Week 02 HW: DNA Read, Write, & Edit
‘Week 2 — DNA Read, Write, & Edit’
Documentation
Make sure to document every step of the in-silico and lab experiments. Make sketches, screenshots, notes, drawings… anything that helps you - and others - understand the experiment.
Your documentation should help you - and others - to understand the topic. Don’t be afraid to add things that don’t work. Show your failures - and how you overcame them. Your Documentation should be a description of the amazing journey you are on!
Part 0: Basics of Gel Electrophoresis
Attend or watch all lecture and recitation videos. Optionally watch bootcamp.
DONE
Part 1: Benchling & In-silico Gel Art
See this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis()” for details. Overview:

Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
CL: Optional
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose. (自分が興味深いと思うタンパク質を1つ選んで、その理由を説明し、NCBI・UniProt・Googleなどのツールを使ってそのタンパク質配列を取得してください)
[Example from our group homework, you may notice the particular format — The example below came from UniProt]
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
Luciferase refers to a group of oxidative enzymes that produce bioluminescence and are composed of many protein groups that differ genetically. These enzymes are named “luciferase” and are found in many organisms, including fireflies and marine animals, where they are involved in bioluminescent phenomena. They are classified as oxidoreductases (EC 1.13.12.-) and catalyze reactions that incorporate molecular oxygen.
Basic Information about Firefly Luciferase:
Basic Characteristics:
Species: Mainly studied in Photinus pyralis (common eastern firefly)
Enzyme Class: Classified as oxidoreductase (EC 1.13.12.7)
Function: Reacts with the substrate luciferin and oxygen to produce light
Mechanism:
- Luciferase binds to luciferin.
- Luciferin is oxidized at the enzyme’s active site, forming oxidized luciferin (oxyluciferin)
- Oxidized luciferin dissociates, releasing energy and producing light (luminescence)
Structure and Data:
PDB: 1LCI (three-dimensional structure data of luciferase)
UniProt: P08659 (protein sequence information)
https://en.wikipedia.org/wiki/Luciferase
Basic Information about Firefly Luciferase
Formal Name: Photinus-luciferin 4-monooxygenase (ATP-hydrolysing)
Common Name: Firefly luciferase
Function: An oxidoreductase enzyme that catalyzes chemiluminescence in fireflies.
Firefly Light Control Mechanism
Light Control: Firefly light flickers, and it has been proposed that this flashing is controlled by nitric oxide (NO)
NO Production:
- Nitric oxide synthase (NOS), located between the nerve endings and light-emitting cells, produces NO
- The produced NO inhibits the activity of cytochrome c oxidase within the mitochondria of the light-emitting cells
Increase in Oxygen Levels:
- Inhibition of cytochrome c oxidase activity increases the oxygen concentration inside the peroxisomes where luciferase is localized
- The increased oxygen concentration in the peroxisomes promotes the light-emitting reaction and directly contributes to the flickering of the firefly’s light
Reaction Mechanism of Firefly Luciferase
First Stage:
- The carboxyl group of luciferin attacks the α-phosphate site of ATP, forming the luciferyl AMP intermediate within the enzyme
Second Stage:
- After the enzyme reacts with the luciferyl AMP intermediate, excited oxyluciferin is produced
- As the excited oxyluciferin returns to the ground state, it releases energy as yellow-green light
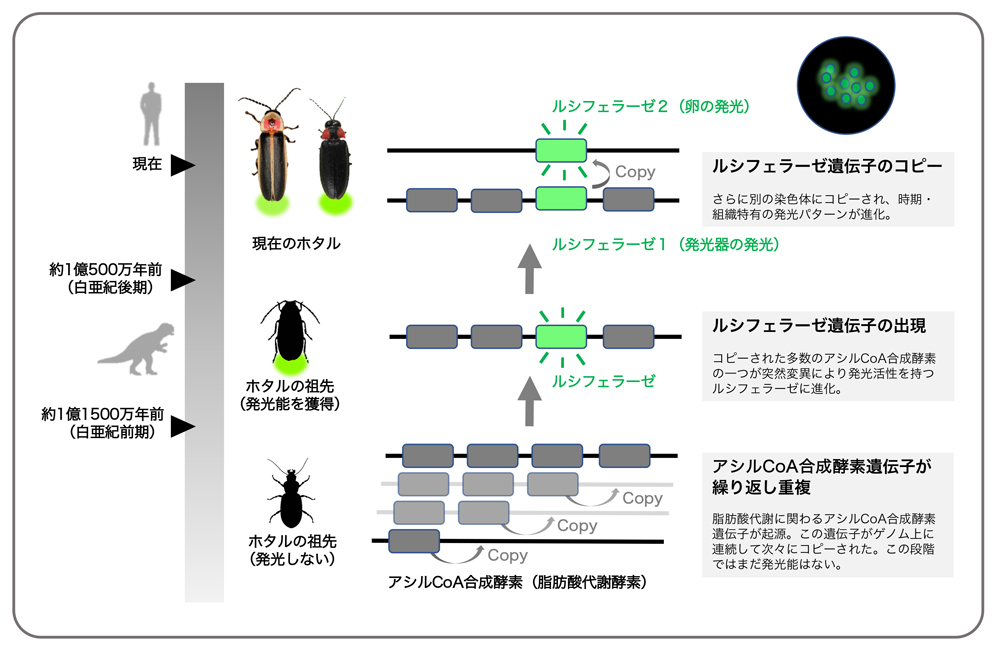 [https://www.nibb.ac.jp/press/2018/10/16.html]
[https://www.nibb.ac.jp/press/2018/10/16.html]UniProt: Luciferin 4-monooxygenase (P08659)
[https://www.uniprot.org/uniprotkb/P08659/entry]
The Protein Sequence of Luciferin 4-monooxygenase
[https://rest.uniprot.org/uniprotkb/P08659.fasta]
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
[Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI]
Lysis protein DNA sequence atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
DNA Sequence of Luciferin 4-monooxygenase
According to the NCBI website:
https://www.ncbi.nlm.nih.gov/nuccore/NW_022170249.1?report=fasta&from=28340362&to=28342515
Photinus pyralis isolate 1611_PpyrPB1 unplaced genomic scaffold, Ppyr1.3 Ppyr1.4_LG1, whole genome shotgun sequence NCBI Reference Sequence: NW_022170249.1
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
[Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI]
Lysis protein DNA sequence with Codon-Optimization ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
https://www.vectorbuilder.jp/tool/codon-optimization.html
We have optimized the codons for efficient protein expression of Luciferin 4-monooxygenase in a specific host organism. For this time, human cells were chosen as the host. Codon optimization is necessary to align with the abundance of the host’s tRNA. Optimized codons enhance the stability of mRNA and facilitate smoother translation processes.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
https://benchling.com/s/seq-wAvxKb4XH5gFiaRWyoN1?m=slm-x4jcGnqtraY7xJvhX2gz
- 適切なプライマーを設計する
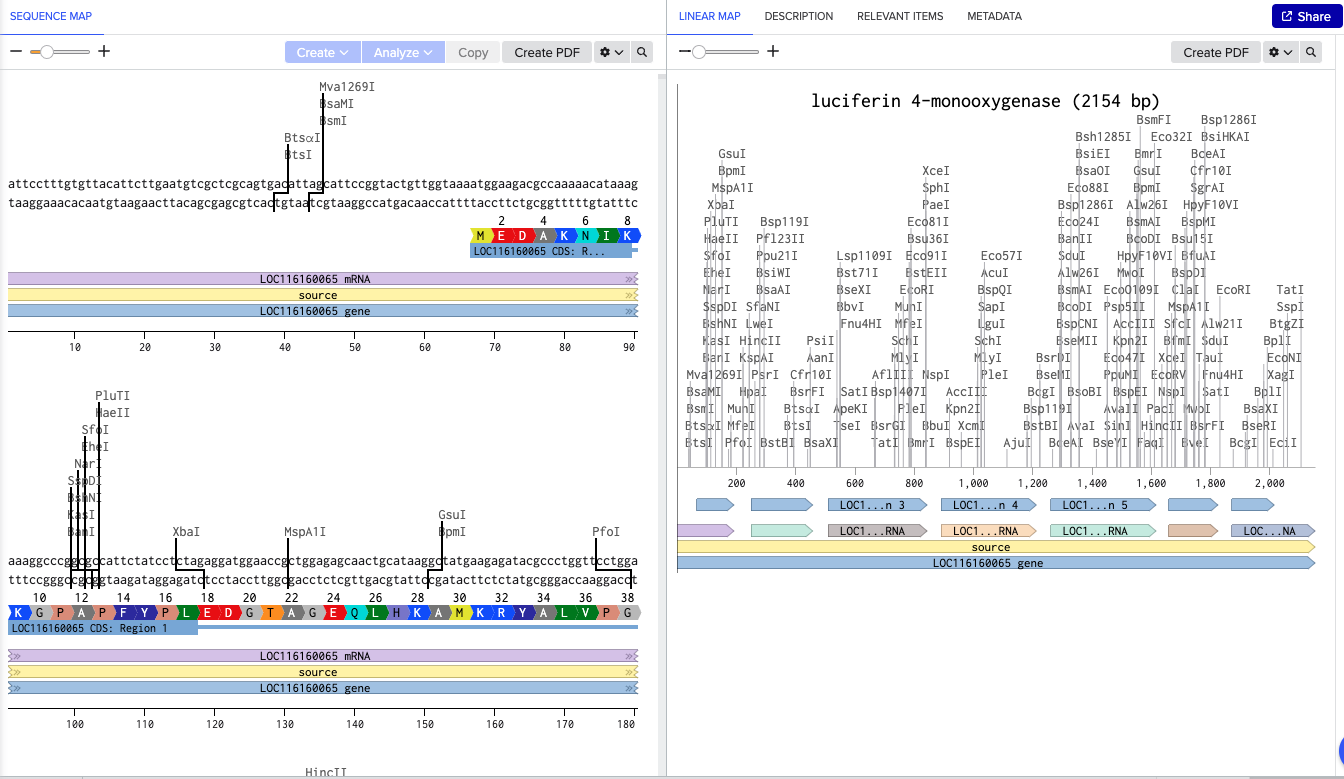
まず、Photinus-luciferin 4-monooxygenase の先端にあるBtsⅠを制限酵素を用いたPrimerとして新規で設計する

https://rebase.neb.com/rebase/enz/BtsI.html
BtsⅠのカットサイト「gcagtg」だが、長さを18~25bpに設計する必要があるため、「gcagtg」の前後10塩基づつ長くする
「gcagtg」の頭「g」が31にあるため、13~43の塩基を使用する
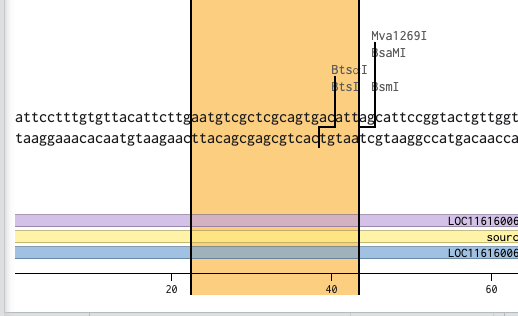
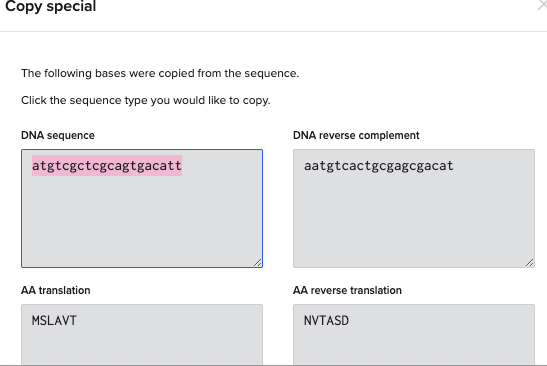
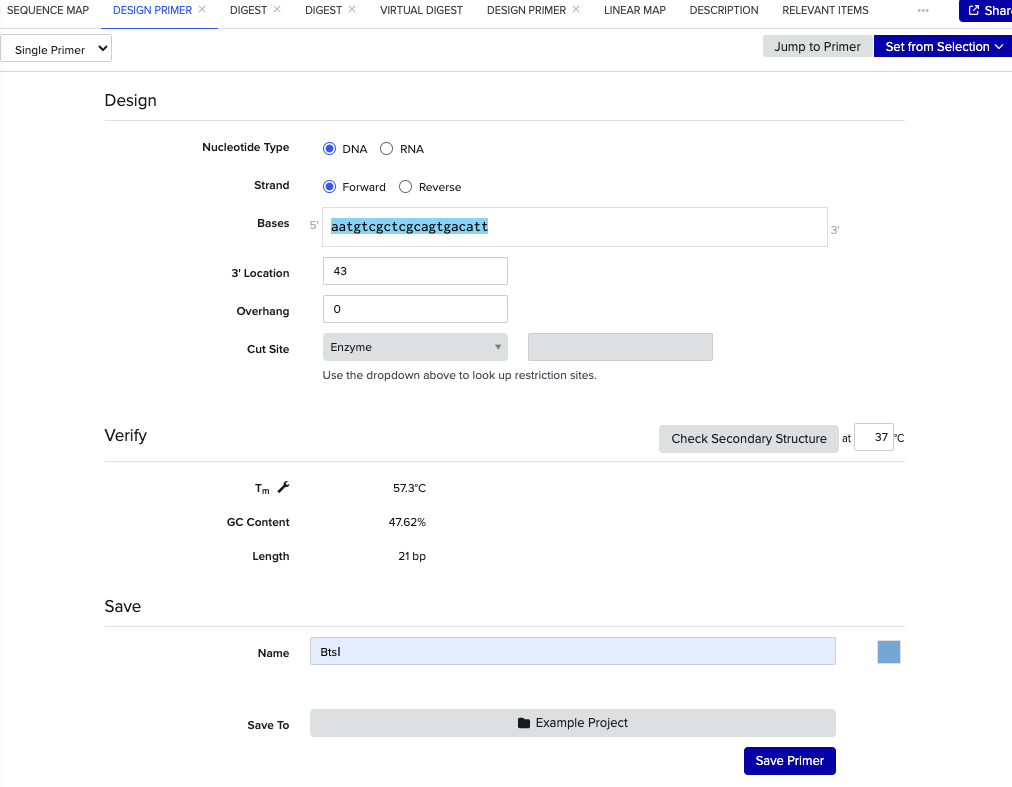
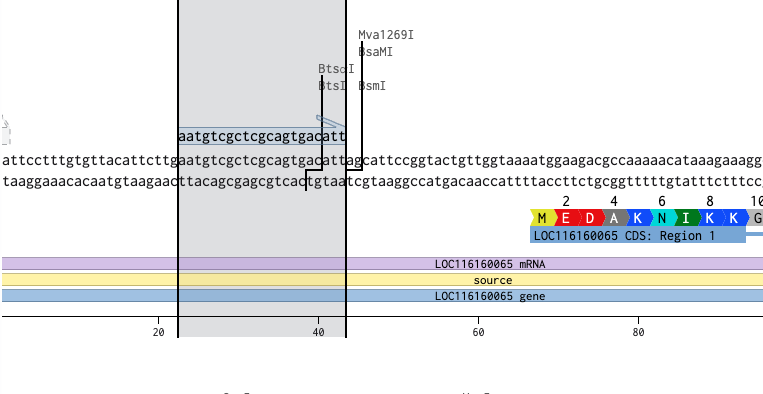
これでプライマーのフォワードプライマーができたので同様にリバースプライマーを設計する。リバースプライマーは2108にいるTatⅠを使用する。

リバースにして、3’ Locationの場所を最後の2113にしたが
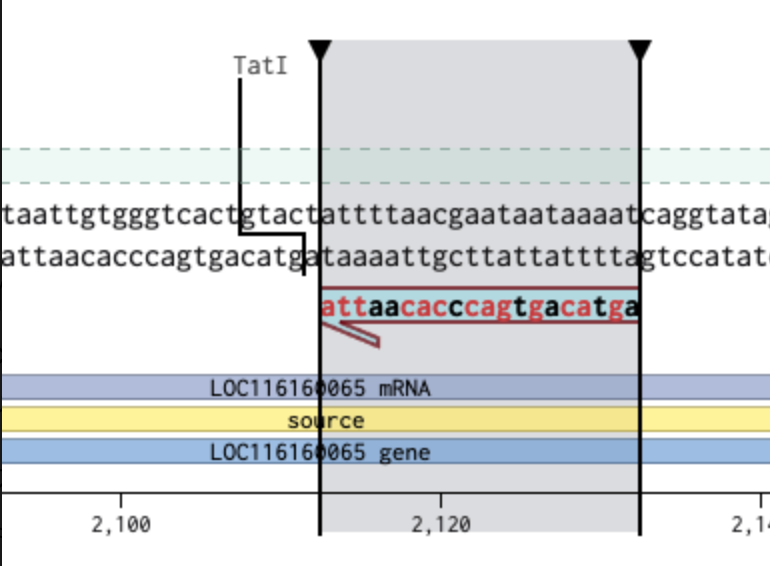
ずれた。そこで、3’ Locationを想定の頭のサイト(2093)に変更したところ…
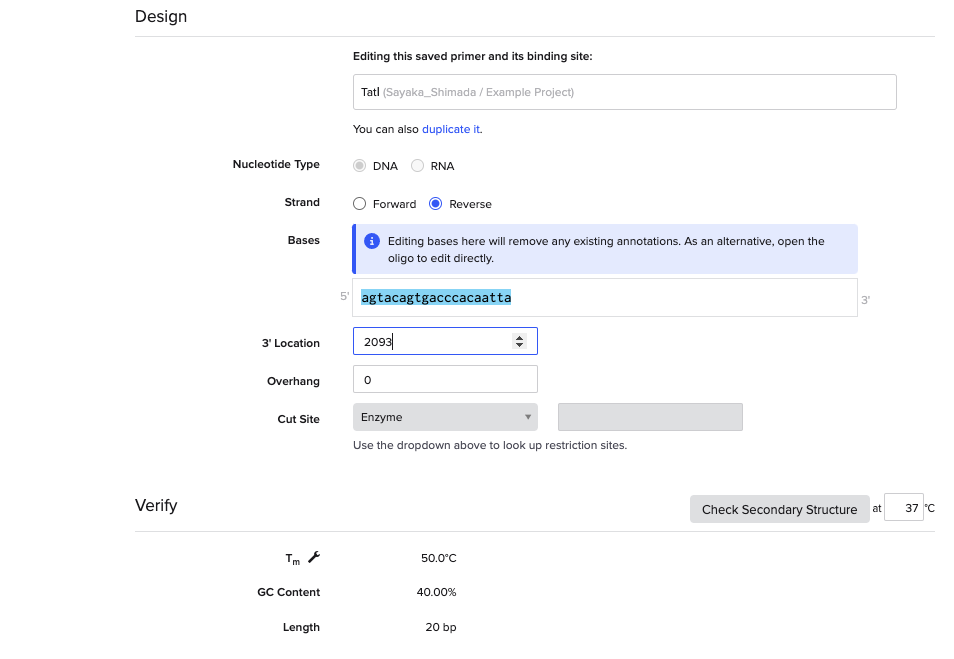

成功した。
- プライマーを使用してPCRを行い、増幅したDNAの断片を生成する。特定のDNAシーケンスを大量に複製する
- ベクターに挿入する。PCRによって増幅したDNA断片を制限酵素サイトを使ってベクターに挿入する。プラスミドが形成される。
3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level. Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below. [Example shows the biomolecular flow in central dogma from DNA to RNA to Protein] Special note that all “T” were transcribed into “U” and that the 3-nt codon represents 1-AA.
NA
Part 4: Prepare a Twist DNA Synthesis Order
This is a practice exercise, not necessarily your real Twist order!
4.1. Create a Twist account and a Benchling account Yes
4.2. Build Your DNA Insert Sequence For example, let’s make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein):
In Benchling, select New DNA/RNA sequence
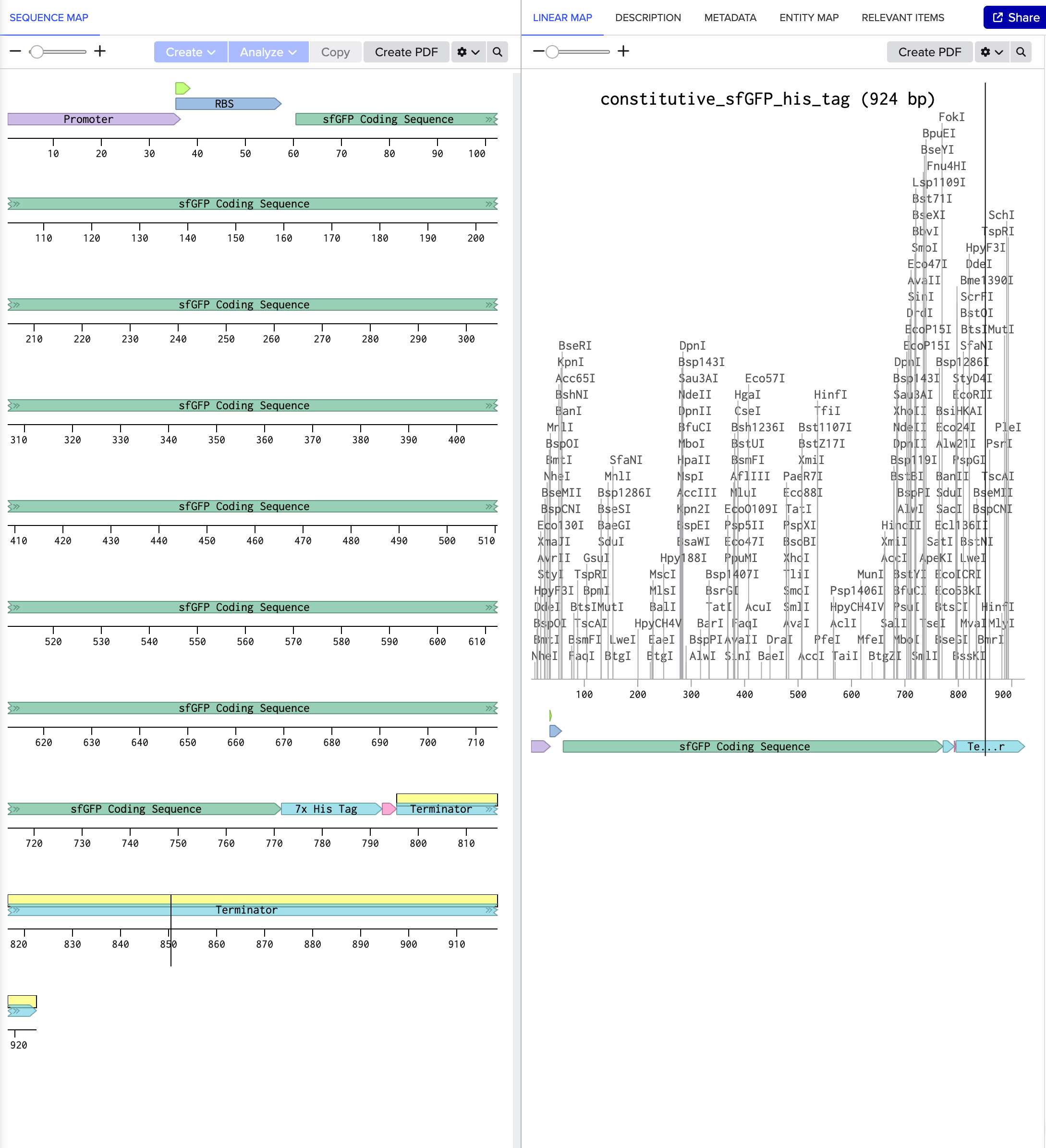
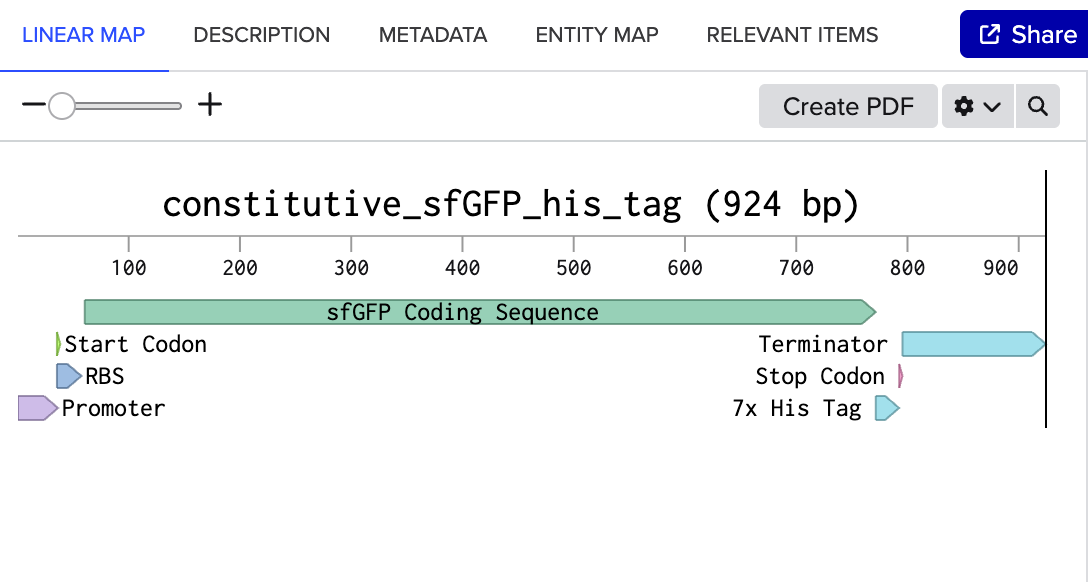
[https://benchling.com/s/seq-WzXC0VtZQgv3NjmBV9eD?m=slm-lDALmve76xdyPzXXyvGW]
4.3. On Twist, Select The “Genes” Option
yes
4.4. Select “Clonal Genes” option
For this exercise, I selected Clonal Genes.
4.5. Import your sequence
yes
4.6. Choose Your Vector
sfGFPのfastaデータ constitutive_sfGFP_his_tag.fasta
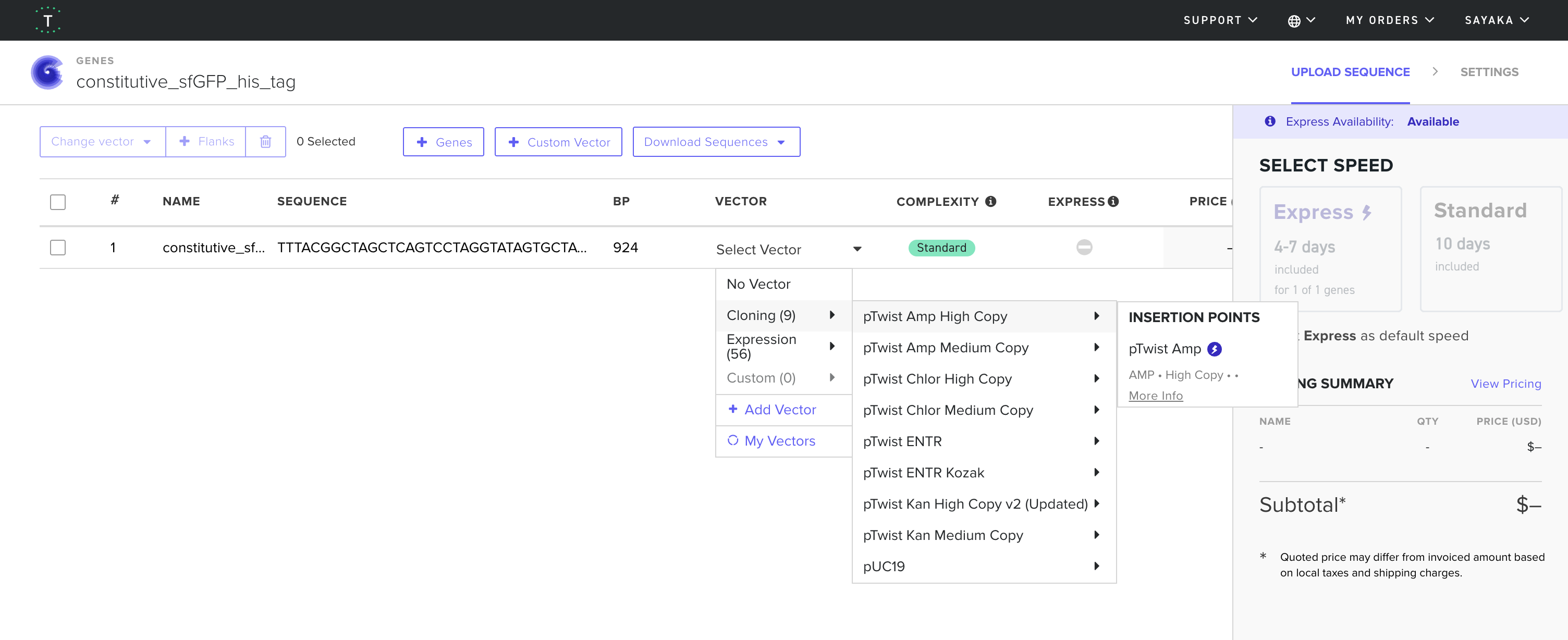
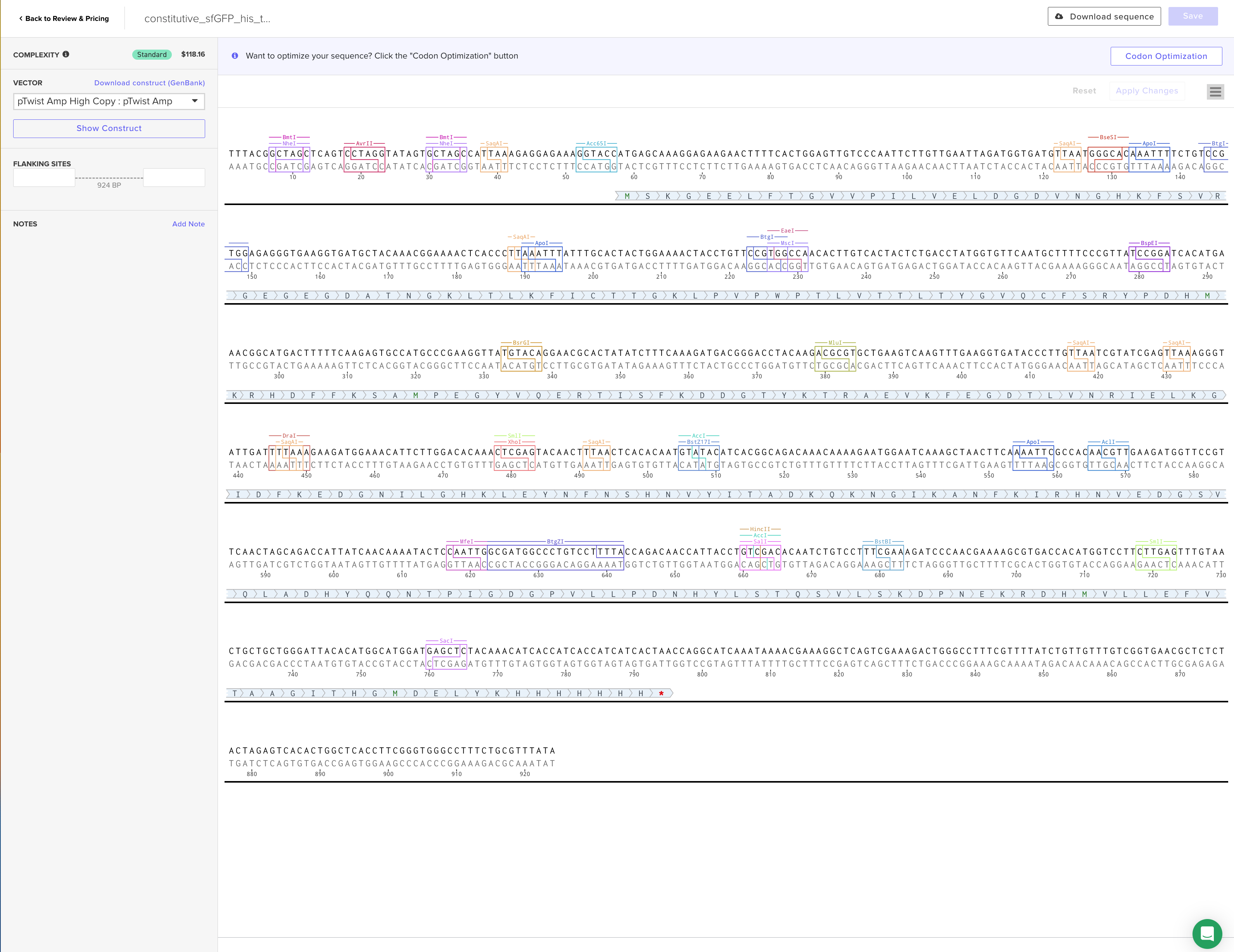
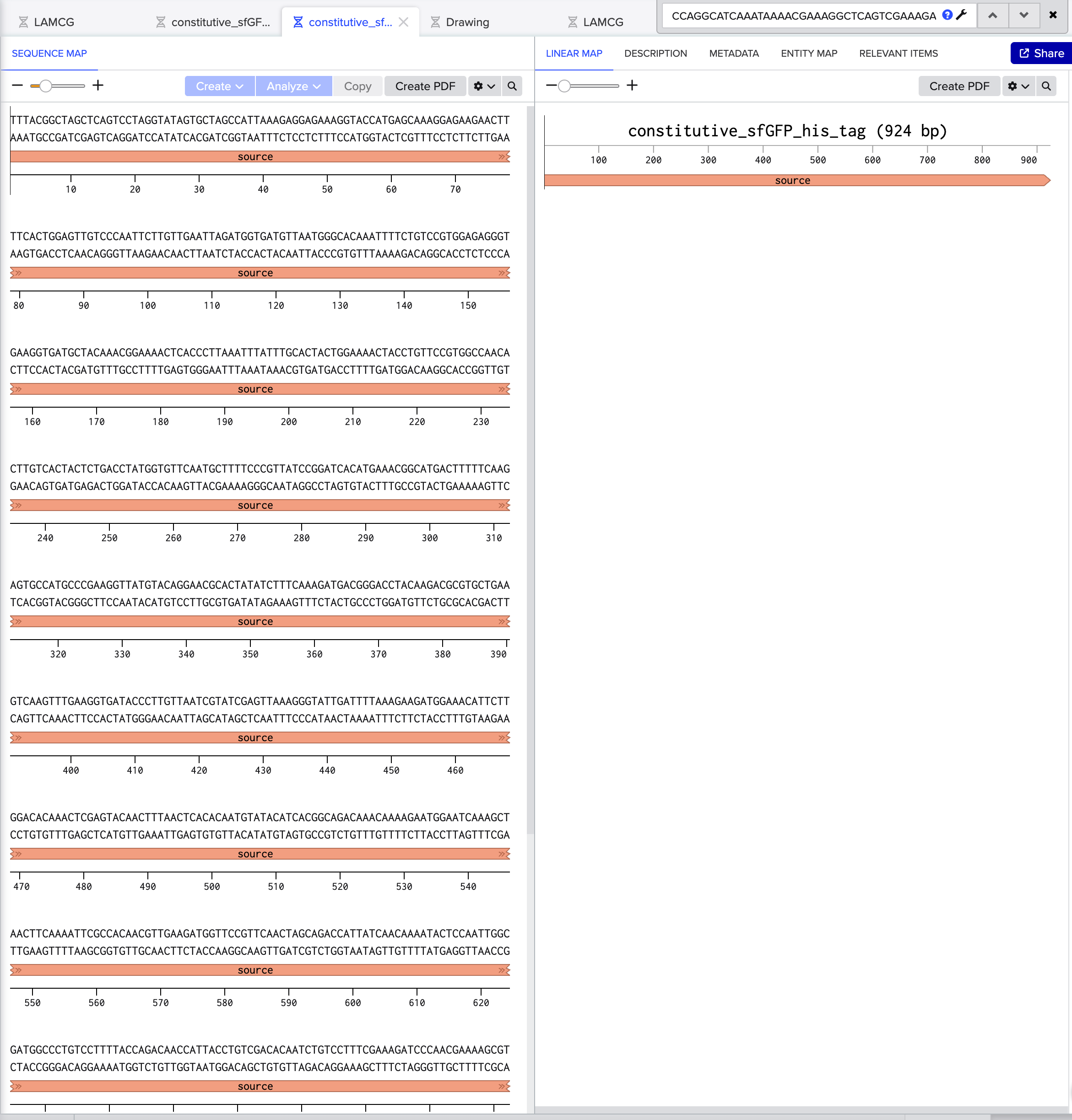
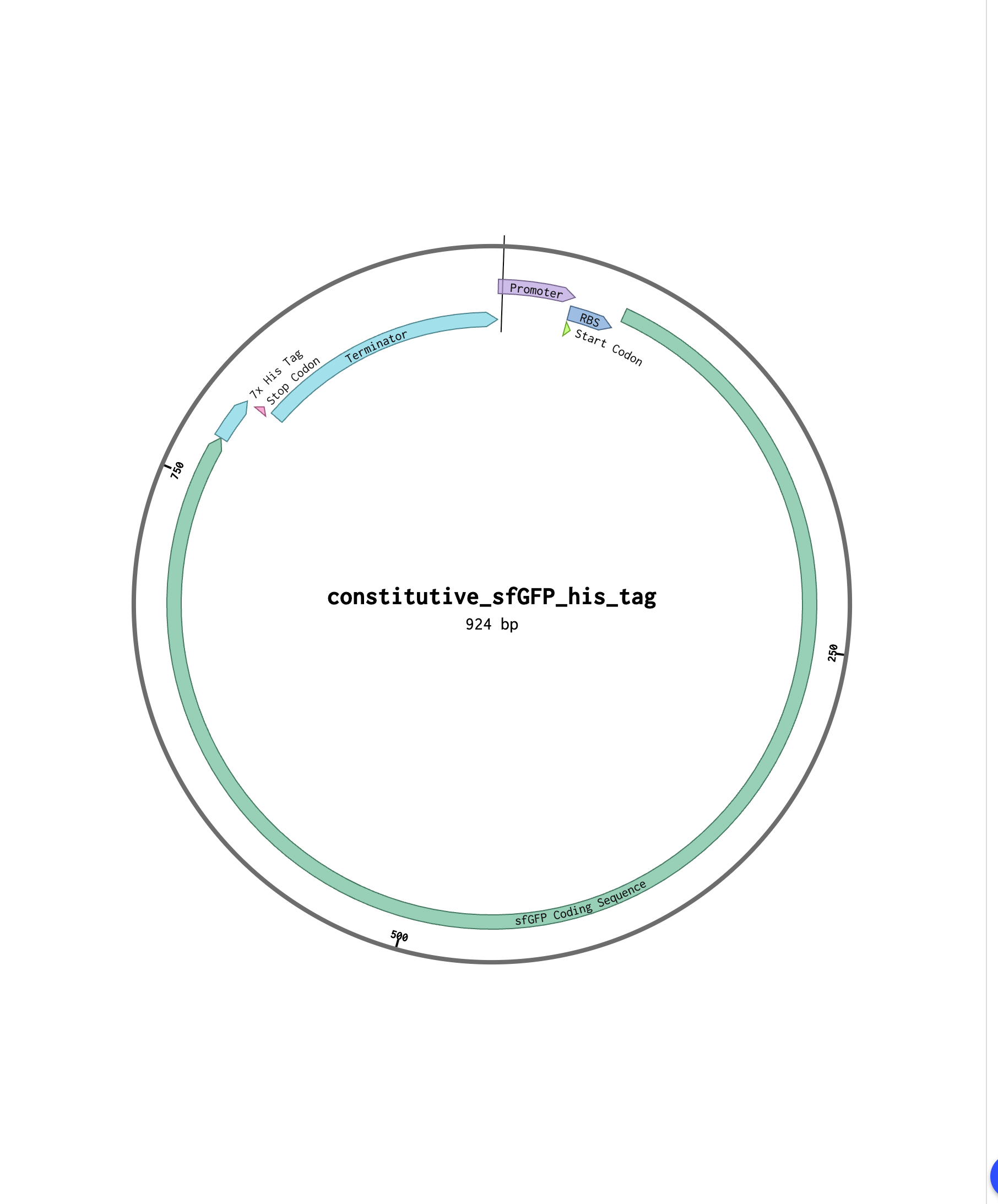
Part 5: DNA Read/Write/Edit
5.1 DNA Read
I am interested in sequencing the DNA of organisms and bacteria that thrive under extreme or specialized conditions, such as those capable of converting CO₂ into O₂ or withstanding intense heat, as they may be relevant to my “firework art” project aimed at improving the environment
- CO₂-to-O₂ Converting Bacteria
- Cyanobacteria, for example, perform photosynthesis by fixing CO₂ and releasing O₂
- Understanding their genes and metabolic pathways could pave the way for applications in environmental cleanup or carbon reduction
- Heat-Resistant Microorganisms
- Some bacteria found in volcanic areas or hot springs are highly thermotolerant
- Sequencing their genomes may reveal genes that produce heat-stable enzymes or proteins, which could be applied to create new materials or sustainable technologies—even in high-temperature settings like firework art
By analyzing the DNA of these organisms, I hope to discover novel approaches or materials that can contribute to environmental improvement and ultimately realize an eco-friendly “firework art”
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
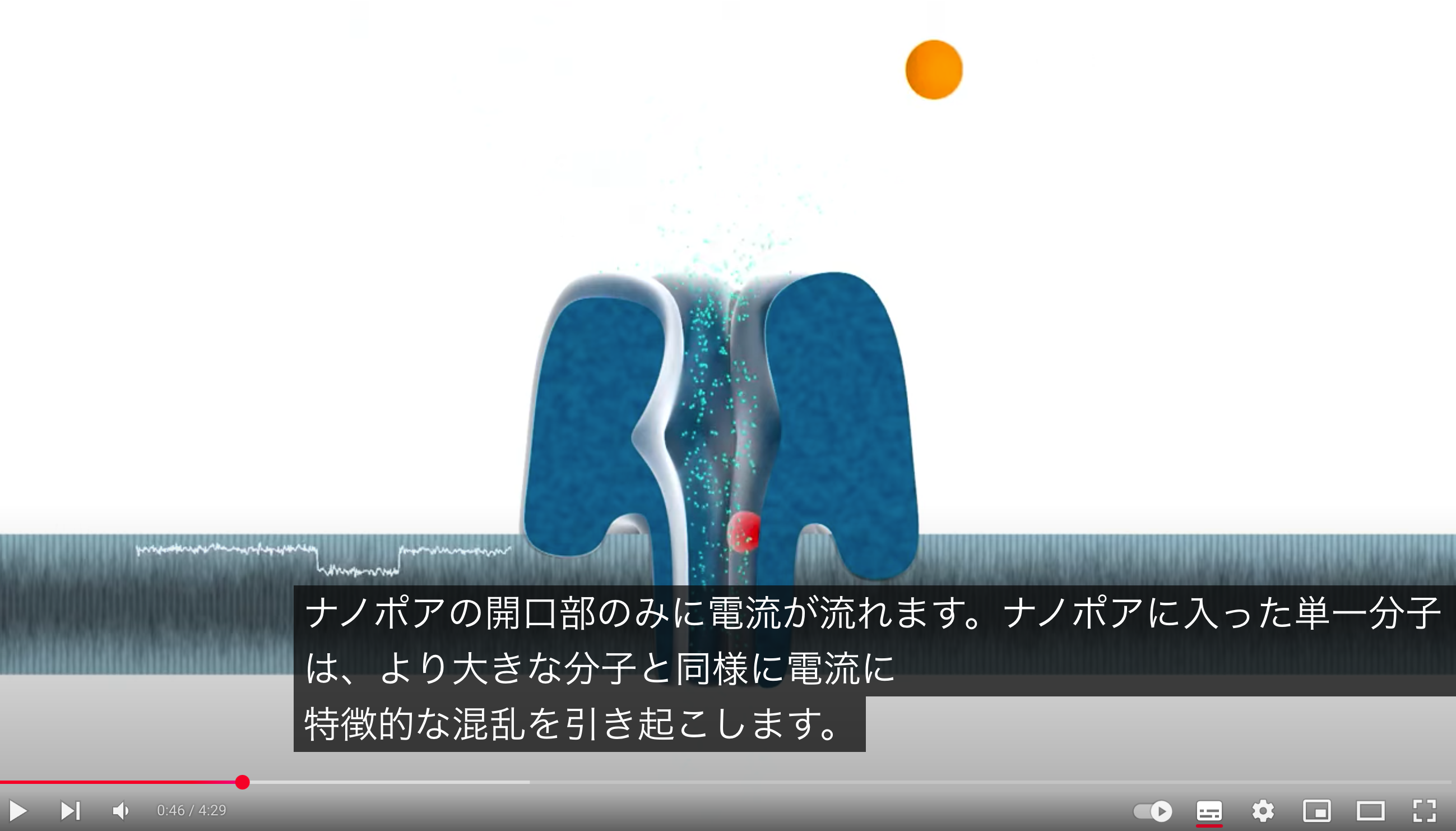
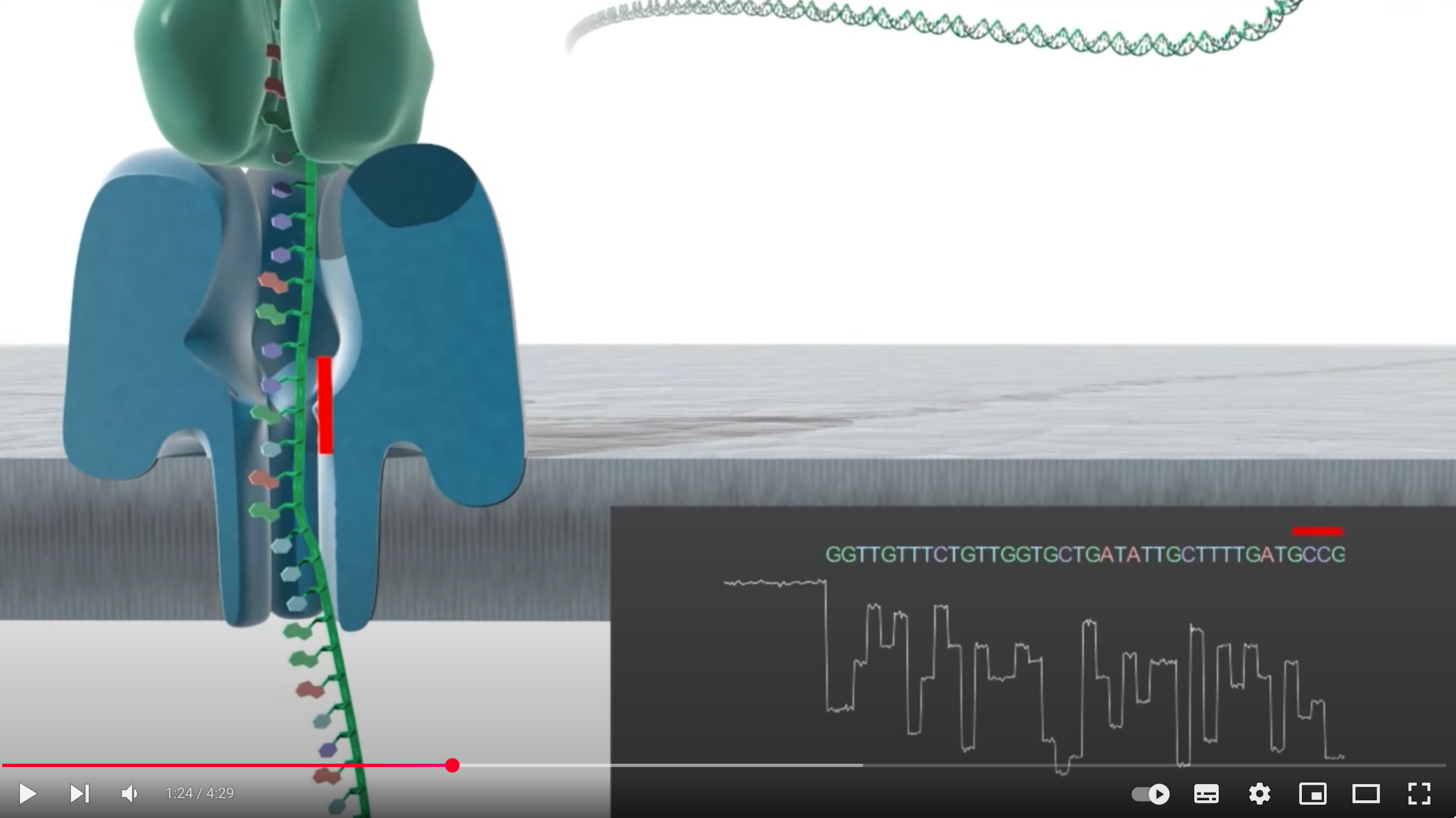
Chosen Technology: Oxford Nanopore (3rd generation) due to its ability to provide long reads and real-time sequencing, ideal for de novo assembly of bacterial genomes
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
Input and Preparation: High-quality genomic DNA is extracted, adapters are ligated, and the library is loaded onto the nanopore device
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Core Steps (Base Calling): Single-stranded DNA passes through a nanopore, causing changes in electrical current that are measured and decoded by software into base sequences
What is the output of your chosen sequencing technology?
Output: Long-read sequences in FASTQ format, facilitating detailed genomic analyses
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
DNA to be Edited (Designed)
I intend to engineer a DNA aptamer system that can detect human metabolism–derived substances (trace gases from breath or skin) with high sensitivity.
I call this non-invasive sensor the “Vital Nano-Sniffer”
Concept Overview
- Utilize genome editing techniques (e.g., expanded nucleobases) to enhance and optimize DNA aptamers that bind to volatile organic compounds (VOCs) and small molecules originating from living organisms
- Implement these aptamers in a cell-free chip device that detects the characteristic metabolic byproducts released when a human passes by, triggering a signal
- AI robots would find it difficult to replicate the exact same metabolites as humans, so this helps in determining “human identity”
Why This Is Necessary
- In the near future, the number of humanoid AI robots may increase to the point where visual appearance alone cannot distinguish them from humans
- Traditional methods (e.g., iris recognition) can be bypassed, but real-time biological metabolism is harder to fake
- By combining DNA aptamers and synthetic biology, we can potentially develop a new high-sensitivity, rapid security technique for detecting VOCs
Advantages and Potential
- Non-invasive and Rapid Detection: Unlike conventional chemical sensors, DNA sequences can be freely edited to improve specificity and sensitivity
- If it can capture the complex metabolic patterns unique to humans, it could be applied not only for personal identification but also for health checkups and disease screening
- Starting from an SF scenario of distinguishing AI from humans, this technology could actually advance the development of biosensors in the real world
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
Enzymatic Assembly (Gibson Assembly, Golden Gate, etc.)
- What: Uses enzymes to join multiple short DNA fragments (from chemical synthesis) into a longer construct
- Why :
Longer Sequences: Suitable for kilobase-scale or more (genes, libraries)
Seamless Cloning: Can avoid leaving restriction sites, allowing flexible designs
Complex Constructs: Perfect for assembling aptamer libraries or multi-fragment genetic circuits
Next-Generation Sequencing (Illumina, etc.) https://jp.illumina.com/
Library Prep: Fragment if necessary, add adapters, possibly PCR
Cluster Generation: DNA binds to a flow cell, forming millions of clusters
Sequencing-by-Synthesis: Each nucleotide incorporation produces a fluorescent signal
Data Processing: Base-calling, quality checks, and assembly if needed
5.3 DNA Edit
(i) What DNA would you want to edit and why?
“Antiphoton microbe”
Editing Target:
A dark-hued fungus or slime mold as the base organism (since it already exhibits some degree of black coloration)
Editing Objective:
Endow it with extremely high light-absorption capability so that it reflects almost no light, essentially becoming a “living darkness”
Bio-Art Aspect:
By producing a “writhing black mass,” we can provide an immersive experience of living darkness – a dramatic piece of bio-art
Optical Application:
There is potential to research this as a “biologically derived jet-black coating” that suppresses stray light, possibly benefiting optical instruments or novel material development
(ii) What technology or technologies would you use to perform these DNA edits and why?
Chosen Technology: CRISPR/Cas9
Reason for Choice:
CRISPR/Cas9 allows relatively precise targeting of specific genes with fewer off-target effects
It has increasingly been applied to black fungi and slime molds, making it suitable for modifying pigment pathways and morphological genes to enhance darkness and introduce nano-scale structures
Goal:
Use CRISPR/Cas9 to edit genes involved in pigment synthesis and surface morphology
By doing so, transform an already dark-hued microbe into an “ultra-black” organism with potential nano structures
Designing Guide RNA (gRNA)
Identify target genes responsible for light absorption (pigment synthesis) or structural morphology, then select the guide sequences
Introducing Cas9
Deliver the Cas9 protein or a plasmid expressing Cas9 into the microbial cells, enabling the gene-cutting process
(At this stage, this is about as much detail as I can address)
Planning/Design
- Identify which pigment biosynthesis genes to upregulate or which inhibitory factors to knock out
- Determine the morphological control genes that affect cell-surface nano-structures
Inputs
- Guide RNAs (gRNAs)
- Cas9 Protein or Cas9-Expressing Plasmid
Editing Efficiency
- There’s a risk the microbe may die off if it experiences excess heat or oxidative stress from absorbing extreme amounts of light
Biosafety
- If these microbes escape into the environment, unforeseen ecological impacts could occur. Proper containment and safety measures are essential
Reading & Resources (click to expand)
Resources
・DNA Sequencing at 40: Past, Present, and Future (2017) Shendure, J., Balasubramanian, S., Church, G. et al.
https://doi.org/10.1038/nature24286
・DNA Synthesis Technologies to Close the Gene Writing Gap (2023), Hoose, A., Vellacott, R., Storch, M. et al.
https://doi.org/10.1038/s41570-022-00456-9
・Recombineering and MAGE (2021), Wannier T, et al. Nat Rev Methods Primers,
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9083505/
・CRISPR Technology: A Decade of Genome Editing is Only the Beginning, Wang, Doudna, et al.,
https://www.science.org/doi/10.1126/science.add8643
Databases
・GenBank overview: https://www.ncbi.nlm.nih.gov/genbank/
・NCBI: https://www.ncbi.nlm.nih.gov/genome/
・Ensembl: https://useast.ensembl.org/index.html
・UCSC Genome Browser: https://genome.ucsc.edu/
・Protective and Enhancing Alleles: https://arep.med.harvard.edu/gmc/protect.html
Editors and tutorials
・CRISPR/Cas9
・Short tutorial for designing gRNAs: https://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing
・Benchling specific tutorial for designing gRNAs: https://www.benchling.com/blog/how-to-design-grnas-to-target-your-favorite-gene
・List of Cas editors and their PAM sites: https://www.synthego.com/guide/how-to-use-crispr/pam-sequence
・Base Editors
・Base editors contain a nicking or dead Cas9 enzyme fused to a deaminase. a.) PAM requirement: Base editors contain a nicking or dead Cas9 enzyme fused to a deaminase. For designing your guide RNA for base editing you will therefore have a PAM requirement like you would have for any Cas9 experiment. b.) Deamination window: An additional design constraint is that the sequence window in which deamination occurs is only a few base pairs long. You can find information on the deamination windows in the review below (even though some new editors are not included).
・BE4 and ABE7.10 are good starting points and both use SpCas9 with NGG Pam requirement. Base editors with other PAM sites have been constructed too.
・Review of base editors (2018) including a list of all base editors, their editing window and PAM requirement:
https://www.nature.com/articles/s41576-018-0059-1?WT.feed_name=subjects_animal-biotechnology
・Other editors:
・Prime editor https://www.nature.com/articles/s41586-019-1711-4
・Tutorials/tools:
・https://primeedit.nygenome.org/
・https://www.nature.com/articles/s41551-020-00622-8
・http://pegfinder.sidichenlab.org/
・TALEN For TALENs, you can assume no sequence restrictions – One of the technology’s previous restrictions was a T starting base, but this has since been overcome. In contrast to the CRISPR/Cas technologies above, your DNA sequence is recognized through interactions between the DNA and the TALEN: each TAL in the array recognizes one base. (Note: In order to introduce a double strand break, you will need to design to TALENs targeting the opposing strands.)
・Short guide: https://www.addgene.org/talen/guide/
・One of the available design resources: https://tale-nt.cac.cornell.edu/node/add/talen
・Directed evolution for overcoming starting base restriction:https://academic.oup.com/nar/article/41/21/9779/1276340
Additional Resources:
・Gel Purification of DNA: after DNA gel electrophoresis, cutting a band of DNA out of the agarose gel allows isolation and purification of a specific DNA fragment:
・Addgene: Protocol - How to Purify DNA from an Agarose Gel
・Overview of synthetic, unnatural organisms using recoding:
・Synthetic genomes with altered genetic codes (2020)
・DNA recorders, Sense+Read+Write:
・Lineage tracing and analog recording in mammalian cells by single-site DNA writing (2021)
・Molecular electronics, integrating single molecules into electronic chips:
・Molecular electronics sensors on a scalable semiconductor chip: A platform for single-molecule measurement of binding kinetics and enzyme activity (2022)
・Review of genome editors (zinc finger nucleases, TALENs, CRISPR) at the time CRISPR was emerging as editing technology:
https://www.cell.com/trends/biotechnology/pdf/S0167-7799(13)00087-5.pdf
・Clinical trials of genome-editing therapies: https://www.nature.com/articles/d41573-020-00096-y