Sarvin Farhangi—HTGAA Spring 2026

About me
Contact info
Homework
- Week 3 – Homework: Lab automation
- Week 2 – Homework: DNA Read, Write, & Edit
- Week 1 – Homework: Principles and Practices
Week 2 – Homework: DNA Read, Write, & Edit
Watercolor and then photoshopped image of the abstract architectural facade. Part 1: Benchling & In-silico Gel Art Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. For the gel art design, I arranged different restriction enzyme digests of Lambda DNA to create an abstract architectural façade. By combining single and triple digests, I created variations in band density and spacing that resemble structural columns, central voids, and layered material systems.
Week 1 – Homework: Principles and Practices
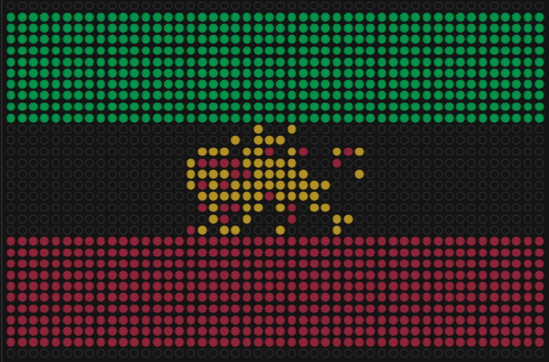
 Watercolor and then photoshopped image of the abstract architectural facade.
Watercolor and then photoshopped image of the abstract architectural facade.
For the gel art design, I arranged different restriction enzyme digests of Lambda DNA to create an abstract architectural façade. By combining single and triple digests, I created variations in band density and spacing that resemble structural columns, central voids, and layered material systems.

I also experimented with arranging digest patterns to form a heart-like shape, using band density and symmetry to create a subtle organic silhouette within the gel layout. However, it looks a bit wonky …
I chose Cyanoexosortase B (CrtB) from Nostoc commune. This protein is involved in extracellular protein processing and surface-associated systems in cyanobacteria. Because Nostoc forms protective biofilms and extracellular matrices, CrtB plays a role in how the organism organises and stabilises its external environment. I am interested in how genetic information translates into material behaviour at environmental interfaces, so this protein connects molecular design to structural and ecological function. The amino acid sequence was obtained from UniProt (A0ABN6PZL9).

Image source: Benchling
For this exercise, I used the “EMBOSS BACKTRANSEQ” tool from www.ebi.ac.uk.
To reverse translate my protein, I used an online reverse translation tool. Since each amino acid is coded by three DNA bases (a codon), the tool converts the amino acid sequence back into a possible DNA sequence. Because multiple codons can code for the same amino acid, there isn’t only one correct DNA sequence. The tool selects codons based on the organism chosen and generates a DNA sequence that could realistically produce my protein.
ATGGTTTTACAACAACAAATTAAAAATCGTAATGCTTCTGGTTTATTAAATTTAGCTATTTTAGGTGTTTTATTATTATTATATGCTCCAATTTTATTACATTGGTTAGATGGTTGGTTATATAAAAATATTTCTACTGAACATGAATATTTTTCTCATGGTATTATTGGTTTACCATTTGCTGCTTATTTAGGTTGGATGAATCGTAAAAAATGGAAACGTTTACCAGATAATATTCATCCATTAGGTGCTGTTTTTTTATTATTAGGTGCTGTTTTTTATTTATCTGGTGTTACTGAATGGGTTAATTTATCTTTACCAGTTATTTTAGTTGGTTTATGTTTATGGTTTAAAGGTATTTCTGGTTTACGTTCTCAAGGTTTTCCATTATTATTAGTTTTTTTAGCTACTCCAACTGCTTTACCATATTTAATTGCTCCATATACTTTACCATTACAATCTTTTATTGCTGGTACTGCTGGTTTTATTTTAAATCAATTTGGTATGGAAGTTACTGTTGATGAAATTAATTTATATGTTGGTGGTCGTATTGTTGAAGTTGCTCCATATTGTGCTGGTTTAAAAATGTTATTTCTACTTTATATGTTGGTTTAATGTTATTATATTGGACTGATGCTTTATCTTCTCGTCGTACTGTTATTTCTTTTTTTCTTTAGCTGCTATTGTTTCTATTATTGCTAATATTATTCGTAATACTTTATTAACTTTTTTTCATGGTACTGGTCAAGAAGCTGCTTTTAAATGGTTACATGATGGTTGGGGTGGTGATGTTTATTCTGCTTGTATGTTAGTTTCTTTAGTTCCATTATTAAATGGTATTAATTCTTATTTTTCTGCTTCTTTAGAAACTGAACAAGAAGGTGAATCT
I used the “Codon Optimization Tool” from eu.idtdna.com for this exercise and a custom organism table from www.kazusa.or.jp (Nostoc commune DRH1).
Codon optimisation is needed because different organisms prefer different DNA codons for the same amino acids. Even though multiple codons can code for the same amino acid, some are used more often in certain species. If the DNA sequence uses codons that are rare in the host organism, the protein may not be made efficiently. I optimised my sequence for Nostoc commune so that the codons better match what this bacterium naturally prefers, which should help improve protein expression.
ATGGTTTTGCAACAACAAATCAAGAACCGCAATGCTTCTGGTTTACTTAATTTAGCGATTCTAGGAGTTCTATTACTACTATATGCGCCTATTTTGCTTCATTGGTTGGATGGTTGGCTATATAAGAATATTTCCACAGAACACGAATACTTTAGTCACGGCATTATCGGTCTACCATTTGCGGCGTATCTTGGATGGATGAATCGTAAAAAATGGAAGCGCCTGCCCGATAATATCCACCCTCTGGGAGCGGTTTTTTTACTGTTAGGAGCTGTATTCTATCTTTCTGGTGTTACTGAATGGGTCAATCTTAGTCTGCCGGTCATCTTAGTTGGTTTGTGCCTGTGGTTCAAGGGTATTTCTGGTTTGCGATCTCAAGGTTTTCCTCTGCTATTGGTATTTTTGGCAACTCCAACTGCCTTGCCTTACCTGATCGCACCATACACATTGCCCTTACAATCTTTCATTGCAGGTACTGCGGGTTTTATCTTAAACCAATTCGGCATGGAAGTTACCGTCGATGAAATTAACCTATATGTTGGTGGTAGAATTGTGGAAGTAGCGCCATACTGTGCAGGTTTGAAAATGTTGTTCACTACATTATATGTGGGCCTTATGCTGCTATACTGGACAGACGCTCTGTCTTCTAGACGTACTGTCATTTCTTTTCTTTCCCTAGCTGCAATTGTATCTATTATTGCCAATATTATCCGTAACACTCTGTTGACCTTTTTCCACGGCACTGGTCAAGAAGCTGCGTTTAAATGGTTACATGATGGATGGGGTGGTGACGTGTACTCAGCTTGTATGTTAGTCTCATTGGTGCCATTACTGAACGGTATTAATTCTTACTTTTCAGCAAGTCTTGAAACAGAGCAAGAAGGTGAATCT
The following restriction enzyme sites have been found in the selected reading frame: AseI (ATTAAT) BbsI (GAAGAC) DraI (TTTAAA) MfeI (CAATTG) SspI (AATATT) XbaI (TCTAGA)
After designing and codon-optimising the DNA sequence, the gene could first be chemically synthesised using modern DNA synthesis technologies. The synthesised gene would then be assembled into a plasmid vector containing a promoter, ribosome-binding site, and terminator. This plasmid could be introduced into Nostoc commune using standard transformation techniques. Inside the cell, RNA polymerase transcribes the DNA into mRNA, and ribosomes translate the mRNA into Cyanoexosortase B according to the genetic code.
Alternatively, the DNA could be used in a cell-free transcription–translation system, where purified molecular machinery produces the protein outside of living cells. Both approaches rely on the same central dogma process: DNA is transcribed into RNA, and RNA is translated into protein.
I would like to sequence environmental DNA from biological soil crusts, with a focus on Nostoc commune and related cyanobacteria. The goal would be to identify genes involved in extracellular matrix production, stress tolerance, and surface adhesion. This would allow me to better understand which genetic pathways contribute to soil stabilisation and how microbial community composition changes under environmental stress.
To sequence environmental DNA from biological soil crust samples, I would use Illumina sequencing, potentially combined with Oxford Nanopore sequencing. Illumina provides high accuracy and depth for microbial community profiling, while Nanopore offers long reads that help resolve full-length genes and structural variation.
Illumina is a second-generation (next-generation) sequencing technology. It performs massively parallel sequencing of short DNA fragments, enabling the simultaneous reading of millions of sequences. Oxford Nanopore is considered a third-generation sequencing technology, as it sequences single DNA molecules in real-time without the need for amplification.
The input for Illumina sequencing would be environmental DNA extracted from soil crust samples. The DNA is prepared by adding sequencing adapters and amplifying the fragments to create a sequencing library. During sequencing, the DNA binds to a flow cell and is copied while fluorescently labelled nucleotides are incorporated one base at a time. Each incorporated base is detected by its fluorescence signal and converted into a nucleotide sequence. The output consists of millions of short DNA reads that can be analysed to identify genes and microbial composition.
I would synthesise a codon-optimised version of the Cyanoexosortase B (CrtB) gene from Nostoc commune. The goal would be to study and potentially enhance extracellular matrix organisation in cyanobacteria, which is relevant to soil stabilisation and biofilm formation.
To synthesise the CrtB gene, I would use phosphoramidite-based oligonucleotide synthesis, followed by gene assembly.
The essential steps include stepwise chemical addition of protected nucleotides to build short oligonucleotides on a solid support, cleavage and deprotection, purification, and assembly of overlapping oligos into a full-length gene using PCR-based methods. The assembled gene can then be sequence-verified before use.
The main limitations of this method are synthesis errors that accumulate with increasing length, practical limits on direct synthesis length (typically ~200 nt per oligo), and the need for assembly and error correction for longer constructs. Although scalable and highly automated, accuracy and yield decrease as sequence length increases.
Beaucage, S.L. and Caruthers, M.H., 1981. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Letters, 22(20), pp.1859–1862.
Gibson, D.G., Young, L., Chuang, R.-Y., Venter, J.C., Hutchison, C.A. and Smith, H.O., 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods, 6, pp.343–345.
Kosuri, S. and Church, G.M., 2014. Large-scale de novo DNA synthesis: Technologies and applications. Nature Methods, 11(5), pp.499–507.
LeProust, E.M., Peck, B.J., Spirin, K., McCuen, H.B., Moore, B., Namsaraev, E. and Caruthers, M.H., 2010. Synthesis of high-quality libraries of long (150-mer) oligonucleotides by a novel depurination-controlled process. Nucleic Acids Research, 38(8), pp.2522–2540.
I would focus on editing regulatory regions of genes involved in extracellular matrix production in Nostoc commune, rather than altering core metabolic pathways. For example, modifying promoter regions could allow increased or more controlled expression of genes associated with surface adhesion or polysaccharide production. The goal would be to fine-tune existing biological functions rather than introduce foreign traits, maintaining ecological compatibility.
I would use CRISPR-Cas9 to edit regulatory regions of the Nostoc commune genome. CRISPR-Cas9 uses a guide RNA to direct the Cas9 nuclease to a specific DNA sequence, where it introduces a double-strand break. The cell then repairs the break either through non-homologous end joining, which can introduce small mutations, or through homology-directed repair if a donor template is supplied.
The preparation includes designing a guide RNA targeting the promoter region, constructing a plasmid carrying Cas9 and the guide RNA, and introducing it into the host cells. If precise edits are required, a repair template DNA is also provided.
Limitations include variable editing efficiency, dependence on cellular repair pathways, and potential off-target effects where unintended DNA regions may be modified.
Cong, L. et al., 2013. Multiplex genome engineering using CRISPR/Cas systems. Science, 339(6121), pp.819–823.
Jinek, M. et al., 2012. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), pp.816–821.
Doudna, J.A. and Charpentier, E., 2014. The new frontier of genome engineering with CRISPR-Cas9. Science, 346(6213), 1258096.
I want to develop a site-attuned microbial engineering approach to accelerate the formation of biological soil crusts (biocrusts) in erosion-prone or degraded soils, to reduce topsoil loss and support early plant establishment. The core biological intervention is a minimal, targeted enhancement of extracellular polymeric substance (EPS) production within site-derived or ecologically matched microbial consortia.
BSCs play a critical role in stabilising soil surfaces, binding particles, retaining moisture at the microscale, and creating conditions that allow plants to establish. However, in many degraded environments, natural biocrust formation is slow and fragile, leaving soils vulnerable to wind and water erosion. Over the past year, I have explored material and process-based strategies (such as surface carriers, textures, and hydration regimes) to support crust formation, but these approaches have shown limited and inconsistent success. This suggests that a key bottleneck lies in the rate and robustness of microbial aggregation, rather than the absence of microbes or materials themselves.
EPS is central to soil particle aggregation and early crust cohesion. By modestly increasing EPS production in microbial consortia that are native to, or in this case, well-matched with a given site, this project aims to accelerate an existing ecological function rather than introduce a new one. Genetic modification is therefore used not to create novel traits or new ecological roles, but to tune a single, well-understood mechanism that directly contributes to soil stability.
The application is designed to fit within a circular economy framework, using biodegradable carriers and locally appropriate inputs that safely reintegrate into the soil system. Ultimately, the goal is to develop a tool that improves soil resilience and plant establishment while remaining environmentally compatible, reversible, and ethically governable.
Ensure that the system does not cause unintended biological, ecological, or social harm.
1.1 Prevent biological misuse or diversion
Ensure that organisms, genetic constructs, and protocols cannot be easily repurposed for harmful or unintended applications.
1.2 Minimise accidental harm
Reduce risks associated with lab work, handling, transport, and early deployment through clear safety practices, documentation, and incident reporting norms.
Ensure that the intervention supports soil function without destabilising local ecosystems or creating irreversible environmental effects.
2.1 Preserve existing ecological roles
Ensure that genetic modification enhances an existing microbial function (EPS production for aggregation) rather than introducing new ecological roles or behaviours.
2.2 Constrain spatial and temporal impact
Limit how long and how far the modified microbes can exert their enhanced function, reducing the risk of unintended persistence or spread beyond the target site.
2.3 Detect and respond to unintended effects
Establish monitoring and response mechanisms to identify ecological changes early and enable corrective action or remediation if adverse effects emerge.
Ensure that synthetic biology is applied only where necessary, at an appropriate level of intervention, and with explicit recognition of its added ethical and governance burden.
3.1 Necessity-based justification
Require clear evidence that genetic modification is addressing a specific, well-defined functional limitation (e.g., insufficient EPS-mediated aggregation) that cannot be adequately resolved through non-genetic approaches alone.
3.2 Scope and intensity limitation
Constrain genetic modification to the minimum effective change, focused on a single functional trait, and avoid escalating intervention complexity without renewed justification.
3.3 Ongoing re-evaluation
Periodically reassess whether genetic modification remains justified as materials, ecological understanding, or any alternative methods improve, rather than treating engineering choices as permanent.
Ensure that responsibility for the development and deployment of the system is clearly assigned, traceable, and sustained over time, rather than diffused across actors or stages of work.
4.1 Clear attribution of responsibility
Define who is responsible for organism sourcing, genetic modification decisions, deployment oversight, and post-deployment monitoring at each stage of the project’s LC.
4.2 Documentation and traceability
Maintain accessible records of design decisions, genetic modifications, deployment contexts, and monitoring plans so actions and outcomes can be reconstructed if concerns arise.
4.3 Knowledge stewardship
Ensure that insights gained from development and deployment, including negative or inconclusive results, are preserved and shared in ways that support collective learning rather than isolated success narratives.
Ensure that decisions about development and deployment respect local agency and that the benefits and risks of technology are not unfairly distributed.
5.1 Local decision-making authority
Ensure that land stewards and affected communities have meaningful influence over whether, where, and how the system is tested or deployed, rather than being treated solely as sites of experimentation.
5.2 Fair allocation of risk and benefit
Avoid situations in which environmental or social risks are borne locally while scientific, economic, or reputational benefits accrue primarily to external actors.
5.3 Accessibility and non-exclusivity
Design governance and deployment pathways that do not restrict participation or benefit to well-resourced institutions, regions, or communities.
To support the ethical development and potential deployment of an EPS-enhanced, site-derived microbial system, I propose three complementary governance actions. Together, these combine design constraints, regulatory staging, and incentive-based oversight and involve different actors across the research, deployment, and accountability landscape.
Purpose
Environmental synthetic biology often relies on general biosafety frameworks not tailored to open ecosystems. This action introduces a formal safety-by-design requirement that explicitly constrains the use of genetic modification in soil-interacting systems.
Design
Actors: Researchers, university biosafety committees, funders, and journals.
Mechanisms:
Analogy: Privacy-by-design or chemical safety-by-design frameworks.
Assumptions
Early design constraints reduce downstream risk and are adopted when tied to funding and publication norms.
Risk of failure and success
Failure: Box-ticking compliance with weak enforcement.
Success risk: Compliance burden disadvantages smaller research groups.
(Regulatory staging; regulators and local governance)
Purpose
Transitions from lab to environment are currently inconsistent. This action introduces a tiered pathway where oversight scales with ecological exposure and deployment size.
Design
Actors: Environmental regulators, land managers, research institutions.
Mechanisms:
Analogy: Phased clinical trials or drone flight regulation.
Assumptions
Regulators have sufficient capacity, and researchers will not bypass formal pathways.
Risk of failure and success
Failure: Delays lead to work being pushed into informal channels.
Success risk: Overly rigid processes slow low-risk research.
(Incentive-based governance; funders, journals, institutions)
Purpose
Accountability often weakens after laboratory development. This action extends responsibility into real-world contexts through traceability and monitoring incentives.
Design
Actors: Funders, journals, universities, standards bodies.
Mechanisms:
Analogy: Post-market surveillance in medical devices or financial reporting requirements.
Assumptions
Incentives can encourage compliance without heavy enforcement.
Risks of failure and success
Failure: Low participation or superficial monitoring.
Success risk: Administrative complexity favours large institutions.
| Policy goal / consideration | Safety-by-design | Tiered pathway | Traceability & incentives |
|---|---|---|---|
| Enhance biosecurity – Prevent incidents | 2 | 2 | 2 |
| Enhance biosecurity – Help respond | 2 | 2 | 1 |
| Foster lab safety – Prevent incidents | 1 | 2 | 2 |
| Foster lab safety – Help respond | 2 | 2 | 1 |
| Protect the environment – Prevent incidents | 2 | 1 | 2 |
| Protect the environment – Help respond | 2 | 2 | 1 |
| Minimise costs/burdens | 2 | 3 | 2 |
| Feasibility | 2 | 2–3 | 2 |
| Not impede research | 2 | 3 | 2 |
| Promote constructive applications | 1–2 | 2 | 2 |
Rationale (brief):
Based on the scoring, I would prioritise Option 1 (safety-by-design requirements) and Option 3 (traceability and post-deployment monitoring incentives), with Option 2 (tiered field testing) applied only to higher-risk or larger-scale deployments. Constraining genetic modification at the design stage is the most effective way to prevent harm, while traceability and monitoring ensure accountability and learning once systems interact with real environments. Although tiered regulatory pathways are important as ecological exposure increases, applying them universally would risk slowing low-risk research. This approach balances innovation with restraint and is best implemented by MIT research leadership and funding bodies through grant conditions and review processes, while acknowledging ongoing uncertainty around long-term ecological effects.
From this week’s slides, DNA polymerase is highly efficient but not perfectly accurate. Its intrinsic error rate is approximately 1 error per 10⁵ nucleotides incorporated. When compared to the size of the human genome (~3 × 10⁹ base pairs), replication at this fidelity alone would introduce an unsustainable number of mutations during each cell division. Biology resolves this discrepancy through a layered system of error correction. Replicative polymerases perform 3′→5′ proofreading, substantially lowering the error rate. This is followed by post-replicative mismatch repair, which further reduces errors to approximately 10⁻⁹–10⁻¹⁰ per nucleotide per division. Together, these mechanisms allow genomes as large as the human genome to be copied reliably, while still permitting rare mutations necessary for evolution. (Kunkel, 2004; Loeb and Monnat, 2008)
Proteins are encoded using triplet codons, with 61 sense codons specifying 20 amino acids. For an average human protein of roughly 300 amino acids, this implies an astronomically large number of possible DNA sequences, on the order of 61³⁰⁰, that could encode the same amino acid sequence. In practice, however, only a small subset functions effectively. Synonymous codons are not functionally equivalent due to several constraints:
As a result, although the genetic code is formally redundant, functional coding space is highly constrained by cellular, regulatory, and biophysical factors.(Plotkin and Kudla, 2011; Zhou et al., 2009)
The most widely used method for oligonucleotide synthesis is solid-phase phosphoramidite chemistry. DNA is synthesised one nucleotide at a time on a solid support through a cyclic process involving coupling, capping, oxidation, and deprotection. This method remains the industry standard for producing short DNA oligos due to its automation, reliability, and scalability.
Each coupling step in phosphoramidite synthesis is efficient but not perfect (~99–99.5%). Errors accumulate multiplicatively with length, causing the fraction of full-length, error-free molecules to drop rapidly as oligos get longer. Longer syntheses are also affected by side reactions such as depurination, incomplete deprotection, and strand truncation. By ~200 nucleotides, the yield of correct sequences becomes very low relative to truncated or error-containing products, making purification inefficient and costly.
A 2000 bp gene would require thousands of sequential coupling steps. Even with high per-step efficiency, the cumulative probability of producing a completely correct sequence becomes vanishingly small. Instead, long DNA sequences are constructed by assembling shorter oligos (60–200 nt) using enzymatic methods such as PCR-based assembly or homologous recombination, which leverage biological error correction and amplification.(Beaucage and Caruthers, 1981)
Animals lack the metabolic pathways required to synthesise certain amino acids and therefore depend on external sources. The essential amino acids generally recognised across animals are:
These amino acids represent fixed biological constraints. No genetic optimisation removes the requirement for their external supply. Viewed through this lens, the lysine contingency becomes structural rather than speculative. Lysine is essential, required in relatively large amounts, and cannot be synthesised by animals. Control over lysine availability directly influences growth and viability. At the same time, recognising lysine as part of a broader pattern reframes contingency as embedded dependency. Synthetic biology often works by formalising such dependencies rather than creating entirely new ones. This raises questions about power, access, and governance, specifically who defines and controls the availability of essential resources.(Wu, 2009; Young and Pellett, 1994)
Kunkel, T.A. (2004). DNA replication fidelity. Journal of Biological Chemistry, 279(17), pp.16895–16898.
Loeb, L.A. and Monnat, R.J. (2008). DNA polymerase fidelity and human disease. Nature Reviews Genetics, 9(8), pp.594–604.
Plotkin, J.B. and Kudla, G. (2011). Synonymous but not the same: the causes and consequences of codon bias. Nature Reviews Genetics, 12(1), pp.32–42.
Zhou, M., et al. (2009). Non-optimal codon usage affects expression, structure, and function of proteins. Biochemical Society Transactions, 37(2), pp.417–421.
Beaucage, S.L. and Caruthers, M.H. (1981). Deoxynucleoside phosphoramidites. Tetrahedron Letters, 22(20), pp.1859–1862.
Wu, G. (2009). Amino acids: metabolism, functions, and nutrition. Amino Acids, 37(1), pp.1–17.
Young, V.R. and Pellett, P.L. (1994). Plant proteins in relation to human protein and amino acid nutrition. American Journal of Clinical Nutrition, 59(5), pp.1203S–1212S.