Week 10 HW -Advanced Imaging & Measurement Technology
‘week-10-hw-imaging-and-measurement’
Documentation
Homework: Advanced Imaging & Measurement Technology
Homework: Final Project
What I will measure in my final project (Final Projectで測定するもの)
In my final project, I would like to measure several aspects of the transformation from body-derived materials into detectable or recoverable substances.(身体由来物質が検出可能、または回収可能な物質へ変換される過程を測定したい。)
Body-derived ions and trace metals(身体由来イオンと微量金属)
I would measure ions and trace metals contained in body-derived materials such as sweat, hair, nails, and menstrual blood.
Examples include K⁺, Na⁺, Ca²⁺, Mg²⁺, Fe, Cu, and Zn.汗、髪、爪、経血などの身体由来物質に含まれるイオンや微量金属を測定する。
例として、K⁺、Na⁺、Ca²⁺、Mg²⁺、Fe、Cu、Zn などがある。Protein expression(タンパク質発現)
If I use a GFP reporter or metallothionein construct, I would measure whether the target protein is expressed.GFP reporter や metallothionein construct を使う場合、目的タンパク質が発現しているかを測定する。
Fluorescent signal(蛍光シグナル)
I would measure GFP fluorescence to test whether body-derived ions such as K⁺ can be converted into a visible biological signal.K⁺ などの身体由来イオンが、目に見える生物学的シグナルへ変換されるかを調べるため、GFP蛍光を測定する。
Metal binding or concentration(金属結合・濃縮)
If I use metallothionein, I would measure whether trace metals are bound or concentrated after biological treatment.metallothionein を使う場合、生物学的処理後に微量金属が結合・濃縮されたかを測定する。
DNA construct accuracy
I would confirm whether the ordered DNA constructs have the correct sequence and size.注文した DNA construct が、正しい配列とサイズを持っているかを確認する。。
・Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
測定したい要素をすべて説明し、それをどのように測定するかも説明してください。
自分のプロジェクトで測りたいものをリストアップし、それぞれについて、どの方法で測定するのかを説明してください。
I would like to measure the following elements(測定したい要素は以下の通りである。):
Body-derived ions and trace metals(身体由来イオンと微量金属)
I would measure K⁺, Na⁺, Ca²⁺, Mg²⁺, Fe, Cu, and Zn in body-derived samples such as sweat, hair, nails, and menstrual blood. These would be measured by ion / metal analysis such as ICP-MS, ion chromatography, or mass spectrometry. 汗、髪、爪、経血などの身体由来サンプルに含まれる K⁺、Na⁺、Ca²⁺、Mg²⁺、Fe、Cu、Zn などを測定する。ICP-MS、ion chromatography、mass spectrometry などによって測定する。GFP fluorescence(GFP蛍光)
I would measure GFP fluorescence to test whether body-derived ions such as K⁺ can be converted into a visible biological signal. This would be measured using fluorescence microscopy, a plate reader, or a fluorescence viewer.K⁺などの身体由来イオンが、目に見える生物学的シグナルに変換されるかを確認するため、GFP蛍光を測定する。蛍光顕微鏡、plate reader、fluorescence viewer などを使う。
Protein expression(タンパク質発現)
If I use a GFP reporter or metallothionein construct, I would measure whether the target protein is expressed. This could be checked by SDS-PAGE or fluorescence measurement.GFP reporter や metallothionein construct を使う場合、目的タンパク質が発現しているかを測定する。SDS-PAGE や蛍光測定によって確認する。
DNA construct accuracy(DNA construct の正確性)
I would confirm whether the ordered DNA constructs have the correct size and sequence. This would be measured by gel electrophoresis and DNA sequencing.注文した DNA construct が正しいサイズと配列を持っているかを確認する。Gel electrophoresis と DNA sequencing によって測定する。
Metal binding or concentration(金属結合・濃縮)
If I use metallothionein, I would compare metal levels before and after biological treatment to test whether trace metals are bound or concentrated.metallothionein を使う場合、生物学的処理の前後で金属量を比較し、微量金属が結合・濃縮されたかを調べる。
・What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
どの技術を使いますか? 詳しく説明してください。
使用する測定技術を書いてください。
たとえば:
・gel electrophoresis ・DNA sequencing ・mass spectrometry ・その他の測定技術
それぞれ、何のために使うのかを詳しく説明してください。
For my final project, I would use several measurement technologies.
Final Projectでは、いくつかの測定技術を使う予定である。
Gel electrophoresis
To confirm the size of DNA constructs and, if needed, protein expression by SDS-PAGE.(DNA construct のサイズを確認するために使う。また必要に応じて、SDS-PAGE によってタンパク質発現を確認する。)
DNA sequencing
To verify that the ordered DNA constructs, including promoter, RBS, coding sequence, and terminator, are correct.(注文した DNA construct の配列が正しいかを確認するために使う。promoter、RBS、coding sequence、terminator などが正しく含まれているかを確認する。)
Fluorescence measurement
To measure GFP expression and test whether body-derived ions such as K⁺ can be converted into a fluorescent signal.(GFP の発現を測定し、K⁺ などの身体由来イオンが蛍光シグナルに変換されるかを確認するために使う。)
Mass spectrometry
To analyze proteins or trace metals in body-derived samples before and after biological treatment.(生物学的処理の前後で、身体由来サンプルに含まれるタンパク質や微量金属を分析するために使う。)
Ion / metal analysis
To quantify ions and trace metals such as K⁺, Na⁺, Ca²⁺, Mg²⁺, Fe, Cu, and Zn from sweat, hair, nails, or menstrual blood.(汗、髪、爪、経血などに含まれる K⁺、Na⁺、Ca²⁺、Mg²⁺、Fe、Cu、Zn などのイオンや微量金属を定量するために使う。)
Homework: Waters Part I — Molecular Weight
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
(下に示されている eGFP の予測アミノ酸配列 と、既知の修飾をもとに、計算上の分子量 はいくつになりますか?
ExPASy の Compute pI/Mw のようなオンライン計算ツールを使ってもかまいません。)
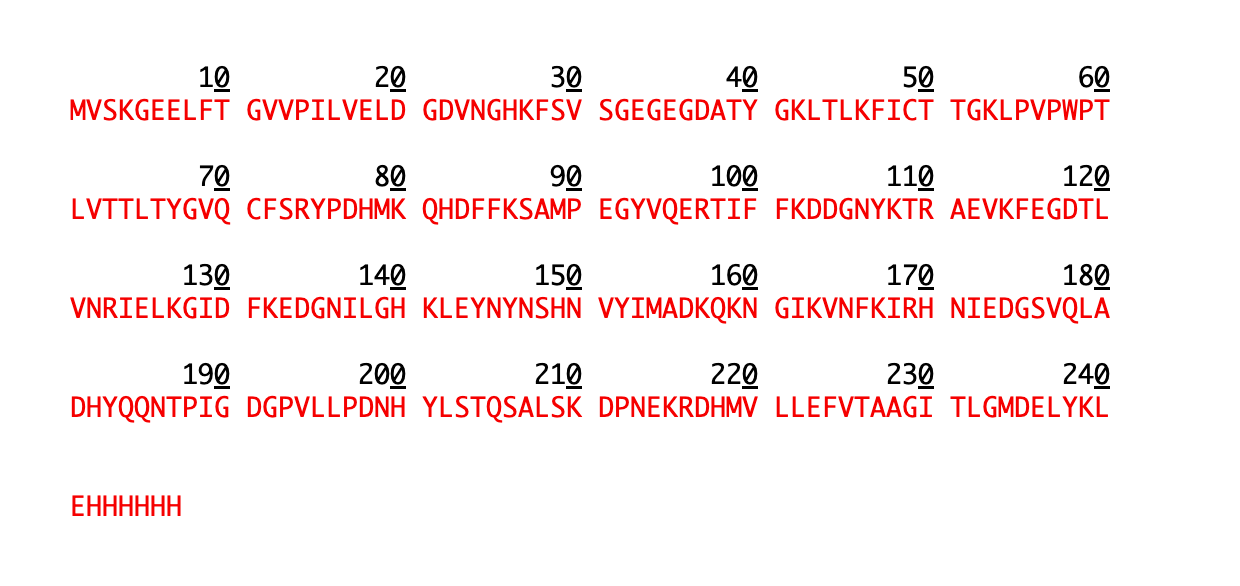
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT
GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF
KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV
YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY
LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHHNote: This contains a His-purification tag (
HHHHHH) and a linker (LEbefore it).

MVSKGEELFTG = 11 amino acids
VVPILVELDG = 10
DVNGHKFSVS = 10
GEGEGDATYG = 10
KLTLKFICTT = 10
GKLPVPWPTL = 10
VTTLTYGVQC = 10
FSRYPDHMKQ = 10
HDFFKSAMPE = 10
GYVQERTIFF = 10
KDDGNYKTRA = 10
EVKFEGDTLV = 10
NRIELKGIDF = 10
KEDGNILGHK = 10
LEYNYNSHNV = 10
YIMADKQKNG = 10
IKVNFKIRHN = 10
IEDGSVQLAD = 10
HYQQNTPIGD = 10
GPVLLPDNHY = 10
LSTQSALSKD = 10
PNEKRDHMVL = 10
LEFVTAAGIT = 10
LGMDELYKLE = 10
HHHHHH = 6
Total: 247 amino acids
Calculated molecular weight: 28,006.6 Da
= 28.01 kDa
Based on the predicted amino acid sequence of eGFP, including the LE linker and the C-terminal 6xHis tag, the calculated molecular weight is approximately 28,006.6 Da, or 28.01 kDa.
The sequence contains 247 amino acids.
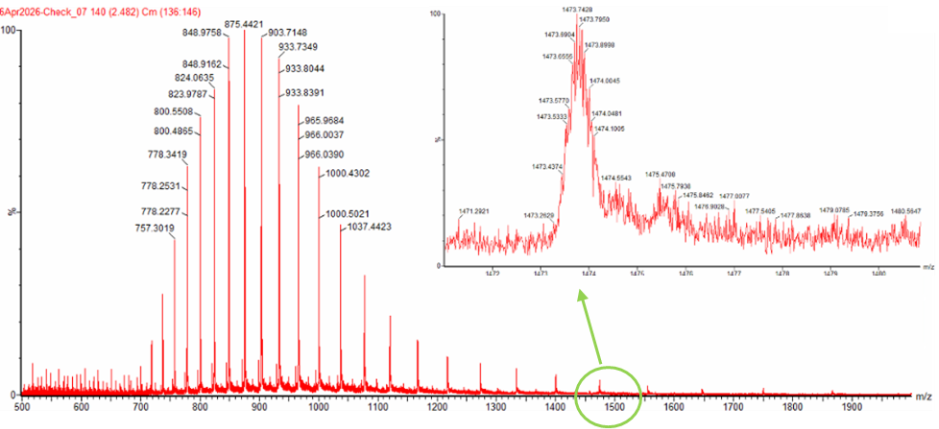
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
(Recitation で説明された adjacent charge state approach(隣接する電荷状態を使う方法) を用いて、eGFP の分子量を計算しなさい。 Intact LC-MS data、つまり Figure 1 の分解されていない eGFP の LC-MS データ から、隣り合う2つの charge state のピークを選び、以下を行いなさい。)
2-1. Determine $z$ for each adjacent pair of peaks $(n, n+1)$ using:
$$ {\large z} = {\Large \frac{\frac{m}{z_{n+1}}}{\frac{m}{z_n} - \frac{m}{z_{n+1}}}} $$
隣り合うピークのペア (n,n+1) について、次の式を使って z(charge state / 電荷数) を求めなさい。
From Figure 1, I selected two adjacent peaks and calculated the charge state of each peak.
Figure 1のピーク2つを選んで、「このピークは何価のイオンか?」を計算する。
The two adjacent peaks selected from Figure 1 are:
Figure 1から、隣り合う2つのピークとして
m/z 1000.4302
m/z 965.9684
を選んだ。
First, I calculated (z) using the adjacent charge state equation:
まず、隣接するcharge stateの式を使って z を計算した。
z = 965.9684 / (1000.4302 − 965.9684) z = 965.9684 / 34.4618 z ≈ 28.0
Therefore,
m/z 1000.4302 のピーク = 28+
m/z 965.9684 のピーク = 29+
These two peaks are considered to correspond to adjacent charge states.
に対応すると考えられる。
2-2. Determine the MW of the protein using the relationship between $\frac{m}{z_n}$, $MW$, and $z$
m/z、MW、z の関係を使って、タンパク質の MW(molecular weight / 分子量) を求めなさい。
Using the charge state (z) calculated in 2-1, I calculated the experimental molecular weight of eGFP from the LC-MS data.
2-1で求めた charge state z を使って、LC-MSデータからeGFPの実測分子量を計算する。
Next, I calculated the molecular weight using:
次に、以下の計算式も使って分子量を求めた。
MW = z × (m/z − 1.0073)
Using the peak at m/z 1000.4302
MW = 28 × (1000.4302 − 1.0073)
MW = 28 × 999.4229
MW ≈ 27,983.8 Da
Using the peak at m/z 965.9684:
MW = 29 × (965.9684 − 1.0073)
MW = 29 × 964.9611
MW ≈ 27,983.9 Da
Therefore, the experimental molecular weight of eGFP calculated from the LC-MS data is: したがって、LC-MSデータから求めた eGFP の実測分子量は
MWexperiment ≈ 27,984 Da = 27.984 kDa
The experimental molecular weight is approximately 27.984 kDa. 27.984 kDa である。
2-3. Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
$$ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $$
2-2で求めた実測分子量と、1で求めた予測分子量を使って、測定の accuracy(正確さ/誤差率) を計算しなさい。
Compared with the theoretical molecular weight calculated in Question 1, 28,006.6 Da: 1で求めた理論分子量 28,006.6 Da と比較すると、
Accuracy = |27,983.9 − 28,006.6| / 28,006.6
Accuracy = 22.7 / 28,006.6
Accuracy ≈ 0.00081
Therefore, the error is approximately 0.081%. つまり、誤差は約 0.081% である。
- Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
intact eGFP の mass spectrum で、拡大表示されたピークの charge state を観察できますか? もし観察できるなら、それは何ですか? もし観察できないなら、なぜ観察できないのですか?
ここでいう charge state は、さっき計算したような 28+ や 29+ のこと。
ただし、zoomed-in peak で直接見えているのは charge state ではなく、通常は isotope pattern / isotopic peaks になる。
タンパク質の質量スペクトルでは、同じ分子でも自然同位体の違いによって、少しずつ m/z が異なる isotopic peaks が現れる。
電荷が 1+ の場合、同位体ピークの間隔は約 1 m/z になる。
しかし、電荷が z+ の場合、同位体ピークの間隔は:
1 / z
になる。
したがって、28+ の場合は:
1 / 28 ≈ 0.036 m/z
となる。
そのため、zoomed-in peak の中で isotope peaks の細かい間隔が見えれば、その間隔から charge state を推定できる。
Figure 1 の zoomed-in peak が m/z 1000.4302 周辺のピークであれば、前の計算からこのピークは 28+ charge state に対応すると考えられる。
Yes, the charge state can be inferred from the zoomed-in peak by looking at the isotope spacing.
The zoomed-in peak shows an isotope pattern, and the spacing between isotopic peaks is approximately 1/z.
For the peak around m/z 1000.4302, the adjacent charge state calculation indicates that this peak corresponds to the 28+ charge state.
For a 28+ ion, the expected isotope spacing is about 1/28 = 0.036 m/z.
Homework: Waters Part II — Secondary/Tertiary structure
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
eGFP を native / folded state(自然に折りたたまれた状態) で分析し、それを denatured / unfolded state(変性してほどけた状態) と比較する。 分析には quadrupole time-of-flight MS を使う。
今回は LC、つまり液体クロマトグラフィーは使わず、MS-only analysis を行う。(= direct infusion experiment とも呼ばれる)
装置は Waters Xevo G3-QToF MS を使用。
1. Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
ラボで学んだことをもとに、native protein conformation と denatured protein conformation の違いを説明しなさい。
たとえば:
・タンパク質が unfolding すると何が起こるのか?
・それは mass spectrometer でどのように判断できるのか?
・native protein analysis と denatured protein analysis の間で、mass spectrum にどのような違いが見えるか?
※ Figure 2 を参照
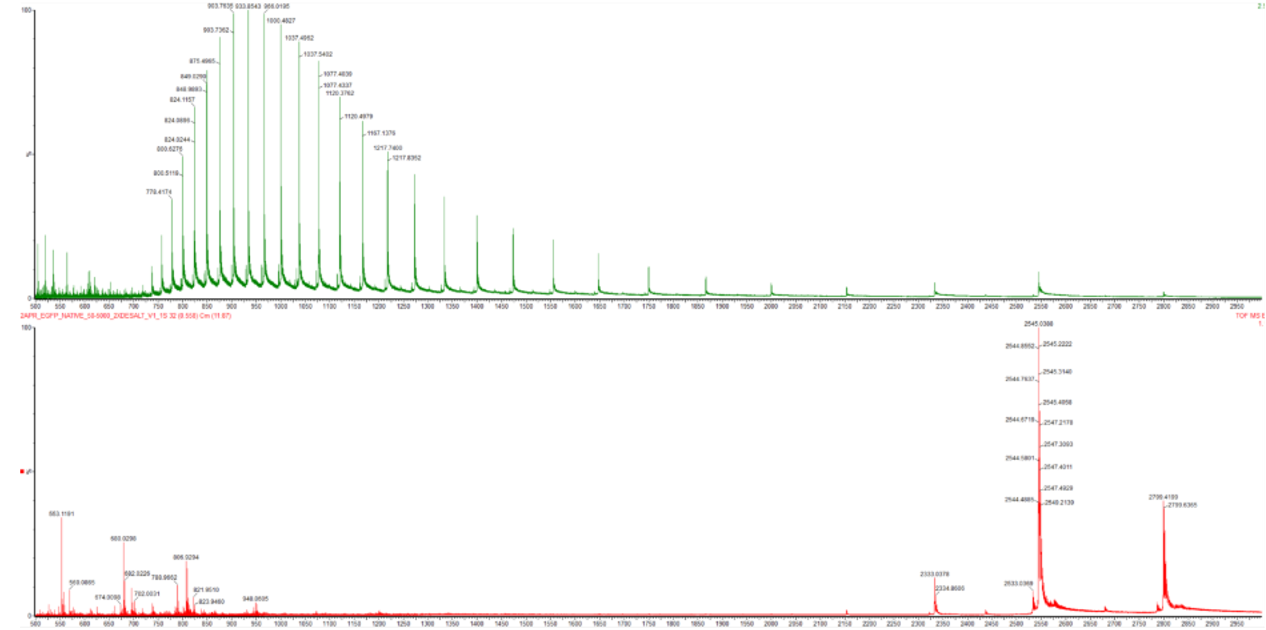
Native state では、タンパク質は自然に折りたたまれたコンパクトな構造をとっている。
この状態では、プロトンが結合できる部位があまり露出していないため、比較的低い charge state で検出される。
そのため、mass spectrum では高い m/z 側に少数のピークとして現れやすい。
一方、denatured state では、タンパク質の折りたたみ構造がほどけ、より伸びた状態になる。
内部に隠れていたアミノ酸やプロトン化可能な部位が露出するため、より多くのプロトンを受け取り、高い charge state で検出される。
その結果、mass spectrum では低い m/z 側に、多数の charge state のピークとして現れる。
Figure 2 では、native eGFP では m/z 2500〜2800 付近に大きなピークが見られ、低い charge state を示している。
一方、denatured eGFP では m/z 700〜1300 付近に多くのピークが分布しており、高い charge state distribution を示している。
この違いから、native eGFP は foldedでコンパクトな状態、denatured eGFP は unfoldedでより多くの電荷を持つ状態として観察されていると考えられる。
Native eGFP is folded and compact, so fewer protonation sites are exposed.
As a result, it carries fewer charges and appears at higher m/z values.
Denatured eGFP is unfolded, exposing more protonation sites, so it carries more charges and appears as many peaks at lower m/z values.
In Figure 2, the native spectrum shows strong peaks around m/z 2500–2800, while the denatured spectrum shows many peaks around m/z 700–1300.
This shift in charge state distribution shows the difference between folded and unfolded eGFP.
2. Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 $\frac{m}{z}$? What is the charge state? How can you tell?
Figure 3 の native eGFP mass spectrum を拡大して見たとき、m/z 約2800のピークの charge state は判別できますか?
もし判別できるなら、その charge state は何ですか?また、どうやってそれがわかりますか?
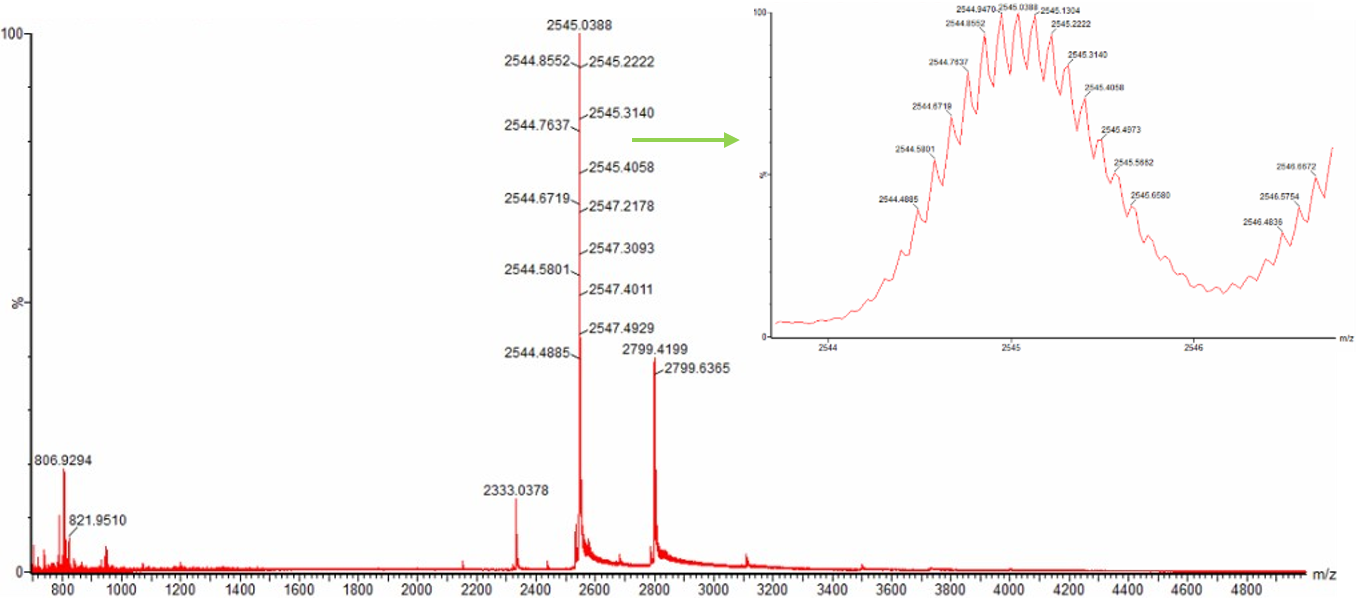
Yes, it can be determined.判別できる。
The peak around m/z 2800 is likely to correspond to approximately the 10+ charge state, because the molecular weight of eGFP is about 27,984 Da. m/z 約2800 のピークは、eGFP の分子量がおよそ 27,984 Da であることから、約 10+ の charge state に対応すると考えられる。
Since MW / z ≈ m/z:
27,984 / 10 ≈ 2,798
This matches the peak around m/z 2800. m/z 約2800 のピークと一致する。
The charge state can also be confirmed from the isotope spacing in the zoomed-in spectrum.
When the charge state is z+, the isotope peak spacing is approximately 1/z.
For a 10+ ion, the expected isotope spacing is about:
また、zoomed-in spectrum で isotope peaks が見える場合、同位体ピーク間隔からも charge state を確認できる。
charge state が z+ のとき、isotope peak spacing は 1/z になる。
10+ の場合、間隔は約 0.1 m/z になるため、ピーク内の isotope spacing が約 0.1 m/z であれば、このピークは 10+ charge state と判断できる。
1 / 10 = 0.1 m/z
Therefore, if the isotope peaks within the zoomed-in peak are spaced by about 0.1 m/z, the peak can be assigned as the 10+ charge state.
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
(eGFP タンパク質標準品を trypsin を使ってペプチドに分解します。 Trypsin は、Lysine(K) と Arginine(R) の後ろのペプチド結合を選択的に切断する酵素です。生成されたペプチドは、Waters BioAccord LC-MS で分析されます。 これにより、それぞれのペプチドの分子量を測定し、さらに断片化して、各ペプチド内のアミノ酸配列を確認します。 この一連のプロセスによって peptide map が作成されます。この方法は、タンパク質の primary structure(一次構造) を確認するために使われます。 タンパク質の分子量を計算したり、trypsin digest によって生成されるペプチドのリストを予測したりするためのオンラインツールはいくつかあります。 今回は、Swiss Institute of Bioinformatics(SIB)の bioinformatics resource portal である ExPASy のツールを使い、eGFP から生成される tryptic peptides のリストを予測します。)
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
(eGFP には、Lysine(K) と Arginine(R) がいくつ含まれていますか? Waters Part I Question 1 で与えられた eGFP 配列の中で、それらを 丸で囲む、または ハイライトしてください。 (注:Benchling にアミノ酸ファイルとして配列を追加し、biochemical properties タブをクリックすると、各アミノ酸の数を確認できます。))
I could not use the highlight function properly, so I made the K and R residues bold instead.
MVSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTLTYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYKLEHHHHHH
Lysine (K): 20 Arginine (R): 6
There are 26 total K/R residues that can be potential trypsin cleavage sites.
2. How many peptides will be generated from tryptic digestion of eGFP? (eGFP を trypsin digestion した場合、いくつのペプチドが生成されますか?)
Navigate to https://web.expasy.org/peptide_mass/
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides. (上にある eGFP 配列を、PeptideMass tool の入力欄にコピー&ペーストする。)
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP. (eGFP から生成されるペプチドを予測するために、下の Figure 4 を参考にして、関連するパラメータを設定する。)
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.(PeptideMass tool の “Perform the Cleavage” (ボタンをクリックし、trypsin を使って digest した場合に生成されるペプチド数を報告する。)
I used the ExPASy PeptideMass tool with the following settings:
- Enzyme: Trypsin
- Missed cleavages: 0
- Cysteines: reduced form
- Methionines: not oxidized
- Mass calculation: monoisotopic
- Ion: [M+H]+
The PeptideMass tool predicted 27 tryptic peptides from the eGFP sequence.
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
(ラボで生成された Peptide Map の LC-MS データをもとに、eGFP peptide map において 0.5分から6分の間に、いくつの chromatographic peaks が見えますか? Figure 5a を参考にしてください。相対存在量が 10%を超えるピークはすべて数えてかまいません。 Figure 5a:eGFP peptide map の total ion chromatogram(TIC)。 2.78分のピークが丸で囲まれており、その MS データは下の Figure 5b の mass spectrum に示されています。)
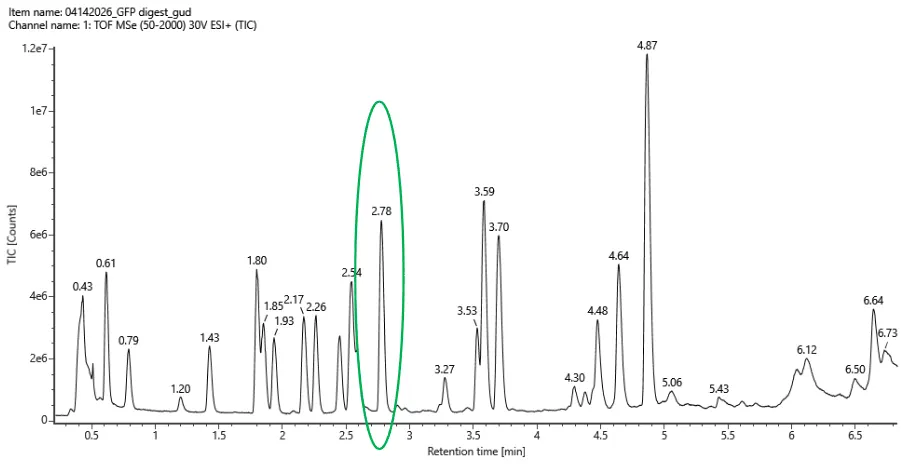
Based on the TIC chromatogram in Figure 5a, I counted approximately 17 chromatographic peaks between 0.5 and 6 minutes that are above 10% relative abundance.
The main peaks I counted are around 0.61, 0.79, 1.43, 1.80, 1.85, 1.93, 2.17, 2.26, 2.54, 2.78, 3.27, 3.53, 3.59, 3.70, 4.48, 4.64, and 4.87 minutes.
Approximately 17
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
(すべてのピークがペプチドであると仮定した場合、chromatogram のピーク数は、上の Question 2 で予測されたペプチド数と一致しますか? chromatogram には、予測されたペプチド数よりも 多くのピークがありますか?それとも 少ないピークがありますか?)
The number of observed peaks does not match the number of predicted peptides.
In Question 2, the ExPASy PeptideMass tool predicted 27 tryptic peptides from the eGFP sequence.
In Figure 5a, I counted approximately 17 chromatographic peaks between 0.5 and 6 minutes above 10% relative abundance.
Therefore, there are fewer peaks in the chromatogram than the number of predicted peptides.
This difference may occur because not every predicted peptide is detected as a separate chromatographic peak. Some peptides may be present at low abundance, ionize poorly, co-elute with other peptides, or fall below the 10% relative abundance threshold.
観察されたピーク数は、予測されたペプチド数とは一致しない。
Question 2 では、ExPASy PeptideMass tool により、eGFP 配列から 27個の tryptic peptides が予測された。
一方、Figure 5a では、0.5分から6分の間で、10%以上の相対存在量を持つ chromatographic peaks を約 17個 数えた。
したがって、chromatogram には、予測されたペプチド数よりも 少ないピーク が見られる。
この違いは、予測されたすべてのペプチドが、それぞれ独立した chromatographic peak として検出されるとは限らないために起こる可能性がある。
一部のペプチドは存在量が低かったり、イオン化効率が悪かったり、他のペプチドと同じ時間に溶出してピークが重なったり、10% relative abundance の閾値を下回ったりする可能性がある。
5. Identify the mass-to-charge ($\frac{m}{z}$) of the peptide shown in Figure 5b. What is the charge ($z$) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ($\small{[M\!\!+\!\!H]^+}$) based on its $\frac{m}{z}$ and $z$.
Figure 5b に示されているペプチドの mass-to-charge ratio、つまり m/z を特定しなさい。 そのペプチドの最も多い charge state、つまり 最も強く観察されている電荷状態 z は何ですか? charge state は、同位体ピーク同士の間隔を使って判断しなさい。 さらに、その m/z と z をもとに、そのペプチドの 一価イオンの質量、つまり [M+H]⁺ を計算しなさい。
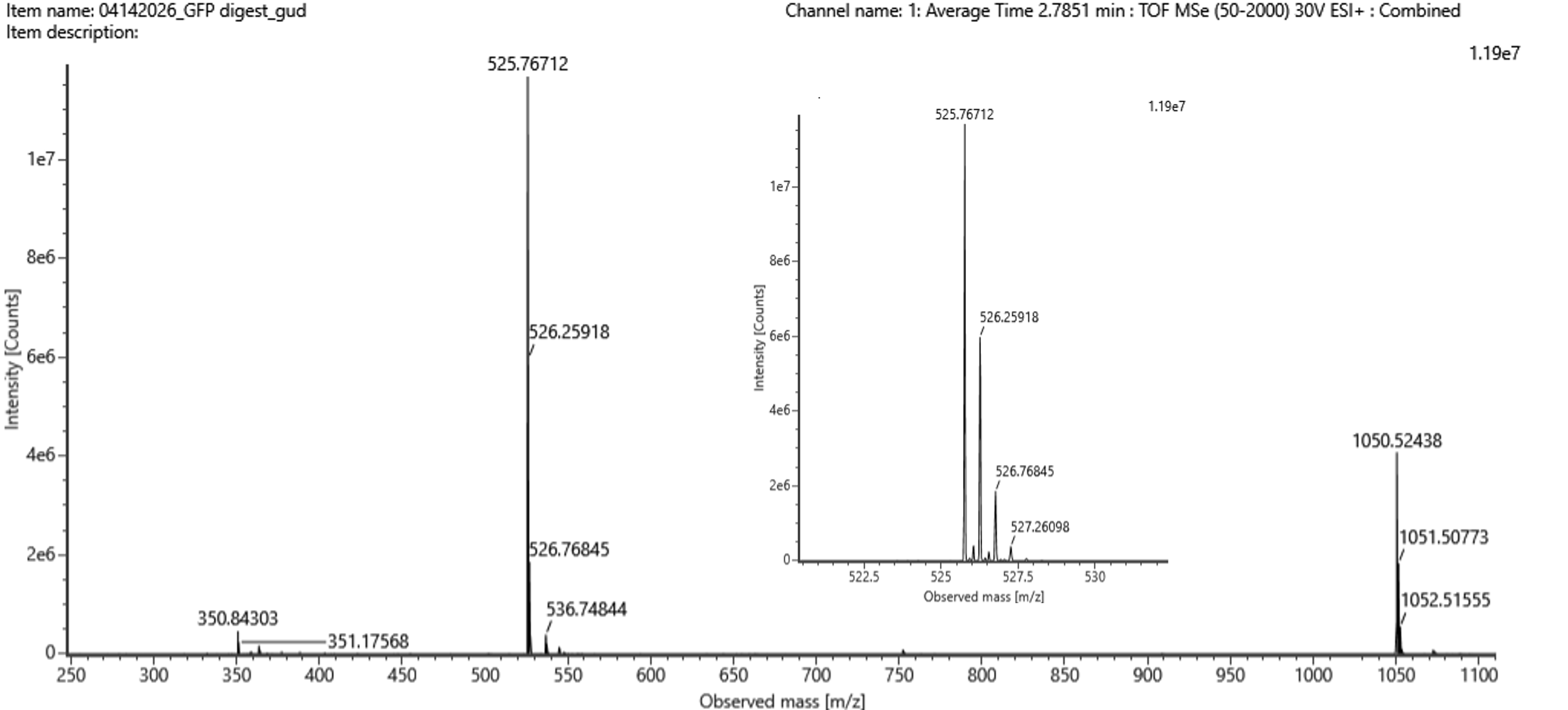
“Figure 5b.
Mass spectrum figure to show $\frac{m}{z}$ for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at M/Z 525.76 to discern the isotope peaks."
2.78分の chromatographic peak に対応する mass spectrum です。 m/z 525.76 のピークが拡大されており、isotope peaks を見分けられるようになっています。
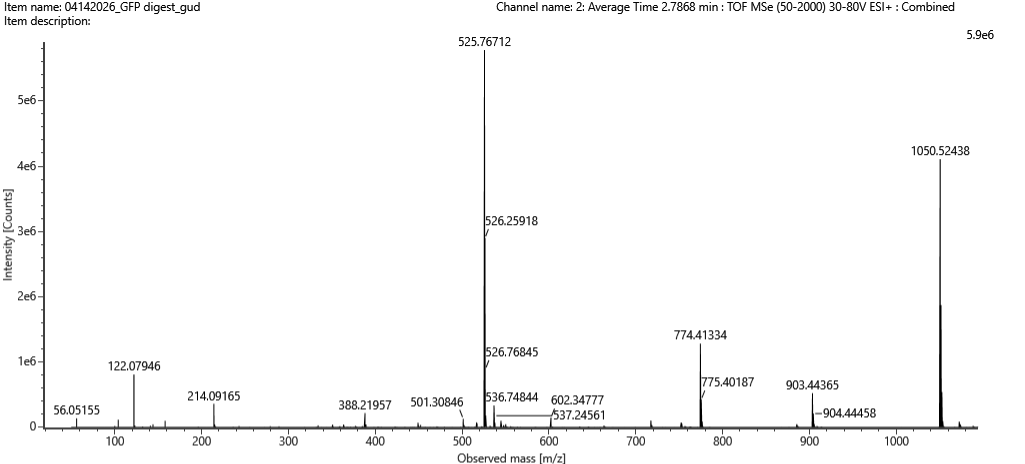
“Figure 5c.
Fragmentation spectrum of the peptide eluting at retention time 2.78 minutes in Figure 5a (above).
Figure 5a の 2.78分に溶出したペプチドの fragmentation spectrum です。
Figure 5b を見ると、
2.78分のピークに対応する主な peptide peak は:m/z = 525.76712
拡大図では、同位体ピークが:
同位体ピークの間隔
526.25918 − 525.76712 = 0.49206
約 0.5 m/z
同位体ピーク間隔は"1 / z"のため
1 / z ≈ 0.5
z ≈ 2
つまり、この peptide の最も多い charge state はz = 2+
観測されたピークは 2価イオンなので:
m/z = 525.76712
z = 2
一価の peptide mass、つまり [M+H]⁺ は:
[M+H]⁺ = z × (m/z) − (z − 1) × 1.0073
[M+H]⁺ = 2 × 525.76712 − 1.0073
[M+H]⁺ = 1051.53424 − 1.0073
[M+H]⁺ ≈ 1050.5269 Da
Figure 5b には実際に m/z 1050.52438 のピークも見えているので、これは同じ peptide の 1+ charge state と考えられる。
The mass-to-charge ratio of the peptide shown in Figure 5b is m/z 525.76712.
The isotope peaks are separated by approximately 0.5 m/z:
526.25918 − 525.76712 = 0.49206
Because isotope spacing is approximately 1/z, this indicates that the most abundant charge state is 2+.
The singly charged peptide mass, [M+H]⁺, can be calculated as:
[M+H]⁺ = z × (m/z) − (z − 1) × 1.0073
[M+H]⁺ = 2 × 525.76712 − 1.0073
[M+H]⁺ ≈ 1050.5269 Da
Therefore, the singly charged form of the peptide is approximately 1050.53 Da.
6. Identify the peptide based on comparison to expected masses in the PeptideMass tool.
What is mass accuracy of measurement? Please calculate the error in ppm.
PeptideMass tool で予測されたペプチドの質量と比較して、Figure 5b のペプチドがどのペプチドかを同定しなさい。 また、その測定の mass accuracy はどのくらいですか?誤差を ppm で計算しなさい。
(Recall that $ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $ )
Note:
Accuracy = |MWexperiment − MWtheory| / MWtheory
ppm で表す場合は、これに 1,000,000 をかける。
ppm error = |MWexperiment − MWtheory| / MWtheory × 1,000,000
The singly charged form calculated in Question 5 was approximately 1050.5269 Da.
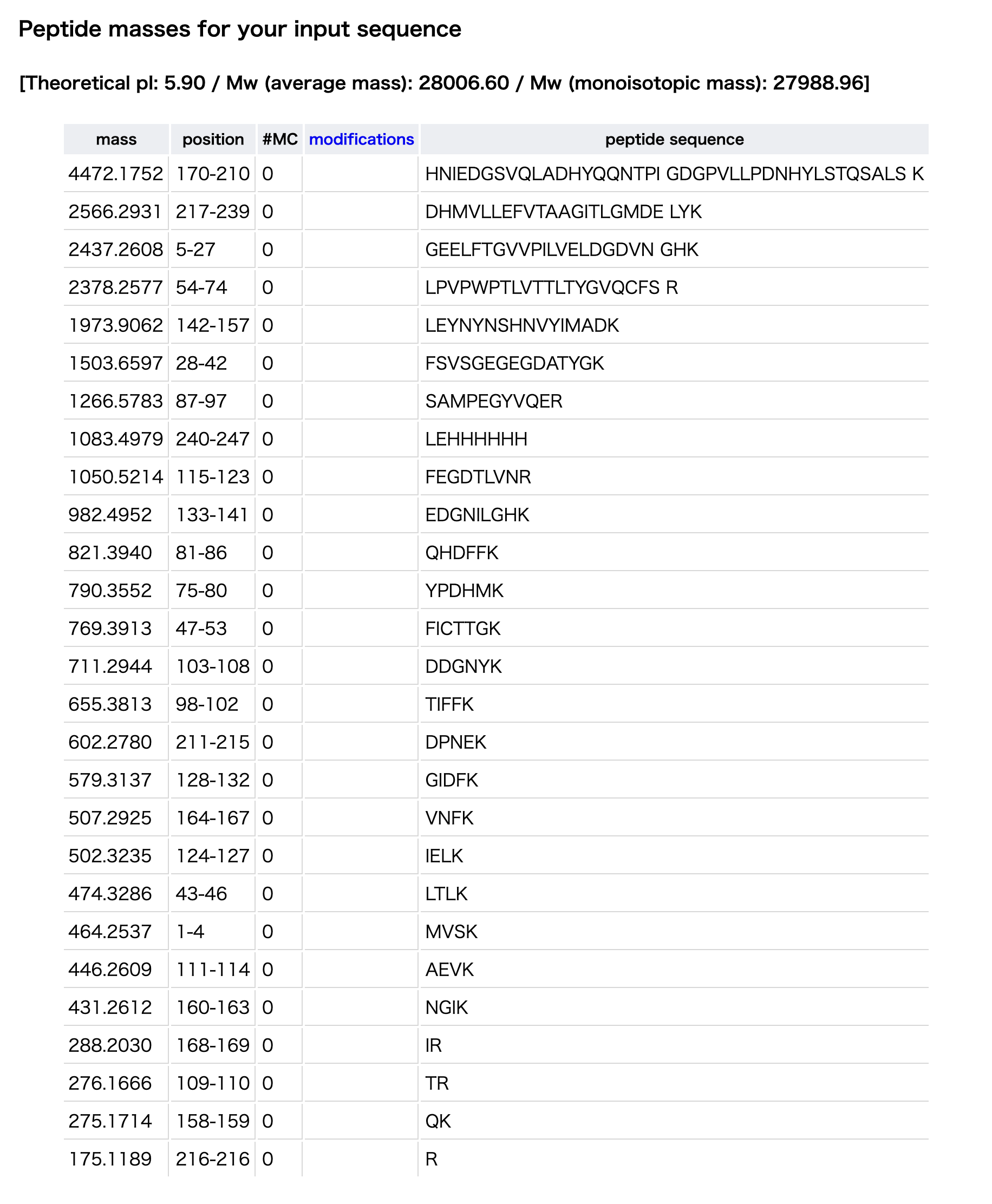
By comparing this value with the expected peptide masses from the ExPASy PeptideMass tool, the closest predicted peptide is FEGDTLVNR, with a theoretical mass of 1050.5214 Da.
Therefore, the peptide can be identified as FEGDTLVNR.
Figure 5b のペプチドについて、設問5で計算した singly charged form は約 1050.5269 Da であった。 PeptideMass tool の予測質量と比較すると、最も近いペプチドは FEGDTLVNR であり、理論質量は 1050.5214 Da である。 したがって、このペプチドは FEGDTLVNR と同定できる。
The peptide is identified as FEGDTLVNR, because its theoretical mass from the ExPASy PeptideMass tool is 1050.5214 Da, which closely matches the experimentally calculated singly charged mass of 1050.5269 Da.
1050.5214 115-123 FEGDTLVNR
The ppm error was calculated as:
ppm error = |1050.5269 − 1050.5214| / 1050.5214 × 1,000,000
ppm error ≈ 5.2 ppm
Therefore, the mass accuracy is approximately 5.2 ppm.
7. What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
ペプチドマッピングによって確認された配列の割合はどれくらいですか?(図6を参照)
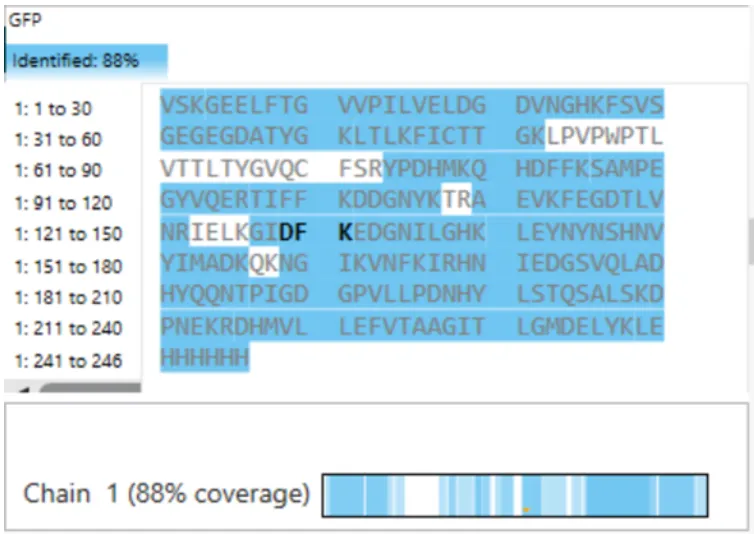
Figure 6 Amino Acid Coverage Map of eGFP based on BioAccord LC-MS peptide identification data.
The percentage of the eGFP sequence confirmed by peptide mapping is 88%.
According to Figure 6, the identified sequence coverage is 88%, meaning that 88% of the eGFP amino acid sequence was confirmed by the peptide map.
Figure 6 では “Identified: 88%” と表示されているため、peptide mapping によって確認された eGFP 配列の割合は 88% である。
Bonus Peptide Map Questions
8. Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c?
Figure 5c に示されている peptide fragmentation spectrum から、ペプチド配列を決定できますか?
(HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
ヒント: 上の Question 2 の結果を使って、Figure 5b に示された peptide molecular weight に最も近いペプチドを探します。 そのペプチド配列を、オンラインの fragmentation prediction tool にコピー&ペーストし、アミノ酸配列にもとづく fragmentation pattern を予測します。 そして、Figure 5c の fragmentation spectrum に最もよく一致する eGFP peptide の配列を答えなさい。
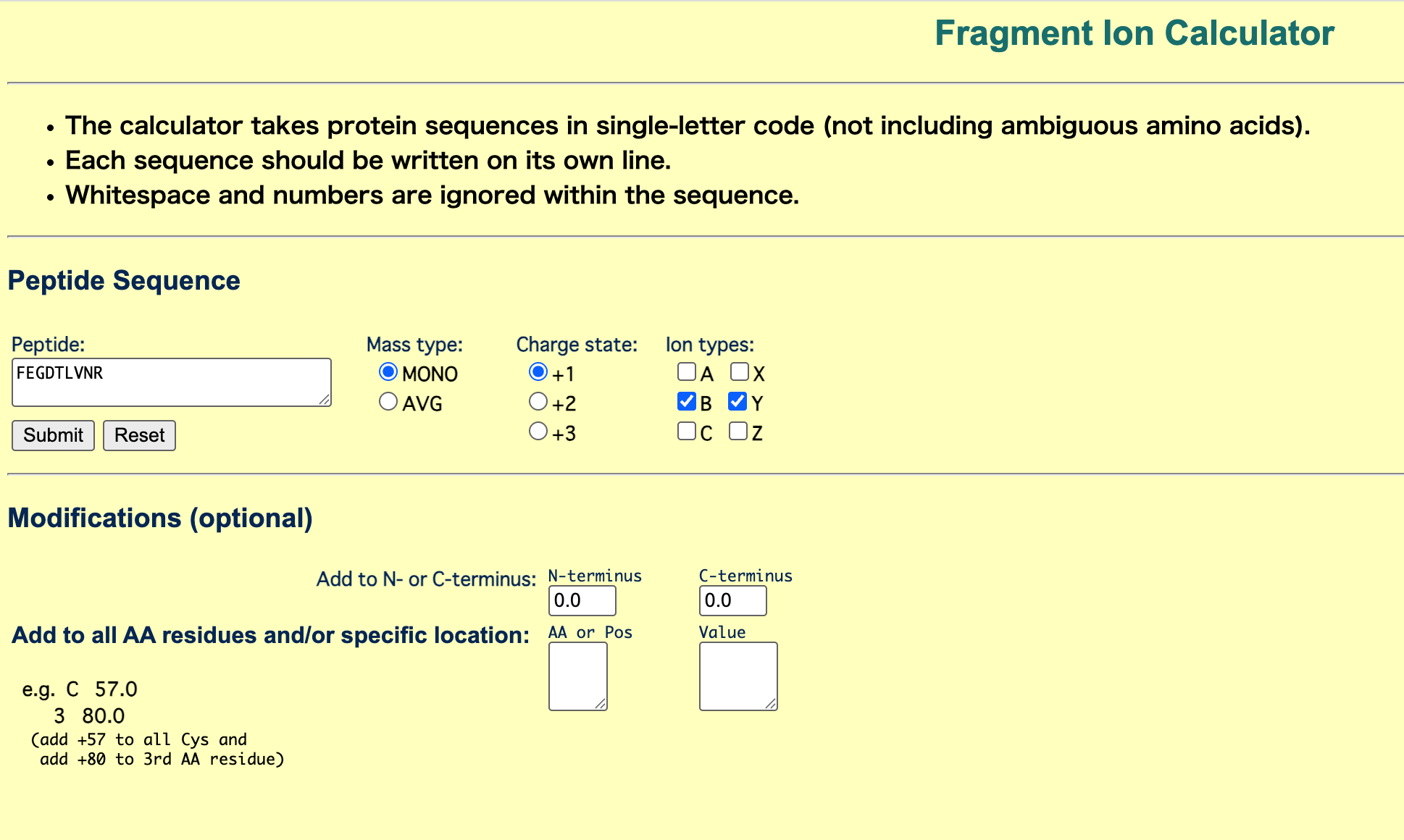
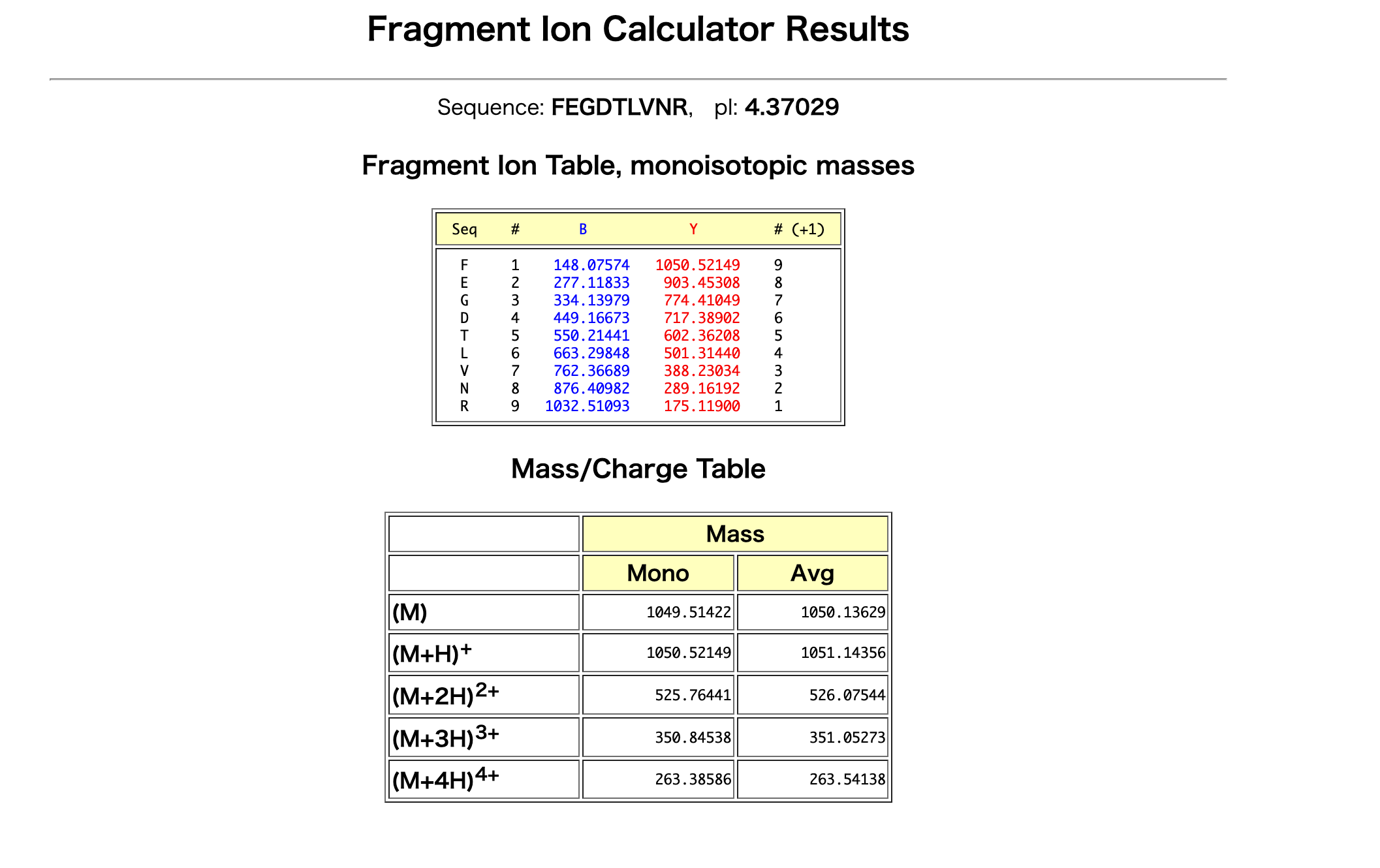
The peptide sequence that best matches the fragmentation spectrum in Figure 5c is FEGDTLVNR.
I entered the peptide sequence FEGDTLVNR into the Fragment Ion Calculator and compared the predicted b/y ions with the observed peaks in Figure 5c.
Several predicted fragment ions match the observed spectrum. For example, the predicted y ions at approximately 903.45, 774.41, and 602.36 m/z match observed peaks around 903.44, 774.41, and 602.35 m/z in Figure 5c.
Therefore, the fragmentation spectrum supports the identification of the peptide as FEGDTLVNR.
Figure 5c の fragmentation spectrum に最もよく一致するペプチド配列は FEGDTLVNR である。
Fragment Ion Calculator に FEGDTLVNR を入力し、予測された b/y ions と Figure 5c のピークを比較した。
予測された y ions のうち、約 903.45、774.41、602.36 m/z のピークが、Figure 5c の 903.44、774.41、602.35 m/z 付近のピークと一致している。
したがって、Figure 5c の fragmentation spectrum は FEGDTLVNR を支持している。
9. Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Peptide map data は妥当ですか?つまり、今回の peptide mapping の結果は、分析したタンパク質が eGFP standard であることを示していますか?
なぜそう言えますか? または、なぜそう言えませんか?
Figure 6 を参照してください。
Figure 6 では、計算された質量と fragmentation pattern によって確実に同定されたペプチドが、eGFPアミノ酸配列の何%をカバーしているかが示されています。
Yes, the peptide map data supports that the protein is the eGFP standard.
Figure 6 shows 88% sequence coverage, meaning that most of the eGFP amino acid sequence was confirmed by peptide mapping. This high coverage indicates that many observed peptides match the predicted eGFP sequence.
In addition, as shown in Questions 6–8, the peptide eluting at 2.78 minutes was identified as FEGDTLVNR based on both its mass and fragmentation pattern. This supports the identification because the peptide matches the expected eGFP sequence.
peptide map data は、サンプルが eGFP standard であることを支持している。 Figure 6 では、eGFP配列の 88% が peptide mapping によって同定されている。これは高い sequence coverage であり、多くのペプチドが予測された eGFP 配列と一致していることを示している。
また、設問6〜8で確認したように、2.78分のピークから得られたペプチドは、質量と fragmentation spectrum の両方から FEGDTLVNR と同定できた。
このように、複数のペプチドが質量と断片化パターンによって確認されているため、分析したタンパク質は eGFP standard であると考えられる。
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
今回は、charge detection mass spectrometry(CDMS) を使って、Keyhole Limpet Hemocyanin(KLH) の oligomeric states を調べます。 CDMS では、KLH の単一粒子を測定することで、直接その質量を測定できます。 それによって、溶液中にどのような oligomeric state、つまり 何個のタンパク質サブユニットが集まった状態 が存在しているかを判断します。 KLH の polypeptide subunit の既知の質量は Table 1 に示されています。
このサブユニット質量を使って、Figure 7 の CDMS spectrum 上で、次の oligomeric species がどこに現れるかを同定しなさい。
- 7FU Decamer
- 8FU Didecamer
- 8FU 3-Decamer
- 8FU 4-Decamer
| Polypeptide Subunit Name | Subunit Mass |
|---|---|
| 7FU | 340 kDa |
| 8FU | 400 kDa |
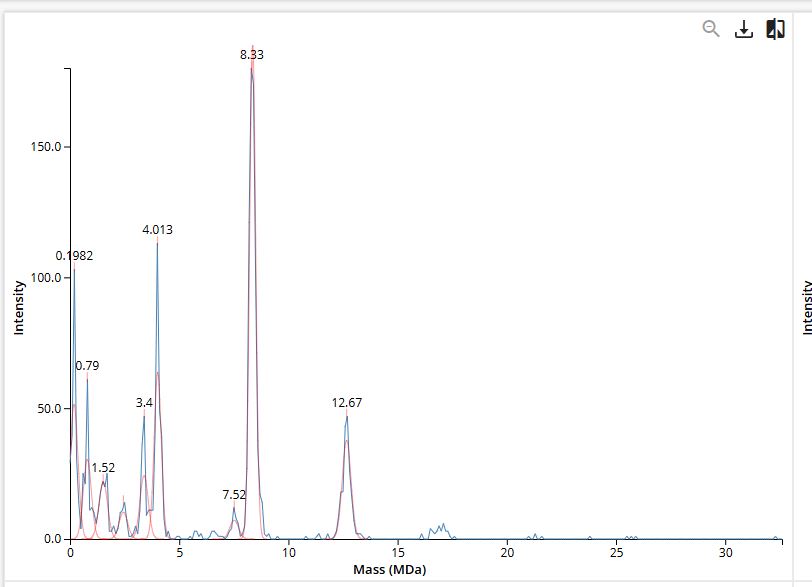
Figure 7
Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Decamer は、10個のサブユニットが集まった状態。なので
で計算する。
7FU Decamer 7FU の subunit mass は 340 kDa。 Decamer は10量体なので:
340 kDa × 10 = 3,400 kDa
7FU Decamer = 3.4 MDa
8FU Didecamer
8FU の subunit mass は 400 kDa。
Didecamer は decamer が2つ、つまり20量体なので:
400 kDa × 20 = 8,000 kDa
8FU Didecamer = 8.0 MDa
8FU 3-Decamer
3-Decamer は decamer が3つ、つまり30量体。
400 kDa × 30 = 12,000 kDa
8FU 3-Decamer = 12.0 MDa
8FU 4-Decamer
4-Decamer は decamer が4つ、つまり40量体。
400 kDa × 40 = 16,000 kDa
8FU 4-Decamer = 16.0 MDa
Therefore, on the CDMS spectrum in Figure 7, these species should appear around 3.4 MDa, 8.0 MDa, 12.0 MDa, and 16.0 MDa, respectively.
したがって、図7のCDMSスペクトル上では、これらの同位体はそれぞれ3.4 MDa、8.0 MDa、12.0 MDa、および16.0 MDa付近に現れるはずである。
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
| Theoretical | Observed/measured on the Intact LC-MS | PPM Mass Error | |
|---|---|---|---|
| Molecular weight (kDa) |
- Theoretical: 28.0066 kDa
- Observed / measured on intact LC-MS: 27.984 kDa
- PPM Mass Error: 約 810 ppm
Theoretical molecular weight は、アミノ酸配列から計算した 28.0066 kDa。
Observed molecular weight は、intact LC-MS の adjacent charge state calculation から求めた 27.984 kDa。
PPM mass error は約 810 ppm。
PPM error = |MWexperiment − MWtheory| / MWtheory × 1,000,000
PPM error = |27.984 − 28.0066| / 28.0066 × 1,000,000 PPM error ≈ 807 ppm
| Theoretical | Observed/measured on the Intact LC-MS | PPM Mass Error | |
|---|---|---|---|
| Molecular weight (kDa) | 28.0066 kDa | 27.984 kDa | ~810 ppm |