Week 2 – Homework: DNA Read, Write, & Edit
 Watercolor and then photoshopped image of the abstract architectural facade.
Watercolor and then photoshopped image of the abstract architectural facade.
Part 1: Benchling & In-silico Gel Art
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
For the gel art design, I arranged different restriction enzyme digests of Lambda DNA to create an abstract architectural façade. By combining single and triple digests, I created variations in band density and spacing that resemble structural columns, central voids, and layered material systems.

I also experimented with arranging digest patterns to form a heart-like shape, using band density and symmetry to create a subtle organic silhouette within the gel layout. However, it looks a bit wonky …
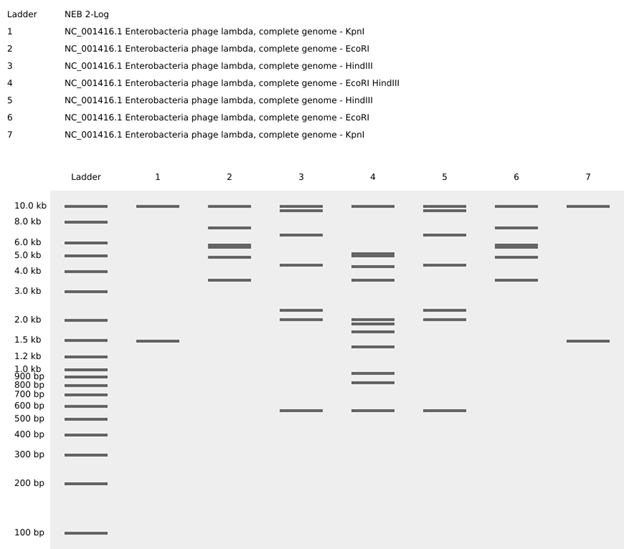
Part 3: DNA Design Challenge
3.1. Choose your protein.
I chose Cyanoexosortase B (CrtB) from Nostoc commune. This protein is involved in extracellular protein processing and surface-associated systems in cyanobacteria. Because Nostoc forms protective biofilms and extracellular matrices, CrtB plays a role in how the organism organises and stabilises its external environment. I am interested in how genetic information translates into material behaviour at environmental interfaces, so this protein connects molecular design to structural and ecological function. The amino acid sequence was obtained from UniProt (A0ABN6PZL9).

Image source: Benchling
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
For this exercise, I used the “EMBOSS BACKTRANSEQ” tool from www.ebi.ac.uk.
To reverse translate my protein, I used an online reverse translation tool. Since each amino acid is coded by three DNA bases (a codon), the tool converts the amino acid sequence back into a possible DNA sequence. Because multiple codons can code for the same amino acid, there isn’t only one correct DNA sequence. The tool selects codons based on the organism chosen and generates a DNA sequence that could realistically produce my protein.
ATGGTTTTACAACAACAAATTAAAAATCGTAATGCTTCTGGTTTATTAAATTTAGCTATTTTAGGTGTTTTATTATTATTATATGCTCCAATTTTATTACATTGGTTAGATGGTTGGTTATATAAAAATATTTCTACTGAACATGAATATTTTTCTCATGGTATTATTGGTTTACCATTTGCTGCTTATTTAGGTTGGATGAATCGTAAAAAATGGAAACGTTTACCAGATAATATTCATCCATTAGGTGCTGTTTTTTTATTATTAGGTGCTGTTTTTTATTTATCTGGTGTTACTGAATGGGTTAATTTATCTTTACCAGTTATTTTAGTTGGTTTATGTTTATGGTTTAAAGGTATTTCTGGTTTACGTTCTCAAGGTTTTCCATTATTATTAGTTTTTTTAGCTACTCCAACTGCTTTACCATATTTAATTGCTCCATATACTTTACCATTACAATCTTTTATTGCTGGTACTGCTGGTTTTATTTTAAATCAATTTGGTATGGAAGTTACTGTTGATGAAATTAATTTATATGTTGGTGGTCGTATTGTTGAAGTTGCTCCATATTGTGCTGGTTTAAAAATGTTATTTCTACTTTATATGTTGGTTTAATGTTATTATATTGGACTGATGCTTTATCTTCTCGTCGTACTGTTATTTCTTTTTTTCTTTAGCTGCTATTGTTTCTATTATTGCTAATATTATTCGTAATACTTTATTAACTTTTTTTCATGGTACTGGTCAAGAAGCTGCTTTTAAATGGTTACATGATGGTTGGGGTGGTGATGTTTATTCTGCTTGTATGTTAGTTTCTTTAGTTCCATTATTAAATGGTATTAATTCTTATTTTTCTGCTTCTTTAGAAACTGAACAAGAAGGTGAATCT
3.3. Codon optimization.
I used the “Codon Optimization Tool” from eu.idtdna.com for this exercise and a custom organism table from www.kazusa.or.jp (Nostoc commune DRH1).
Codon optimisation is needed because different organisms prefer different DNA codons for the same amino acids. Even though multiple codons can code for the same amino acid, some are used more often in certain species. If the DNA sequence uses codons that are rare in the host organism, the protein may not be made efficiently. I optimised my sequence for Nostoc commune so that the codons better match what this bacterium naturally prefers, which should help improve protein expression.
ATGGTTTTGCAACAACAAATCAAGAACCGCAATGCTTCTGGTTTACTTAATTTAGCGATTCTAGGAGTTCTATTACTACTATATGCGCCTATTTTGCTTCATTGGTTGGATGGTTGGCTATATAAGAATATTTCCACAGAACACGAATACTTTAGTCACGGCATTATCGGTCTACCATTTGCGGCGTATCTTGGATGGATGAATCGTAAAAAATGGAAGCGCCTGCCCGATAATATCCACCCTCTGGGAGCGGTTTTTTTACTGTTAGGAGCTGTATTCTATCTTTCTGGTGTTACTGAATGGGTCAATCTTAGTCTGCCGGTCATCTTAGTTGGTTTGTGCCTGTGGTTCAAGGGTATTTCTGGTTTGCGATCTCAAGGTTTTCCTCTGCTATTGGTATTTTTGGCAACTCCAACTGCCTTGCCTTACCTGATCGCACCATACACATTGCCCTTACAATCTTTCATTGCAGGTACTGCGGGTTTTATCTTAAACCAATTCGGCATGGAAGTTACCGTCGATGAAATTAACCTATATGTTGGTGGTAGAATTGTGGAAGTAGCGCCATACTGTGCAGGTTTGAAAATGTTGTTCACTACATTATATGTGGGCCTTATGCTGCTATACTGGACAGACGCTCTGTCTTCTAGACGTACTGTCATTTCTTTTCTTTCCCTAGCTGCAATTGTATCTATTATTGCCAATATTATCCGTAACACTCTGTTGACCTTTTTCCACGGCACTGGTCAAGAAGCTGCGTTTAAATGGTTACATGATGGATGGGGTGGTGACGTGTACTCAGCTTGTATGTTAGTCTCATTGGTGCCATTACTGAACGGTATTAATTCTTACTTTTCAGCAAGTCTTGAAACAGAGCAAGAAGGTGAATCT
The following restriction enzyme sites have been found in the selected reading frame: AseI (ATTAAT) BbsI (GAAGAC) DraI (TTTAAA) MfeI (CAATTG) SspI (AATATT) XbaI (TCTAGA)
3.4. You have a sequence! Now what?
After designing and codon-optimising the DNA sequence, the gene could first be chemically synthesised using modern DNA synthesis technologies. The synthesised gene would then be assembled into a plasmid vector containing a promoter, ribosome-binding site, and terminator. This plasmid could be introduced into Nostoc commune using standard transformation techniques. Inside the cell, RNA polymerase transcribes the DNA into mRNA, and ribosomes translate the mRNA into Cyanoexosortase B according to the genetic code.
Alternatively, the DNA could be used in a cell-free transcription–translation system, where purified molecular machinery produces the protein outside of living cells. Both approaches rely on the same central dogma process: DNA is transcribed into RNA, and RNA is translated into protein.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would like to sequence environmental DNA from biological soil crusts, with a focus on Nostoc commune and related cyanobacteria. The goal would be to identify genes involved in extracellular matrix production, stress tolerance, and surface adhesion. This would allow me to better understand which genetic pathways contribute to soil stabilisation and how microbial community composition changes under environmental stress.
(ii) In the lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA, and why?
To sequence environmental DNA from biological soil crust samples, I would use Illumina sequencing, potentially combined with Oxford Nanopore sequencing. Illumina provides high accuracy and depth for microbial community profiling, while Nanopore offers long reads that help resolve full-length genes and structural variation.
Illumina is a second-generation (next-generation) sequencing technology. It performs massively parallel sequencing of short DNA fragments, enabling the simultaneous reading of millions of sequences. Oxford Nanopore is considered a third-generation sequencing technology, as it sequences single DNA molecules in real-time without the need for amplification.
The input for Illumina sequencing would be environmental DNA extracted from soil crust samples. The DNA is prepared by adding sequencing adapters and amplifying the fragments to create a sequencing library. During sequencing, the DNA binds to a flow cell and is copied while fluorescently labelled nucleotides are incorporated one base at a time. Each incorporated base is detected by its fluorescence signal and converted into a nucleotide sequence. The output consists of millions of short DNA reads that can be analysed to identify genes and microbial composition.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesise a codon-optimised version of the Cyanoexosortase B (CrtB) gene from Nostoc commune. The goal would be to study and potentially enhance extracellular matrix organisation in cyanobacteria, which is relevant to soil stabilisation and biofilm formation.
(ii) What technology or technologies would you use to perform this DNA synthesis, and why?
To synthesise the CrtB gene, I would use phosphoramidite-based oligonucleotide synthesis, followed by gene assembly.
The essential steps include stepwise chemical addition of protected nucleotides to build short oligonucleotides on a solid support, cleavage and deprotection, purification, and assembly of overlapping oligos into a full-length gene using PCR-based methods. The assembled gene can then be sequence-verified before use.
The main limitations of this method are synthesis errors that accumulate with increasing length, practical limits on direct synthesis length (typically ~200 nt per oligo), and the need for assembly and error correction for longer constructs. Although scalable and highly automated, accuracy and yield decrease as sequence length increases.
References:
Beaucage, S.L. and Caruthers, M.H., 1981. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Letters, 22(20), pp.1859–1862.
Gibson, D.G., Young, L., Chuang, R.-Y., Venter, J.C., Hutchison, C.A. and Smith, H.O., 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods, 6, pp.343–345.
Kosuri, S. and Church, G.M., 2014. Large-scale de novo DNA synthesis: Technologies and applications. Nature Methods, 11(5), pp.499–507.
LeProust, E.M., Peck, B.J., Spirin, K., McCuen, H.B., Moore, B., Namsaraev, E. and Caruthers, M.H., 2010. Synthesis of high-quality libraries of long (150-mer) oligonucleotides by a novel depurination-controlled process. Nucleic Acids Research, 38(8), pp.2522–2540.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would focus on editing regulatory regions of genes involved in extracellular matrix production in Nostoc commune, rather than altering core metabolic pathways. For example, modifying promoter regions could allow increased or more controlled expression of genes associated with surface adhesion or polysaccharide production. The goal would be to fine-tune existing biological functions rather than introduce foreign traits, maintaining ecological compatibility.
(ii) What technology or technologies would you use to perform these DNA edits, and why?
I would use CRISPR-Cas9 to edit regulatory regions of the Nostoc commune genome. CRISPR-Cas9 uses a guide RNA to direct the Cas9 nuclease to a specific DNA sequence, where it introduces a double-strand break. The cell then repairs the break either through non-homologous end joining, which can introduce small mutations, or through homology-directed repair if a donor template is supplied.
The preparation includes designing a guide RNA targeting the promoter region, constructing a plasmid carrying Cas9 and the guide RNA, and introducing it into the host cells. If precise edits are required, a repair template DNA is also provided.
Limitations include variable editing efficiency, dependence on cellular repair pathways, and potential off-target effects where unintended DNA regions may be modified.
References:
Cong, L. et al., 2013. Multiplex genome engineering using CRISPR/Cas systems. Science, 339(6121), pp.819–823.
Jinek, M. et al., 2012. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), pp.816–821.
Doudna, J.A. and Charpentier, E., 2014. The new frontier of genome engineering with CRISPR-Cas9. Science, 346(6213), 1258096.