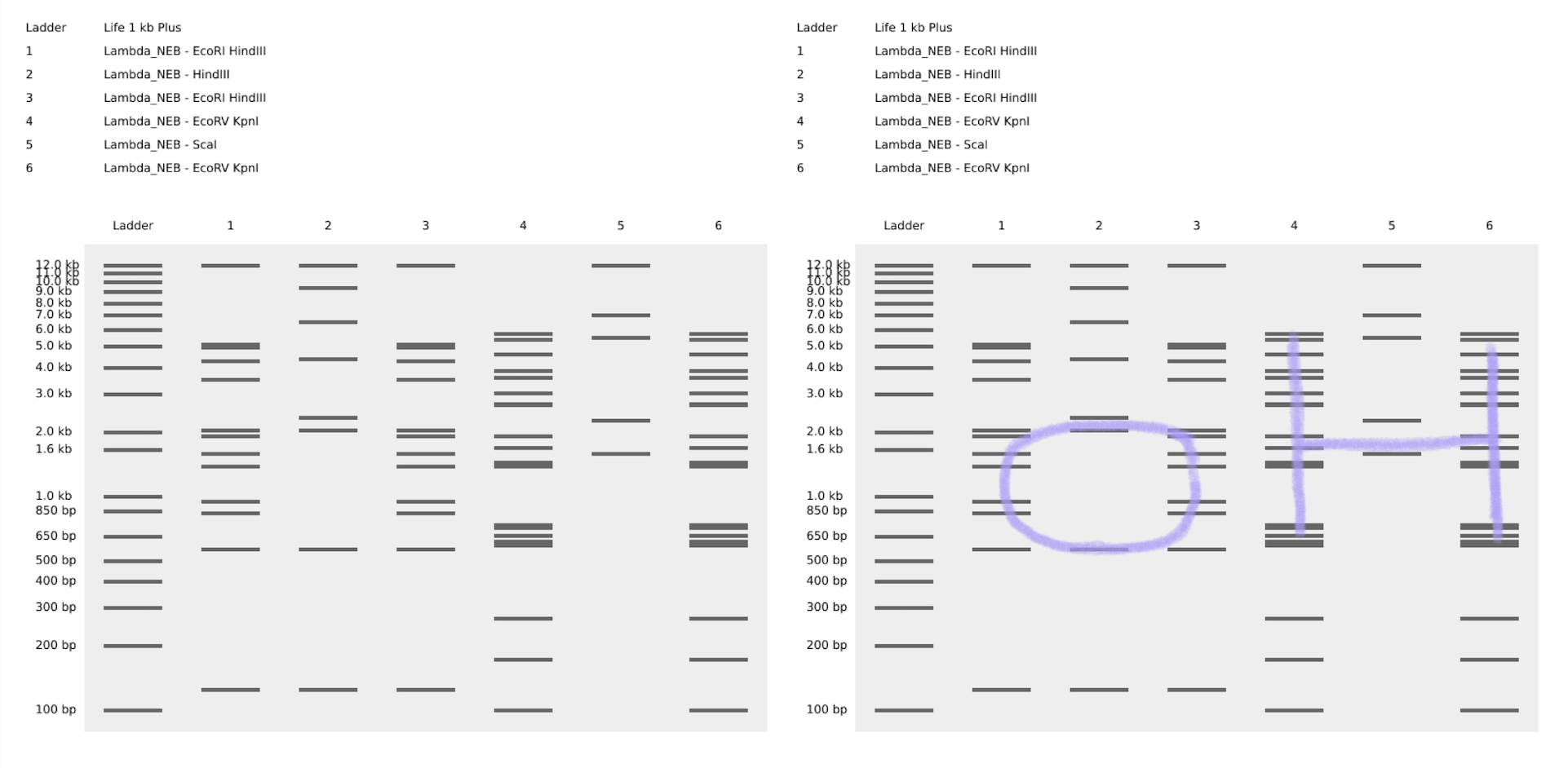
Part 1: Benchling and in silico gel art
Using benchling, and after some time playing around with Ronan’s website for iteration, I decided to created the expression “OH”.
Part 3: DNA Design Challenge
3.1. Choose your protein
I decided to work with the E Protein of the Zika virus Brazil strain.
>tr|A0A060H177|A0A060H177_ZIKV E protein (Fragment) OS=Zika virus OX=64320 GN=E PE=4 SV=1
IRCIGVSNRDFVEGMSGGTWVDVVLEHGGCVTVMAQDKPTVDIELVTTTVSNMAEVRSYCYEASISDMASDSRCPTQGEAYLDKQSDTQYVCKRTLVDRGWGNGCGLFGKGSLVTCAKFACSKKMTGKSIQPENLEYRIMLSVHGSQHSGMIVNDTGHETDENRAKVEITPNSPRAEATLGGFGSLGLDCEPRTGLDFSDLYYLTMNNKHWLVHKEWFHDIPLPWHAGADTGTPHWNNKEALVEFKDAHAKRQTVVVLGSQEGAVHTALAGALEAEMDGAKGRLSSGHLKCRLKMDKLRLKGVSYSLCTAAFTFTKIPAETLHGTVTVEVQYAGTDGPCKVPAQMAVDMQTLTPVGRLITANPVITESTENSKMMLELDPPFGDSYIVIGVGEKKITHHWHRSGSTIGKAFEATVRGAKRMAVLGDTAWDFGSVGGALNSLGKGIHQIFGAAFKSLFGGMSWFSQILIGTLLMWLGLNTKNGSISLMCLALGGVLIFLSTAVSA
3.2 Reverse Translate into DNA
First, I had to check for the correct codon table for Zika virus.
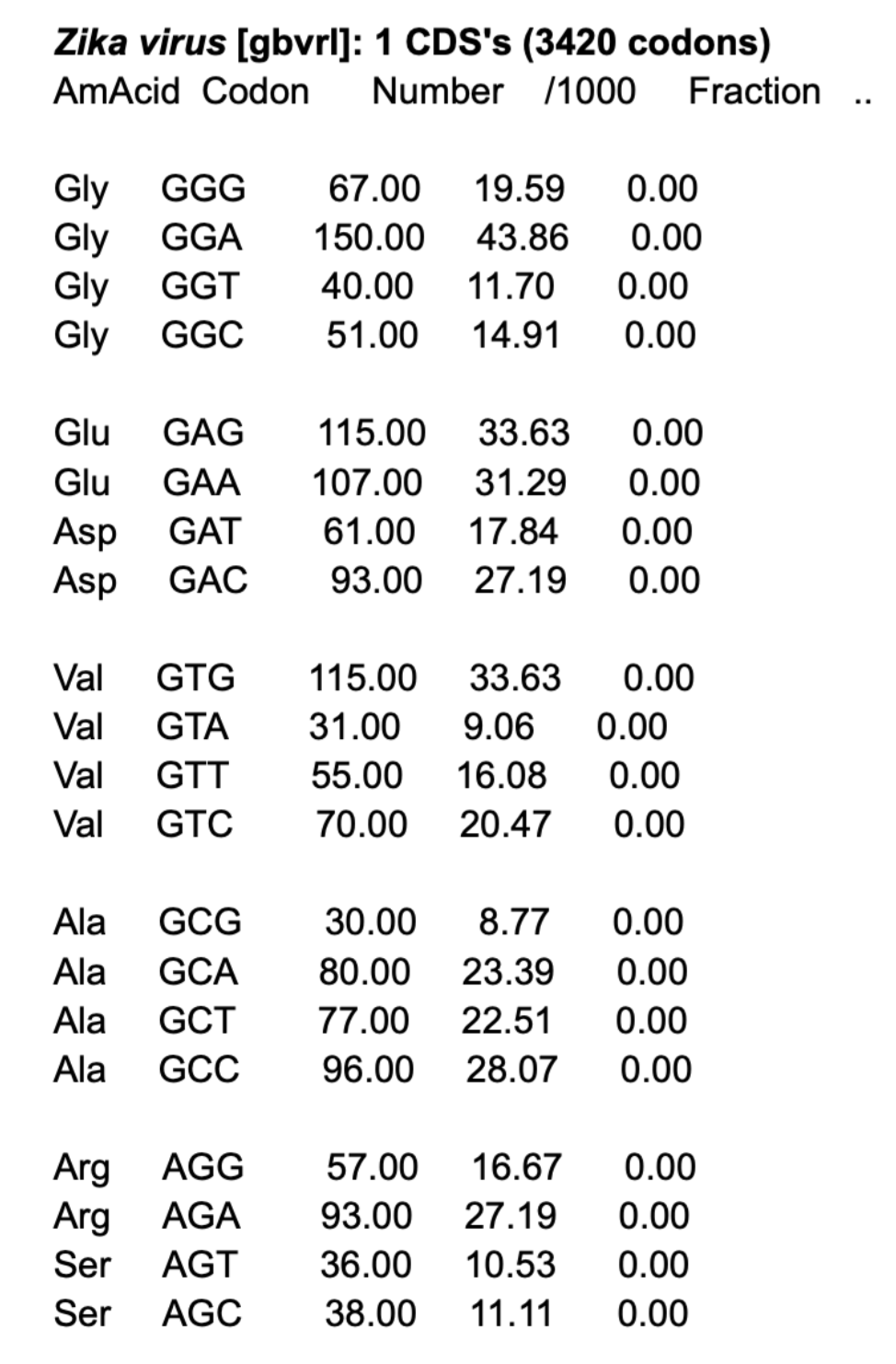
Then, using an online tool, I reverse translated the aa sequence into DNA
>reverse translation of tr|A0A060H177|A0A060H177_ZIKV E protein (Fragment) OS=Zika virus OX=64320 GN=E PE=4 SV=1 to a 1512 base sequence of most likely codons.
atcagatgtatcggagtgtcaaacagagacttcgtggagggaatgtcaggaggaacatgggtggacgtggtgctggagcacggaggatgtgtgacagtgatggcccaggacaagccaacagtggacatcgagctggtgacaacaacagtgtcaaacatggccgaggtgagatcatactgttacgaggcctcaatctcagacatggcctcagactcaagatgtccaacacagggagaggcctacctggacaagcagtcagacacacagtacgtgtgtaagagaacactggtggacagaggatggggaaacggatgtggactgttcggaaagggatcactggtgacatgtgccaagttcgcctgttcaaagaagatgacaggaaagtcaatccagccagagaacctggagtacagaatcatgctgtcagtgcacggatcacagcactcaggaatgatcgtgaacgacacaggacacgagacagacgagaacagagccaaggtggagatcacaccaaactcaccaagagccgaggccacactgggaggattcggatcactgggactggactgtgagccaagaacaggactggacttctcagacctgtactacctgacaatgaacaacaagcactggctggtgcacaaggagtggttccacgacatcccactgccatggcacgccggagccgacacaggaacaccacactggaacaacaaggaggccctggtggagttcaaggacgcccacgccaagagacagacagtggtggtgctgggatcacaggagggagccgtgcacacagccctggccggagccctggaggccgagatggacggagccaagggaagactgtcatcaggacacctgaagtgtagactgaagatggacaagctgagactgaagggagtgtcatactcactgtgtacagccgccttcacattcacaaagatcccagccgagacactgcacggaacagtgacagtggaggtgcagtacgccggaacagacggaccatgtaaggtgccagcccagatggccgtggacatgcagacactgacaccagtgggaagactgatcacagccaacccagtgatcacagagtcaacagagaactcaaagatgatgctggagctggacccaccattcggagactcatacatcgtgatcggagtgggagagaagaagatcacacaccactggcacagatcaggatcaacaatcggaaaggccttcgaggccacagtgagaggagccaagagaatggccgtgctgggagacacagcctgggacttcggatcagtgggaggagccctgaactcactgggaaagggaatccaccagatcttcggagccgccttcaagtcactgttcggaggaatgtcatggttctcacagatcctgatcggaacactgctgatgtggctgggactgaacacaaagaacggatcaatctcactgatgtgtctggccctgggaggagtgctgatcttcctgtcaacagccgtgtcagcc
3.3. Codon Optimization
Using Codon Optimization Tool of Twist Bioscience avoiding Type IIs enzyme (BsaI, BsmBI and Bbs), avoiding Standard MCS sites (BamHI, EcoRI, HindIII, NcoI, and XhoI), and preserving glycosylation sites and fusion loop for protein functionality, I obtained the following sequence:
>EProtein
ATCAGATGTATCGGAGTGTCAAACAGAGACTTCGTGGAGGGAATGTCAGGAGGAACATGGGTGGACGTGGTGCTGGAGCACGGAGGATGTGTGACAGTGATGGCCCAGGACAAGCCAACAGTGGACATCGAGCTGGTGACAACAACAGTGTCAAACATGGCCGAGGTGAGATCATACTGTTACGAGGCCTCAATCTCAGACATGGCCTCAGACTCAAGATGTCCAACACAGGGAGAGGCCTACCTGGACAAGCAGTCAGACACACAGTACGTGTGTAAGAGAACACTGGTGGACAGAGGATGGGGAAACGGATGTGGACTGTTCGGAAAGGCATTACAGGAGATATGTGTCAGGTACGACTTTTCAAAGAGGACGATCGAAAGGTGAACCCTGCTCGTGAGCCCGGGGTTCAAAACCACGCAGTTAGCGCGCGGATCACAGCACTCAGGAATGATCGTGAACGACACAGGACACGAGACAGACGAGAACAGAGCCAGGGCGGCGACCATACGAAACTTACTAAATCCCGGGGACATACAGGCCGCATCCGAATTACGGGCACGGGTCTGTAGGCCAAGAACAGGACTGGACTTCTCAGACCTGTACTACCTGACAATGAACAACAAGCACTGGCTGGTGCACAAGGAGTGGTTCCACGACATCCCACTGCCATGGCACGCCGGAGCCGACACAGGAACACCACACTGGAACAACAAGGAGGCCCTGGTGGAGTTCAAGGACGCCCACGCCAAGAGACAGACAGTGGTGGTGCTGGGATCACAGGAGGGAGCCGTGCACACAGCCCTGGCCGGAGCCCTGGAGGCCGAGATGGACGGAGCCAAGGGAAGACTGTCATCAGGACACCTGAAGTGTAGACTGAAGATGGACAAGCTGAGACTGAAGGGAGTGTCATACTCACTGTGTACAGCCGCCTTCACATTCACAAAGATCCCAGCCGAGACACTGCACGGAACAGTGACAGTGGAGGTGCAGTACGCCGGAACAGACGGACCATGTAAGGTGCCAGCCCAGATGGCCGTGGACATGCAGACACTGACACCAGTGGGAAGACTGATCACAGCCAACCCAGTGATCACAGAGTCAACAGAGAACTCAAAGATGATGCTGGAGCTGGACCCACCATTCGGAGACTCATACATCGTGATCGGAGTGGGAGAGAAGAAGATCACACACCACTGGCACAGATCAGGATCAACAATCGGAAAGGCCTTCGAGGCCACAGTGAGAGGAGCCAAGAGAATGGCCGTGCTGGGAGACACAGCCTGGGACTTCGGATCAGTGGGAGGAGCCCTGAACTCACTGGGAAAGGGAATCCACCAGATCTTCGGAGCCGCCTTCAAGTCACTGTTCGGAGGAATGTCATGGTTCTCACAGATCCTGATCGGAACACTGCTGATGTGGCTGGGACTGAACACAAAGAACGGATCAATCTCACTGATGTGTCTGGCCCTGGGAGGAGTGCTGATCTTCCTGTCAACAGCCGTGTCAGCC
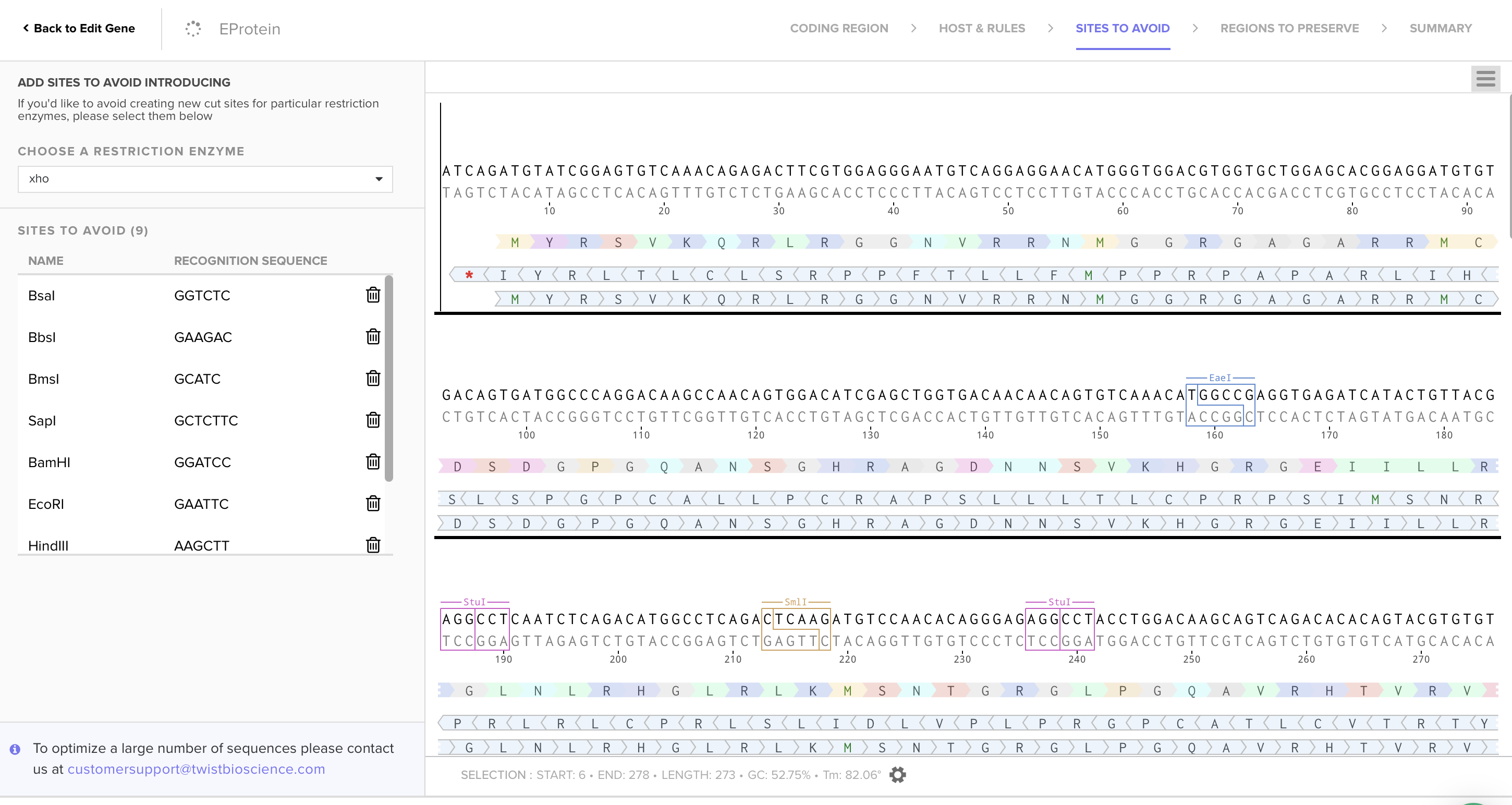
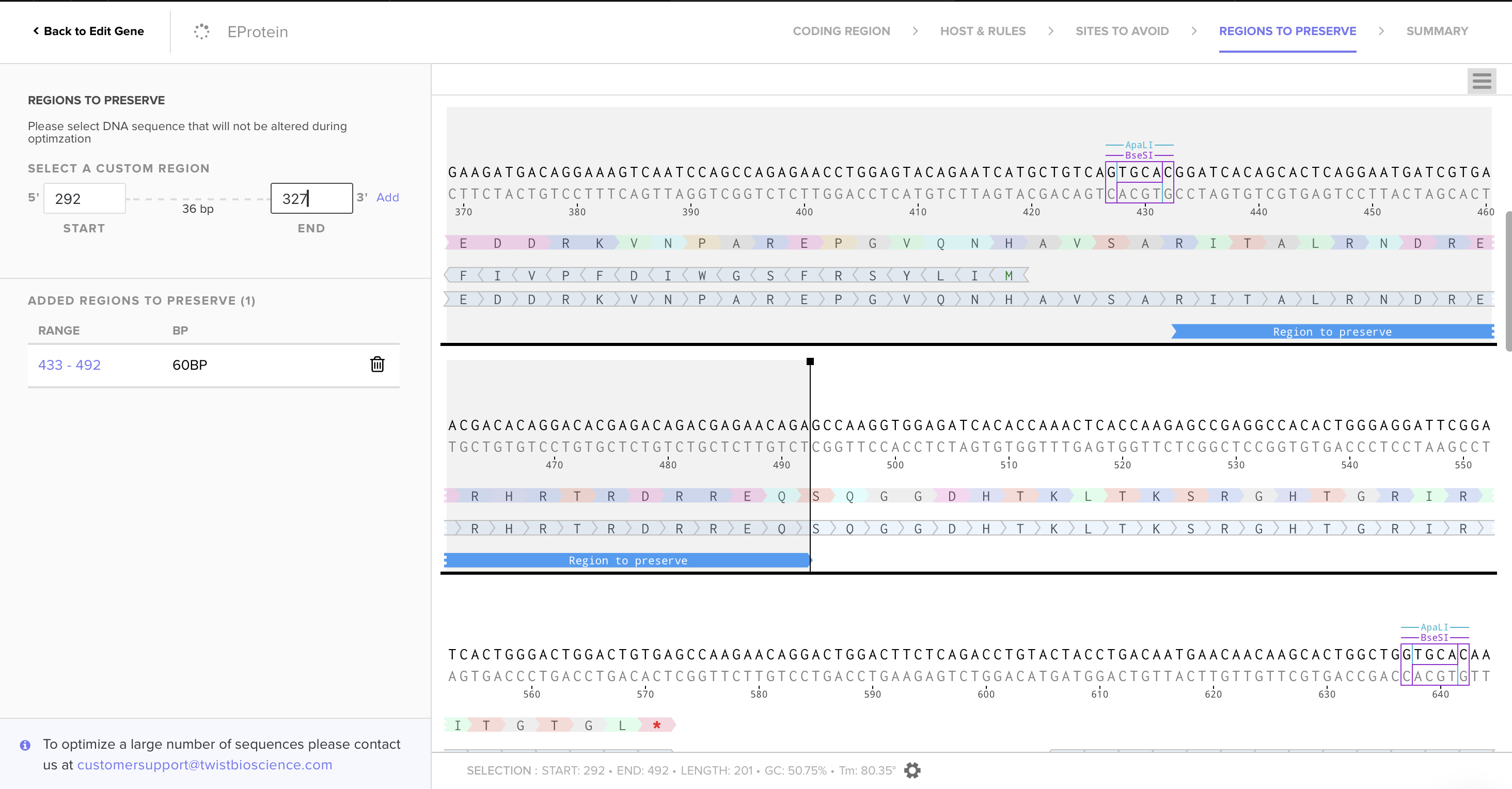
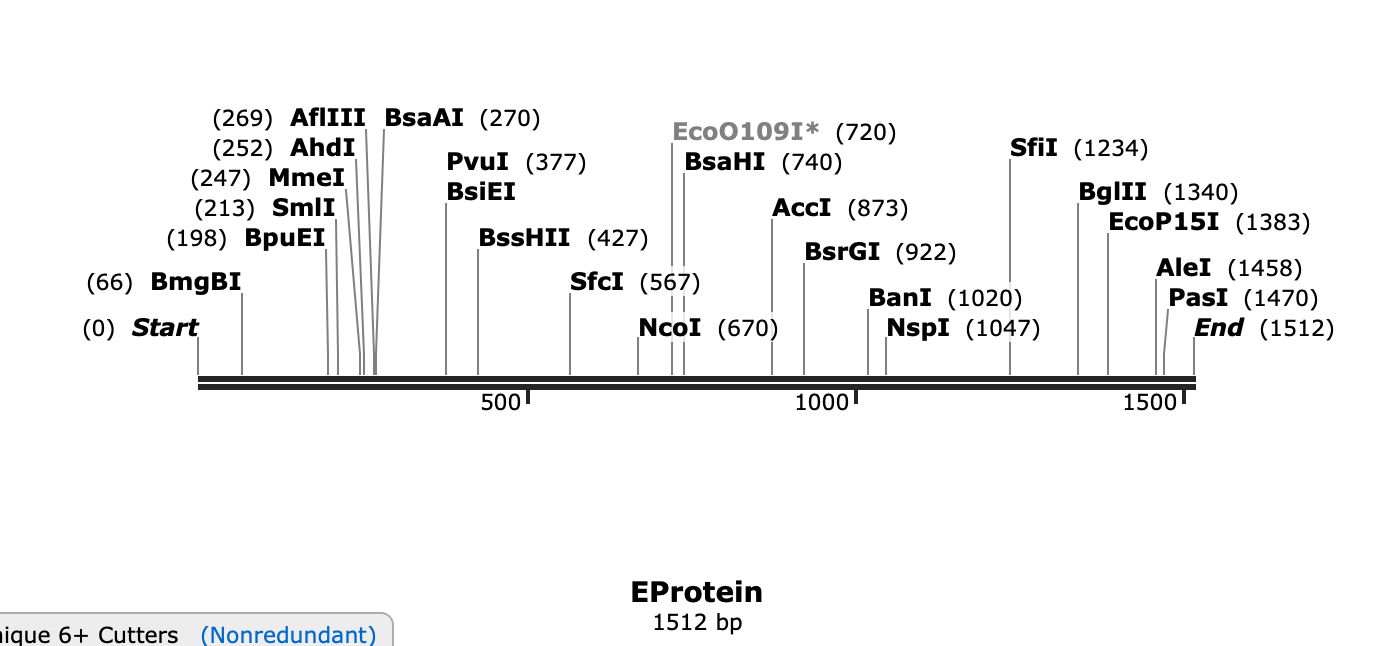
3.4. You have a sequence! Now what?
As it is a viral protein and it has methylations, I would have to transfected into a mammalian cell system such a HECK293, obviously having the optimize sequence on a expression mammalian vector.
3.5. How does it work in nature/biological systems?
A single gene codes for mutiple proteins at the transcriptional level because of the Open Reading Frame and the splicing of genes which can take out some exons.

Part 4: Prepare a Twist DNA Sunthesis Order
4.2-4.6 Build Your DNA Insert Sequence and expression system
As the E protein is a viral surface protein, I must use for expression of this protein the twist CMV plasmid.


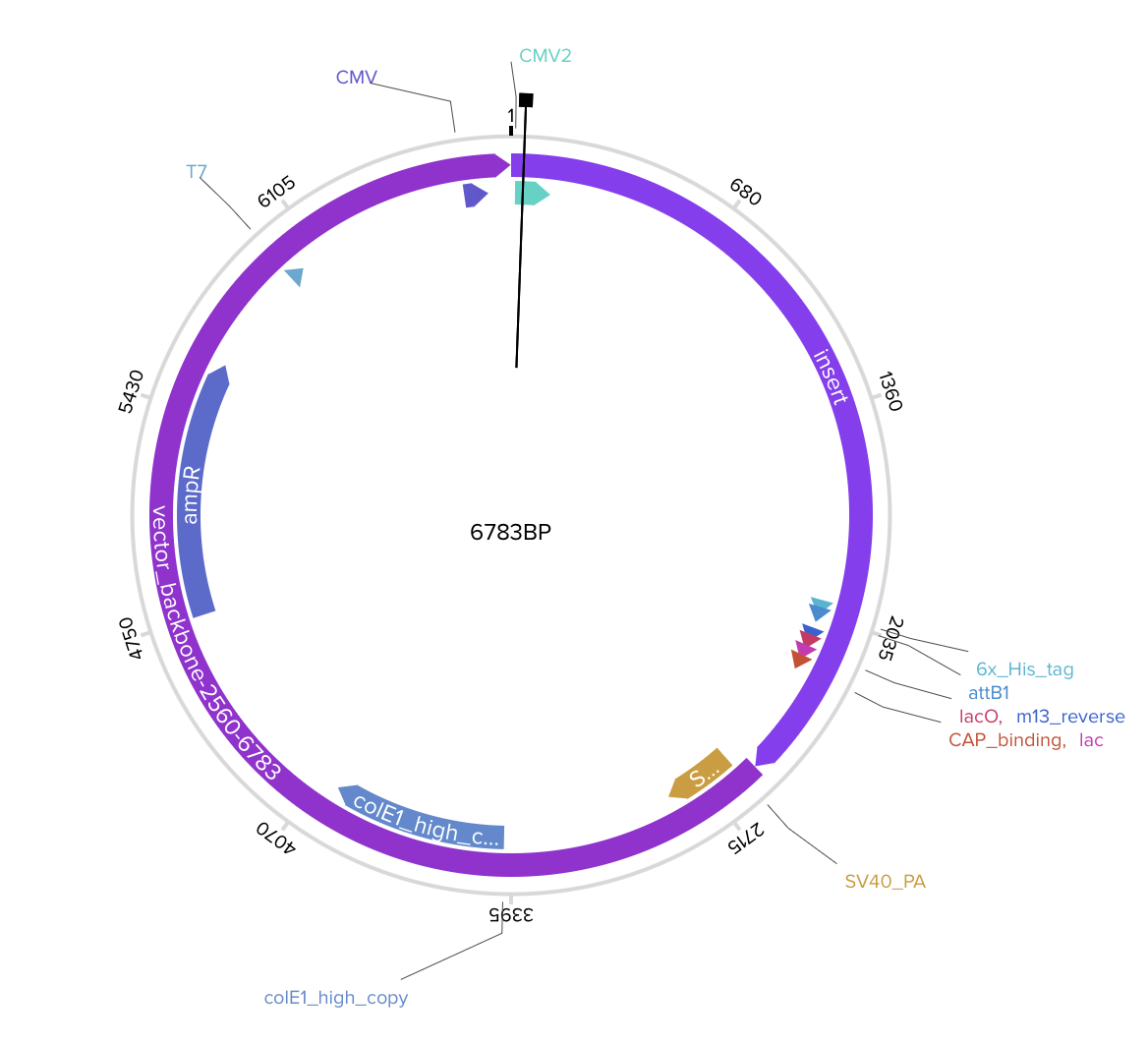
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence and why?
I would prioritize sequencing immunoglobulin G (IgG)–encoding genes, specifically the immunoglobulin heavy chain (IGH) and light chain (IGK or IGL) loci, as well as recombinant plasmids encoding engineered Fab fragments. IgG genes are particularly compelling because they undergo V(D)J recombination, somatic hypermutation, and class-switch recombination, processes that collectively generate extraordinary antibody diversity. Sequencing these loci enables detailed characterization of clonal expansion, affinity maturation, and mutational landscapes during immune responses. Such analyses are highly relevant to therapeutic antibody development, vaccine design, and the study of autoimmune or lymphoproliferative disorders. In parallel, plasmid sequencing is essential for molecular cloning workflows, as it confirms the correct insertion of coding sequences, preservation of reading frames, and absence of deleterious point mutations prior to protein expression.
(ii) What sequencing technology would you use and why?
For plasmid constructs and individual Ig variable regions, I would use Sanger sequencing, a first-generation sequencing technology. Because these targets are relatively short (typically under 3 kb), Sanger sequencing provides sufficient read length, high per-base accuracy, and straightforward validation. The input material would consist of purified plasmid DNA or PCR-amplified variable regions. Preparation involves plasmid isolation, primer design (vector-specific or insert-specific), and a sequencing reaction containing template DNA, a single primer, DNA polymerase, deoxynucleotides (dNTPs), and fluorescently labeled dideoxynucleotides (ddNTPs). During the reaction, stochastic incorporation of ddNTPs terminates DNA elongation, generating fragments of varying lengths. These fragments are separated by capillary electrophoresis, and laser detection of fluorescent signals enables base calling. The output consists of a chromatogram (electropherogram) with color-coded peaks corresponding to A, T, C, and G, along with a base-called sequence and associated quality scores. If full-length immunoglobulin loci or complex repertoires were to be analyzed, I would alternatively employ third-generation long-read sequencing, such as PacBio HiFi sequencing, which provides highly accurate circular consensus reads and resolves repetitive or structurally complex regions more effectively than short-read methods.
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
I would synthesize engineered Fab fragments derived from human IgG, optimized for therapeutic applications. These constructs would include codon-optimized variable heavy (VH) and variable light (VL) domains fused to constant regions (CH1 and CL), with engineered complementarity-determining regions (CDRs) to enhance antigen affinity and specificity. Additional design features could include a secretion signal peptide, affinity purification tags (e.g., His-tag), and optimized linker sequences where appropriate. The rationale for synthesizing such constructs lies in their relevance to antibody therapeutics, where improved binding kinetics, reduced immunogenicity, and controlled expression are critical parameters. Synthetic gene construction allows precise sequence engineering beyond what is achievable through conventional cloning.
(ii) What technology would you use for DNA synthesis and why?
To synthesize these constructs, I would rely on phosphoramidite-based solid-phase DNA synthesis for short oligonucleotides, followed by enzymatic assembly methods such as Gibson Assembly. In this approach, short chemically synthesized oligonucleotides are generated sequentially through nucleotide coupling cycles, cleaved from the solid support, deprotected, and purified. Overlapping fragments are then assembled enzymatically into a full-length gene construct and cloned into an appropriate expression vector. The assembled plasmid is subsequently sequence-verified, typically by Sanger sequencing. Limitations of chemical DNA synthesis include increased error rates with longer sequences, challenges associated with repetitive or GC-rich regions, and cost scalability. However, high-fidelity assembly methods and post-synthesis sequence validation mitigate these concerns and allow reliable production of gene-length constructs.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would focus on editing the genomes of animal models, such as mice, to introduce fluorescent reporters (e.g., EGFP) or precise disease-associated mutations. For example, inserting an EGFP reporter at an endogenous locus would enable real-time visualization of gene expression patterns during development. Alternatively, introducing defined point mutations into genes implicated in cancer or developmental disorders would facilitate mechanistic studies and disease modeling. Genome editing in animal systems provides powerful tools for functional genomics, lineage tracing, and in vivo investigation of gene regulation.
(ii) What technology would you use and how does it work?
To perform these edits, I would use CRISPR-Cas9 genome editing. This system employs a single-guide RNA (sgRNA) to direct the Cas9 endonuclease to a complementary genomic locus, where it induces a double-strand break (DSB). The cell subsequently repairs the DSB through either non-homologous end joining (NHEJ), which can introduce insertions or deletions, or homology-directed repair (HDR), which enables precise sequence insertion when a donor DNA template is supplied. Preparation requires rational guide RNA design to minimize off-target effects, construction or procurement of Cas9 expression systems (plasmid, mRNA, or ribonucleoprotein complex), and, when precise edits are desired, synthesis of a donor DNA template containing homology arms. Delivery methods may include electroporation, viral transduction, or microinjection into embryos. Edited cells or organisms must then be screened and validated via PCR and sequencing. Although CRISPR-Cas9 is highly versatile, limitations include variable editing efficiency, potential off-target cleavage, mosaicism in early embryos, and constraints associated with homology-directed repair efficiency. Despite these challenges, CRISPR-based editing remains the most precise and adaptable platform currently available for targeted genome engineering.