Biological engineering application: PGPR Agriculture in northern climates, such as Canada, faces structural biological constraints, particularly short and unpredictable growing seasons, cold soil temperatures in early spring, and variable nutrient availability. These constraints limit crop maturation time and increase vulnerability to climate change.
I propose developing an engineered plant growth-promoting rhizobacterium (PGPR) to enhance early-stage plant growth, nutrient uptake, and cold-stress resilience. The literature has recognized the effectiveness of PGPR in improving the growth and quality of certain crops and plants (Singh et al, 2023; Zhang et al, 2024). However, the theories have not been applied to improving the early developmental rate in cold climates. Therefore, I believe this biological engineering approach not only leverages rhizosphere ecology but also aligns agricultural productivity and ecological systems thinking.
Part 1: Benchling & In-silico Gel Art
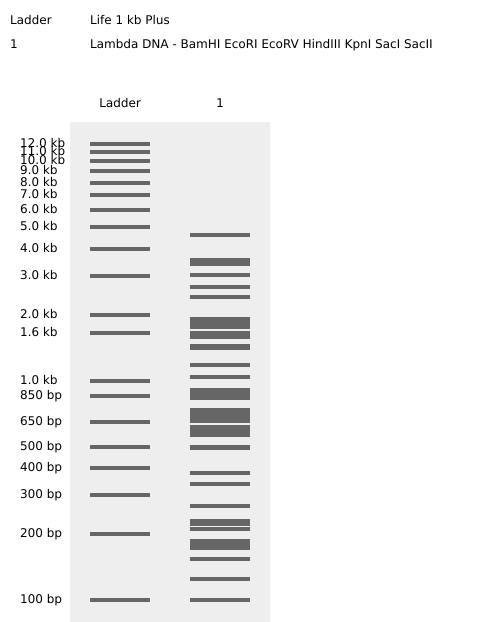
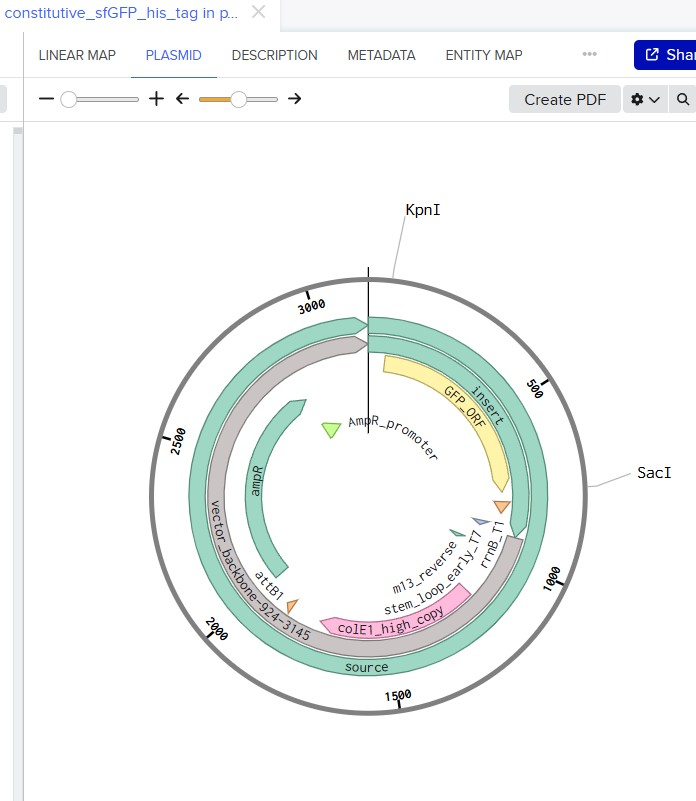
Enzymes Cuts Temp. 1.1 2.1 3.1 4/CS
BamHI 5 37°C 75* 100* 100 100*
EcoRI 5 37°C 25 100* 50 50*
EcoRV 21 37°C 10 50 100 10
HindIII 7 37°C 25 100 50 50
KpnI 2 — Not available for this vendor
SacI 2 — Not available for this vendor
SacII 4 37°C 10 100 10 100
I have difficulty creating a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. I will use this image as a placeholder for now. Sorry about that.
Part A. Conceptual Questions (the responses heavilily relied on Google)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
It depends on the meat. If it is red meat, it is about 67.8e23 amino acid molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Part A: SOD1 Binder Peptide Design (From Pranam)
SRWDVYAGAVKWARK 10.46594,
WWVPPYTAVYAWKKK 17.463782,
SRWGEYVGVYKARAA 12.508359,
WRVDVVVAVKKAKKK 12.361373,
while FLYRWLPSRRGG shows perplexity 8.1.
imTM = 0.37, seems to be a random surface and hardly binds.
imTM = 0.31, a random surface and hardly binds
imTM = 0.4, dimer interface
imTM = 0.32, random surface
imTM = 0.43, dimer interface
The imTM values are small, which returns weak binding. I don’t think that any peptide matches the known binder.
Part 1: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The key components include Phusion DNA polymerase (providing extreme fidelity), optimized reaction buffer (providing the necessary environment for enzyme activity), Magnesium Chloride (supporting DNA polymerase activity), and Deoxynucleotide Triphosphates (dNTPs - building new DNA strands). The purpose of the master mix is to reduce the risk of contamination, save time, and minimize mixing errors.
Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? The advantages include nonlinear and adaptive processing, instead of on/off states.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal. I will use the application I proposed in the Week 1 homework, soil microbes to optimize nitrogen release in crops in cold climates.
Part A: General and Lecturer-Specific Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. According to Biocompare “Advantages of Cell-Free Protein Expression,” Cell-free protein expression uses cellular lysates, instead of living cells as a source of components required for protein synthesis. The advantages include ease of use, speed in protein production, and minimal lab equipment and expertise requirements compared to traditional methods.
Homework: Final Project
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. a. Whether the hydrophobin gene is correctly inserted and expressed. b. the presence of a relative quantity of hydrophobin protein.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. This will be measured from both the DNA and protein levels. a. DNA-level on the presence and correctness of hydrophobine gene by extracting DNA from the system, amplifying target region, and verifying size and sequence. b. Protein-level on hydrophobin expression by extracting proteins, seperate by size, and detect target protein.
The homework requirements are posted under Week 11 homework page.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Mine didn’t work well, and I missed the April 19 due date.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents (used GenAI for answers)
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction. E. coli Lysate:
Week 01 HW: Engineering a Soil Microbe to Support Short-Season Agriculture in Canada
Biological engineering application: PGPR
Agriculture in northern climates, such as Canada, faces structural biological constraints, particularly short and unpredictable growing seasons, cold soil temperatures in early spring, and variable nutrient availability. These constraints limit crop maturation time and increase vulnerability to climate change.
I propose developing an engineered plant growth-promoting rhizobacterium (PGPR) to enhance early-stage plant growth, nutrient uptake, and cold-stress resilience. The literature has recognized the effectiveness of PGPR in improving the growth and quality of certain crops and plants (Singh et al, 2023; Zhang et al, 2024). However, the theories have not been applied to improving the early developmental rate in cold climates. Therefore, I believe this biological engineering approach not only leverages rhizosphere ecology but also aligns agricultural productivity and ecological systems thinking.
Policy goal: Climate Adaptation Equity
To reduce vulnerability and enhance resilience, applying PGPR to crops in harsh climates can help promote agriculture for all Canadians. In particular, Northern, small-scale, and Indigenous agricultural communities face a harsher climate environment, are more vulnerable to climate change, and have fewer resources than industrial agriculture. Therefore, the development of an engineered PGPR can promote equity if the application is cost-effective, easy to adopt, and accounts for geographic, economic, and climatic realities.
Potential governance actions
a. Technical considerations
PGPR has been proposed to be useful in certain climates, but it cannot be assumed that it will be useful in Canada, given the harsh climate. Moreover, soil composition is a complex science that can change the technical and resource requirements for northern communities. Therefore, academic researchers and scientists need to apply current understanding and practices regarding PGPR to Canadian realities and tailor these potential solutions accordingly. To facilitate research, grants should target PGPR-focused research though Natural Sciences and Engineering Research Council of Canada. The risk is that PGPR is not a good fit for Canada due to the low return relative to cost; however, understanding failure is in itself a success, as it prevents further funding or widespread application and ensures that lessons are learned. The US government has an existing 5-year study through the National Institute of Food and Agriculture Annual in 2021. The project concludes this year, and the methodology, findings, and risks can be referenced by Canadian researchers.
b. Community and Indigenous co-governance mechanisms
Formal consultation and co-design frameworks should be established while funding PGPR-focused research. Indigenous agricultural knowledge and perspectives can provide valuable insights that no laboratory can produce. Moreover, respecting soil stewardship and intergenerational responsibility aligns with both ethical guidance and legal obligations. To facilitate this, the federal government, especially the Crown-Indigenous Relations and Northern Affairs Canada, should take the lead. The risk that the Indigenous communities do not support PGPR exists, which will result in the loss of a major market in Canada. However, success is reflected in understanding of the user base, respect for Indigenous communities, adherence to the principle of inclusion, and valuable knowledge on both the scientific and political dimensions.
c. National biosafety standard update
The current edition of the Canadian BiosafetyStandard was published in 2022. Microbe-related mentions are primarily about toxins and laboratory practices. There is no standard for assessing the safety of larger ecosystems for PGPR and potential ecosystem implications. Given that we are now in 2026 in the AI era, the standard is due for an update to consider ecological risk modelling, mandatory post-release surveillance, and data transparency requirements. National regulatory bodies, such as the Canadian Food Inspection Agency, should lead the revision of the standard to include an expanded scope. The risk is that the standard update proceeds slowly and may not apply to PGPR if it is scientifically sound and a wide application becomes feasible. However, regardless of what PGPR can or cannot do, the biosafety standard is due for an update, and such an update would be successful if its scope were expanded to meet current economic and environmental needs.
Score (from 1-3 with, 1 as the best, or n/a) each of your governance actions
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
3
2
1
• By helping respond
1
2
3
Foster Lab Safety
• By preventing incident
2
3
1
• By helping respond
3
2
1
Protect the environment
• By preventing incidents
1
2
3
• By helping respond
1
3
2
Other considerations
• Minimizing costs and burdens to stakeholders
1
2
3
• Feasibility?
2
1
3
• Not impede research
1
2
3
• Promote constructive applications
2
1
3
Combination of options
The three governance options have distinguished focuses on research, collaboration, and policy. A scientific innovation needs a holistic governance structure to ensure its scientific feasibility and credibility, respect for diverse knowledge and legal obligation, and policy alignment to ensure safety. Therefore, I believe all three options should be incorporated without prioritizing. The biggest trade-off is the need and the time invested. No scientific innovation and testing can happen overnight, but uncertainties about theories and feasibility may lead stakeholders to question whether investments are worth the uncertain returns, while many innovative ideas do not require long waits for research on feasibility and can deliver immediate returns. This is misleading, as immediate implementation does not guarantee immediate returns, and understanding the science and technology is essential, even if it takes time. Therefore, the main audience for this approach is the Canadian federal government, which invests in research, understands wait times, leads engagement, and updates standards. Given that the result can potentially benefit the national interest of Canada, especially in today’s trade environment,
I have difficulty creating a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. I will use this image as a placeholder for now. Sorry about that.
Part 3: DNA Design Challenge
3.1. Choose your protein.
I chose the cold shock protein CspA from Bacillus subtilis because it plays a role in cellular adaptation to low temperatures. Since my Week 1 homework focused on enhancing crop growth in short-season Canadian climates, selecting a protein involved in cold resilience aligns conceptually with my broader engineering interests.
I obtained the following protein sequence from Uniprot under the protein name Cold shock protein CspC, gene name cspC, BSU0520, CSPC_BACSU, P39158.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
One result from Bioinformatics.org shows
reverse translation of sp|P39158|CSPC_BACSU Cold shock protein CspC OS=Bacillus subtilis (strain 168) OX=224308 GN=cspC PE=1 SV=1 to a 198 base sequence of most likely codons.
Codon optimization is necessary because different organisms preferentially use specific codons to encode the same amino acid. Although the genetic code is universal, codon usage bias affects translation efficiency and protein yield. I optimized the Bacillus subtilis expression sequence to improve expression efficiency in a soil bacterium relevant to my project proposed in Week 1 homework. The selection of Bacillus subtilis used help from GenAI. The tool is IDT Codon Optimization Tool https://www.idtdna.com/CodonOpt. The product type is gBlocks Gene Fragments. Restriction sites to avoid are Bsal, BsmBL, and Bbsl.
I will explore the cell-free method, as it does not require living organisms, so that the system can be more controlled. In this way, cell-free expression systems could synthesize the protein directly from the DNA template without using living cells.
I will have to revisit this part, once I go through the lecture again. Currently, the answers have relied heavily on Google and GenAI.
5.1 DNA Read
(i) What DNA would you want to sequence and why?
I would want to sequence DNA used in DNA-based digital data storage, which was used in the homework example. This would allow verification of synthesis accuracy, detection of errors introduced during storage or replication, and complete recovery of the original encoded information.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
(I sought help from ChatGPT to answer this question.)
Illumina Sequencing by Synthesis (SBS) due to the parallel sequencing of fragment libraries and accuracy.
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
Second-generation due to the generated short reads.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
DNA molecules (synthetic oligo libraries) as input, adapter ligation to both ends, and PCR to create clusters.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
DNA fragments bind to a flow cell and form clusters by bridge amplification.
What is the output of your chosen sequencing technology?
FASTQ files with short reads.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesize DNA constructs encoding digitally compressed cultural content because it enables long-term digital archiving.
(ii) What technology or technologies would you use to perform this DNA synthesis and why? Also answer the following questions:
I would use chip-based solid-phase oligonucleotide synthesis, such as the platforms developed by Twist Bioscience and others. These use phosphoramidite chemistry in a massively parallel microarray format.
What are the essential steps of your chosen sequencing methods?
-A DNA chain, protected nucleotides, and deprotection and coupling cycles.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
-error and length
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would like to continue my proposed project in Week 1 homework and add this week’s work on Bacillus subtilis.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
I would like to employ CRISPR-Cas9, as mentioned in Professor Church’s slide. It is a tool that is frequently mentioned in the lecture, and I wish to explore more.
How does your technology of choice edit DNA? What are the essential steps?
-CRISPR-Cas9 uses a short guide RNA (gRNA) to direct Cas9 to a specific DNA sequence. Once bound, Cas9 introduces a double-strand break at the target site.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
-1) Guide RNA(s) targeting loci involved in repair or replication; 2) Cas9 or base/prime editor constructs; 3) Donor DNA templates for HDR when introducing specific sequences; and 4) Competent cells capable of taking up editing components
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
-Unintended edits can occur at similar sequences.
Week 1 Homework: Lecture 2 Preparation:
Professor Jacobson’s lecture:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate is 1:100,000 or 1,000,000 throughput: 10mS per Base Addition (according to the slide and Google).
Human Genome Length: A single human diploid cell contains roughly 6 million base pairs. The discrepancy without correction, if the error rate were 100,000, every cell division would introduce roughly 100,000-600,000 errors. This is drawn from Google AI Overview.
One way biology deals with the discrepancy is intrinsic proofreading (3’-5’ exonuclease), according to Google.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
(Directly from Google search)
Different ways: synonymous codons, mathematical combinations, and redundant genetic code.
Reasons not working: condon usage bias, protein folding defects, mRNA stability and decay, mRNA structure, and transcriptional efficiency.
Dr. LeProust’s lecture:
What’s the most commonly used method for oligo synthesis currently?
Solid-phase phosphoramidite chemistry. (Google)
Why is it difficult to make oligos longer than 200nt via direct synthesis?
It is difficult because the product’s yield and purity decrease exponentially with each added base. (Google)
Why can’t you make a 2000bp gene via direct oligo synthesis?
This is because conventional automated phosphoramidite synthesis is limited by the efficiency of nucleotide coupling and error accumulation. (Google)
George Church’s lecture (choose one to answer):
(Using Google & Prof. Church’s slide #4) What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”
The ten essential amino acids are Arginine, Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, and Valine. The effect is that humans may be able to control nature if they control these ten amino acids, which is not true. (Google)
Note: I apologize for heavily relying on Google, but I am struggling to understand the material by myself.
pass this e.g. ‘Red’ and get back a Location which can be passed to aspirate()
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well]
raise ValueError(f"No well found with color {color_string}")
For this lab, instead of calling pipette.dispense(1, loc) use this: dispense_and_detach(pipette, 1, loc)
def dispense_and_detach(pipette, volume, location):
"""
Move laterally 5mm above the plate (to avoid smearing a drop); then drop down to the plate,
dispense, move back up 5mm to detach drop, and stay high to be ready for next lateral move.
5mm because a 4uL drop is 2mm diameter; and a 2deg tilt in the agar pour is >3mm difference across a plate.
"""
assert(isinstance(volume, (int, float)))
above_location = location.move(types.Point(z=location.point.z + 5)) # 5mm above
pipette.move_to(above_location) # Go to 5mm above the dispensing location
pipette.dispense(volume, location) # Go straight downwards and dispense
pipette.move_to(above_location) # Go straight up to detach drop and stay high
The art is published at opentrons-art.rcdonovan.com/?id=89w6rb219717q1z
The code is generated with the help of GenAI.
Post-Lab Questions:
Article:
Bryant, J. A., Kellinger, M., Longmire, C., Miller, R., & Wright, R. C. (2023). AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots. Synthetic Biology (Oxford University Press), 8(1), ysac032. https://doi.org/10.1093/synbio/ysac032
The article presents AssemblyTron, an open-source automation platform that enables flexible and programmable DNA assembly workflows using Opentrons OT-2 liquid-handling robots. The system was developed to overcome common bottlenecks in synthetic biology — especially the manual, repetitive pipetting steps required for assembling DNA constructs.
Week 04 HW: Protein Design Part 1
Part A. Conceptual Questions (the responses heavilily relied on Google)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
It depends on the meat. If it is red meat, it is about 67.8e23 amino acid molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans’ digestive system and immune system identify and destroy foreign DNA, preventing it from being incorporated into the human genome. Therefore, human bodies break down foreign proteins and DNA into basic nutrients, using them to build human cells rather than adopting the food’s genetic structure.
Why are there only 20 natural amino acids?
The 20 natural amino acids are the result of evolution, which has optimized them to create stable, functional, and soluble proteins. In this way, 20 suffices to create all necessary proteins, offering resistance against mutations.
Can you make other non-natural amino acids? Design some new amino acids.
Yes. For example, Photo-crosslinkers, such as the 4-benzoyl-L-phenylalanine (BPA), are used to map protein-protein interactions by creating covalent bonds upon exposure to UV light.
Where did amino acids come from before enzymes that make them, and before life started?
Likely through non-biological, prebiotic chemical reactions driven by energy sources like UV light, lightning, or hydrothermal heat acting on simple atmospheric gases.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A left-handed helix. While natural proteins made of L-amino acids form right-handed
-helices, D-amino acids are mirror images of L-amino acids, causing their stable secondary structures to form the corresponding mirror-image (left-handed) helix
Can you discover additional helices in proteins?
Researchers have discovered that by modifying protein ends, they can create or stabilize specific helices to study their function, such as HBS (Hydrogen Bond Surrogate) helices: Designed to cap the N-terminus of a helix to increase stability, which can act as a “new” type of stable helical structure in design contexts.
Why are most molecular helices right-handed?
Because this structure is energetically more stable and allows for less steric hindrance between amino acid side chains or bases.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Because of their inherent structural propensity to form extensive intermolecular hydrogen bonds. The driving force for this aggregation is a combination of hydrophobic effects, hydrogen bonding, and, most importantly, the dehydration of pre-formed intramolecular hydrogen bonds, which triggers the burial of non-polar groups and the formation of highly stable “cross-β” structures.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
The protein I chose was Nitrogenase iron protein (NifH). Nitrogenase is the enzyme complex responsible for converting atmospheric nitrogen (N₂) into ammonia (NH₃), which plants can use. Because nitrogen availability limits crop productivity in cold climates, improving the efficiency of nitrogen fixation in engineered soil microbes is directly relevant to my proposed final project.
-How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
-Does your protein belong to any protein family?
NifH/BchL/ChlL family.
Identify the structure page of your protein in RCSB
-When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
-Are there any other molecules in the solved structure apart from protein?
ADP or ATP analogs
-Does your protein belong to any structure classification family?
NifH/FrxC-like; SCOP ID: 4003981; PF00142
Open the structure of your protein in any 3D molecule visualization software:
-PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands). Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
-Color the protein by secondary structure. Does it have more helices or sheets?
A mix of alpha helices and beta sheets.
-Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophobic residues cluster in the protein core.
-Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes, it does.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans: Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
-Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The deep mutational scan reveals strong intolerance to mutation at conserved cysteine residues.
Latent Space Analysis: Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
NifH is positioned centrally within the nitrogenase cluster.
C2. Protein Folding
-Folding a protein: Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
There will be small modifications.
-Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
The structure may look different, especially with Large segment mutations to critical residues. However, the function may not be fundamentally shifted by changes in structure.
C3. Protein Generation
-Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
The structure resembles the original.
Part D. Group Brainstorm on Bacteriophage Engineering
I would like to choose the goal, Increased stability. This is because Lysis proteins are often small, membrane-associated, and partially disordered, and can be unstable when expressed at high levels.
One tool to use would be ESMFold, which predicts 3D structure of L protein, and stability engineering requires structural context. Another tool would be ESM2 language model to perform single-residue mutational scanning. The potential pitfall is the stability vs. function tradeoff, as mutations that stabilize fold may reduce lytic activity.
Input: L Protein Sequence → ESMFold: Predict Structure to identify unstable regions (low pLDDT) → ESM2 Deep Mutational Scan to select tolerated stabilizing mutations → ESMFold re-prediction of mutants to tank variants for experimental validation
Week 05 HW: Protein Design Part 2
Part A: SOD1 Binder Peptide Design (From Pranam)
SRWDVYAGAVKWARK 10.46594,
WWVPPYTAVYAWKKK 17.463782,
SRWGEYVGVYKARAA 12.508359,
WRVDVVVAVKKAKKK 12.361373,
while FLYRWLPSRRGG shows perplexity 8.1.
imTM = 0.37, seems to be a random surface and hardly binds.
imTM = 0.31, a random surface and hardly binds
imTM = 0.4, dimer interface
imTM = 0.32, random surface
imTM = 0.43, dimer interface
The imTM values are small, which returns weak binding. I don’t think that any peptide matches the known binder.
Weak binding, soluble, non-hemolytical, 2.46, 1793 Da
Weak binding, soluble, non-hemolytical, 2.76, 1923.3 Da
Weak binding, soluble, non-hemolytical, 1.46, 1712.9 Da
Weak binding, soluble, non-hemolytical, 4.76, 1754.2 Da
Weak binding, soluble, non-hemolytical, 2.76, 1507.7 Da
I see a similar pattern regarding week binding, being soluble, and non-hemolytic. The net charge does not seem to play a role in binding. Based on this, it can be predicted that strong binders will be hemolytic and poorly soluble. In theory, higher ipTM should show stronger predicted affinity, but mine are all weak. I don’t have a peptide of strong binding yet.
I failed to generate peptides on moPPit Colab. Theoretically, I believe that the peptides would be stronger because of the specific instructions (binding locations, criteria…). In this way, peptides would be more chemically balanced, less likely to cause toxicity, and better suited for therapeutic development.
Part C: Final Project: L-Protein Mutants
I can’t finish running the notebook, as I get stuck at from google.colab import files. I tried two different networks and spent hours running them, but I can’t continue. Therefore, in the absence of the notebook scores, I expect variations between the experimental data and the scores, as experience and predictions never align perfectly.
The five mutated sequences could be:
Q to E. METRFPQQSQETPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
S to T. METRFPQQSQQTPASTNTRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
L to I. METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFIAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
F to Y. METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIYLSKFTNQLLLSLLEAVIRTVTTLQQLLT
V to I. METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAIIRTVTTLQQLLT
Week 06 HW: Genetic Circuit Part 1
Part 1: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The key components include Phusion DNA polymerase (providing extreme fidelity), optimized reaction buffer (providing the necessary environment for enzyme activity), Magnesium Chloride (supporting DNA polymerase activity), and Deoxynucleotide Triphosphates (dNTPs - building new DNA strands). The purpose of the master mix is to reduce the risk of contamination, save time, and minimize mixing errors.
What are some factors that determine primer annealing temperature during PCR?
Primer melting temperature is the most crucial factor. Primer length, Guanine and Cytosine content, primer concentration, and primer pair match are also primary factors that determine primer annealing temperature during PCR.
There are two methods from this class that create linear fragments of DNA: PCR and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both PCR and restriction enzyme digests are fundamental to DNA manipulation and analysis. In general, unless PCR, Restriction Enzyme Digest doesn’t have Magnesium or dNTP as reagents, add the enzyme last in a reaction setup, require a lower temperature, and last shorter than RCR. In general, PCR is chosen when the target sequence is unknown or needs to add specific restriction sites. Restriction Digests are chosen when there is already a cloned construct and a need for high fidelity (can’t risk introducing mutations)
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
There are a few considerations of primer design, including high-fidelity amplification, purification, and accurate quantification to ensure optimal overlap and molar ratios.
How does the plasmid DNA enter the E. coli cells during transformation?
By passing through transient pores created in the cell membrane. A heat shock creates a thermal imbalance that facilitates the movement of negatively charged DNA through the pores.
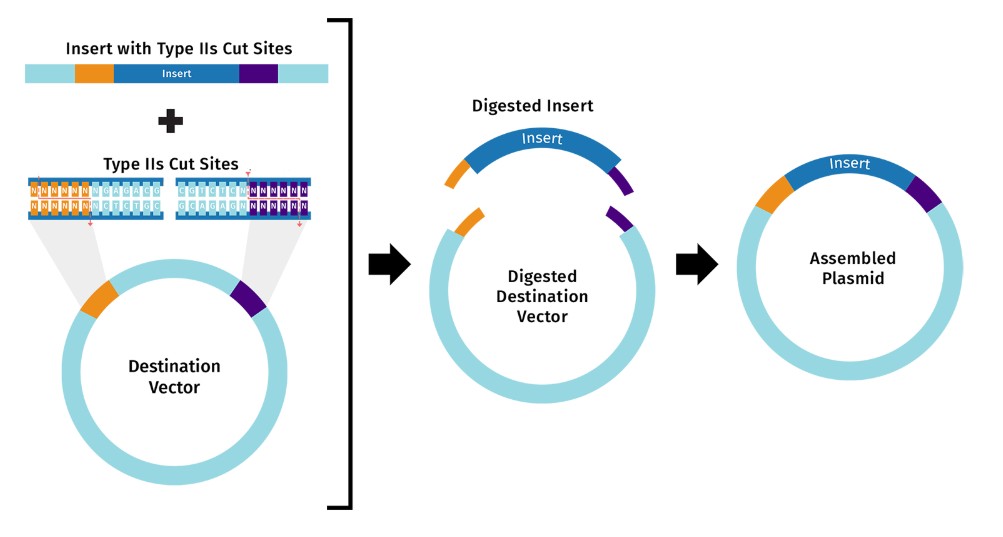
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
This response is based on SnapGene Academy. Golden Gate Assembly is a one-pot, one-reaction cloning procedure created by Carola Engler et al. in 2008. The method takes advantage of Type IIS restriction enzymes (e.g. BsaI), which cleave DNA outside their recognition sequences. The result is an ordered assembly of a vector and one or more DNA fragments.
Golden Gate assembly can be split into two distinct steps that occur within the same reaction: 1) Type IIS restriction enzyme digestion, and 2) DNA ligation. It works by mixing the following components into a single reaction tube: Destination vector
DNA insert(s) (e.g. amplicon or pre-cloned), Type IIS restriction enzyme (e.g. BsaI),
T4 DNA ligase, and Reaction buffer. Golden Gate Assembly.jpg
Model this assembly method with Benchling or Asimov Kernel!
Modeling with benchling is based on the response from Google AI Overview. Modeling Golden Gate Assembly in Benchling is streamlined through their built-in Assembly Wizard, which supports the one-pot, scarless assembly of multiple DNA fragments using Type IIS restriction enzymes (e.g., BsaI, BsmBI). Benchling, in collaboration with New England Biolabs (NEB), allows to simulate digestion and ligation, generate specific 4-base overhangs, and design the necessary primers to add Type IIS sites to inserts.
Part 2: Asimov Kernel
The results are recorded in my notebook “SK”. I am not sure about the third construct, as the results from the first two are not satisfactory.
Week 07 HW: Genetic Circuit Part 2
Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
The advantages include nonlinear and adaptive processing, instead of on/off states.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
I will use the application I proposed in the Week 1 homework, soil microbes to optimize nitrogen release in crops in cold climates.
The input signals include sensing multiple environmental factors, such as soil nitrate levels, plant growth activity, and soil temperature. The IANN processing is weighted and nonlinear. For example, low nitrogen levels combined with active plant behaviours signal strong activation, while cold temperatures signal partial suppression. The output behaviour would regulate the expression of nitrogen-fixation genes under suitable conditions.
The limitations include the inherent noise of gene expression, as well as the limited precision of weights in complex biological systems.
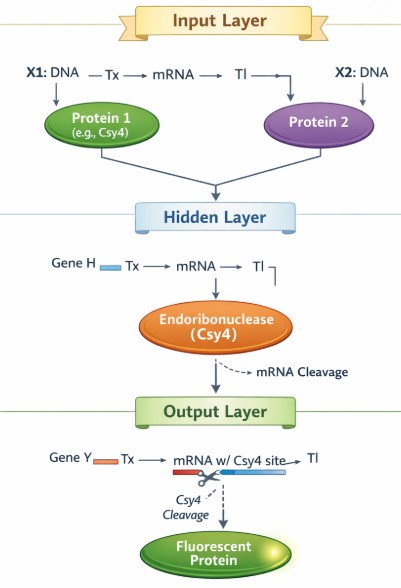
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Got help from GenAI.
Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials are primarily based on mycelium, which is the root structure of mushrooms. Sample existing materials include mycelium-based packaging and insulation. The advantage is that it is more environmentally friendly and sustainable than traditional materials, which are manufactured instead of grown. The disadvantage is its high water absorption, which makes it unsustainable in humid environments, and a lack of scalability, especially in areas that need high mechanical strength, such as buildings.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Genetically engineering fungi enables the development of valuable, organic materials for food and pharmaceuticals. It also provides environmental solutions when producing what we need in industrial settings. It has an advantage over bacteria because of its eukaryotic internal machinery, protein secretion, and environmental tolerance.
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
According to Biocompare “Advantages of Cell-Free Protein Expression,” Cell-free protein expression uses cellular lysates, instead of living cells as a source of components required for protein synthesis. The advantages include ease of use, speed in protein production, and minimal lab equipment and expertise requirements compared to traditional methods.
The first case is the rapid prototyping and high-throughput screening. I think COVID screening was used as an example in the lecture. The second case was also mentioned in the lecture, which used cellular lysates (ribosomes and enzymes) rather than living cells to ensure that the toxic protein couldn’t kill the machine that produces it.
Describe the main components of a cell-free expression system and explain the role of each component.
According to New England Biolabs, Cell-free protein synthesis (CFPS) is a protein expression approach that enables production of a target protein without the use of living cells. In CFPS, a solution containing all the cellular machinery needed to direct protein synthesis (e.g., ribosomes, tRNAs, enzymes, cofactors, amino acids, etc.) is used to transcribe and translate a supplied nucleic acid template (e.g., plasmid DNA, linear DNA or mRNA).
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
This is because the CFPS system always requires a high ATP input, which outperforms traditional ATP-regeneration systems. An article titled “Cell-free Systems to Mimic and Expand Metabolism” indicates that machine learning, design of experiments, and model-based predictions will accelerate the improvement of cell-free metabolism, particularly because in vitro experiments can rapidly generate large sets of training data required for useful algorithms.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems offer high yields and low costs, making them ideal for simple proteins, such as the E. coli system for producing Green Fluorescent Protein. Eukaryotic systems offer complex folding and post-translational modification. For Human Erythropoietin, Eukaryotic lysates provide the machinery that prokaryotic systems can’t perform and ensure the protein is properly folded.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
According to Google AI overview, designing a cell-free experiment to optimize the expression of a membrane protein focuses on maximizing yield by adjusting detergent concentration, optimizing magnesium/potassium ratios, and increasing template concentration. Key challenges include protein aggregation and incorrect folding. To address them, using detergents for solubilization without interfering with translation and adding DnaK, etc., to assist with folding can be considered.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
A common reason is the rapid depletion of energy resources, such as ATP. Using a continuous-exchange cell-free (CECF) system allows for the continuous replenishment of substrates and removal of inhibitory byproducts. Another reason is the improper protein folding. Lowering the incubation temperature can slow down translation and allow more time for proper folding. Inefficient transcription/translation coupling can also be a reason, which can be tackled by optimizing the DNA template concentration and quality.
Part B: Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
The answers heavily relied on GenAI.
Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
My cell is a synthetic minimal cell designed to detect lactate levels. The input would be normal things like sweat or tissues, and the output would be a fluorescent signal when lactate is high.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Maybe, but encapsulation improves stability.
c. Could this function be realized by genetically modified natural cell?
Yes, engineered Escherichia.
d. Describe the desired outcome of your synthetic cell operation.
A visible fluorescent signal proportional to lactate concentration, enabling real-time metabolic monitoring.
Design all components that would need to be part of your synthetic cell.
a. What would be the membrane made of?
Phospholipid bilayer.
b. What would you encapsulate inside? Enzymes, small molecules.
Cell-free transcription/translation (Tx/Tl) system, DNA encoding sensor + reporter
RNA polymerase, ribosomes, Amino acids, ATP, cofactors, and Lactate-responsive transcription factor
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
It will come from a bacterial system such as Escherichia coli, simple, fast, and sufficient for small-molecule sensing.
d. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Lactate can passively diffuse across the membrane.
Experimental details
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
b. How will you measure the function of your system?
Measure fluorescence intensity and compare the signal across different lactate concentrations.
Parc C: Homework question from Peter Nguyen:
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
The answers heavily relied on GenAI.
a. Write a one-sentence summary pitch sentence describing your concept.
A smart textile embedded with freeze-dried cell-free systems that detects sweat biomarkers (e.g., stress or fatigue) and produces a visible colour change in real time.
b. How will the idea work, in more detail? Write 3-4 sentences or more.
A Freeze-dried cell-free transcription/translation system is embedded into fabric fibres or patches. When activated by sweat (moisture), the system rehydrates and detects specific biomarkers such as lactate or cortisol. A biosensor circuit triggers the production of a fluorescent protein, causing the fabric to visibly change color. This enables continuous, non-invasive monitoring during daily activities or exercise.
c. What societal challenge or market need will this address?
This addresses the growing demand for real-time, non-invasive health monitoring, particularly in fitness, mental health, and occupational safety. It enables early detection of fatigue, stress, or overexertion, reducing risks in high-performance or high-risk environments (e.g., athletes, healthcare workers).
d. How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Use sweat as a natural trigger for rehydration. Stability in protective polymers or hydroge is to extend shelf life
Part D: Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
The answers heavily relied on GenAI.
a. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Long-duration spaceflight exposes astronauts to microgravity, which alters metabolism and can lead to muscle fatigue and impaired recovery. Monitoring metabolic stress in space is challenging due to limited lab infrastructure. Cell-free systems, such as BioBits Cell-Free Protein Expression System, offer a lightweight and stable platform for biosensing in resource-constrained environments. Developing a portable system to detect metabolic biomarkers like lactate could enable real-time health monitoring in space. This is significant for maintaining astronaut performance, reducing health risks, and advancing autonomous biomedical diagnostics for deep-space missions.
b. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Lactate and the lactate-responsive regulator LldR, coupled to a GFP reporter system.
c. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Lactate is a key biomarker of metabolic stress and muscle fatigue, both of which are affected by microgravity. The transcription factor LldR responds to lactate levels and can regulate gene expression accordingly. By coupling LldR to a reporter gene, such as GFP, the system can translate metabolic changes into a measurable fluorescent signal. This enables indirect monitoring of astronaut physiological status using a simple biochemical assay, making it highly suitable for space environments where traditional diagnostic tools are limited.
d. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
We hypothesize that a freeze-dried cell-free system incorporating an LldR-regulated genetic circuit can reliably detect lactate levels and produce a quantifiable fluorescent signal under space-relevant conditions. The goal is to demonstrate that cell-free biosensors remain functional after storage and activation in microgravity-like environments. This approach leverages the stability and portability of cell-free systems while avoiding the complexity of maintaining living cells. If successful, this system could provide a scalable platform for monitoring astronaut health biomarkers in real time. The reasoning is based on prior success of cell-free systems in detecting small molecules on Earth, combined with their demonstrated robustness to freeze-drying and rehydration.
e. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Freeze-dried reactions using the BioBits Cell-Free Protein Expression System will be rehydrated with samples containing varying lactate concentrations. A genetic circuit with LldR controlling GFP expression will be tested. Controls include reactions without lactate and without DNA. Fluorescence output will be measured using the P51 Molecular Fluorescence Viewer. If needed, DNA amplification can be performed using the miniPCR thermal cycler. Data will be collected as fluorescence intensity relative to lactate concentration.
Week 10 HW: Imaging and Measurement
Homework: Final Project
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
a. Whether the hydrophobin gene is correctly inserted and expressed. b. the presence of a relative quantity of hydrophobin protein.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
This will be measured from both the DNA and protein levels. a. DNA-level on the presence and correctness of hydrophobine gene by extracting DNA from the system, amplifying target region, and verifying size and sequence. b. Protein-level on hydrophobin expression by extracting proteins, seperate by size, and detect target protein.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
a. DNA sequencing to confirm correct design with no mutations. b. PCR to confirm gene presence.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Theoretical pI/Mw: 5.90 / 28006.60
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
z=22; MW = 32977.8 Da; accuracy = 17.7% error
Homework: Waters Part III — Peptide Mapping - primary structure
K:20; R:9
19 peptides generated
22 peaks between 0.5 and 6 minutes.
There are more peaks in the chromatogram.
m/z = 525. z=1, [M+H]+=523.9
error = -19.1ppm
I am not sure. The sequence appears to be shifted by 1 amino acid to the right relative to the original sequence.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Theoretical: 524
Observed: 523.99
PPM Mass Error: -19.1ppm
Week 11 HW: Building Genomes
HTGAA 2026 Global Pixel Artwork Cloud Lab
Final projects (forms, slides, and orders are complete)
Week 12 HW: Bioproduction
The homework requirements are posted under Week 11 homework page.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Mine didn’t work well, and I missed the April 19 due date.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents (used GenAI for answers)
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate:
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): Provides the core cellular machinery for transcription and translation, including ribosomes, enzymes, and T7 RNA polymerase for high-efficiency expression of genes under T7 promoters.
Salts/Buffer:
Potassium Glutamate: Maintains ionic strength and mimics intracellular conditions to support enzyme activity and protein synthesis.
HEPES-KOH pH 7.5: Acts as a buffering agent to stabilize pH during the reaction, ensuring optimal enzyme function.
Magnesium Glutamate: Essential cofactor for ribosomes and polymerases, supporting transcription and translation processes.
Potassium phosphate monobasic / Potassium phosphate dibasic: Provide additional buffering capacity and help maintain phosphate balance for energy metabolism and nucleic acid stability.
Energy / Nucleotide System:
Ribose / Glucose: Serve as energy sources and metabolic substrates to regenerate ATP and sustain the reaction.
AMP / CMP / GMP / UMP: Nucleotide monophosphates that are phosphorylated to form NTPs required for RNA synthesis.
Guanine: A nucleotide base that supports nucleotide pool balance and efficient transcription.
Translation Mix (Amino Acids):
17 Amino Acid Mix: Provides most of the amino acids required for protein synthesis.
Tyrosine / Cysteine: Added separately due to stability or solubility limitations, completing the full set of amino acids for translation.
Additives:
Nicotinamide: Acts as a cofactor precursor (NAD⁺ related), supporting redox balance and metabolic reactions in the lysate.
Backfill:
Nuclease Free Water: Used to adjust reaction volume and maintain purity by preventing degradation of DNA or RNA.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour PEP-NTP relies on rapic but short-lived protein synthesis. The 20-hour-NMP-ribose-glucose mix relies on metabolic pathways, so it is more sustainable, though slower.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design (used GenAI for answers)
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: Engineered for enhanced folding efficiency and stability, sfGFP folds reliably even in suboptimal conditions, making it highly robust in cell-free systems.
mRFP1: Has a slow maturation time, meaning fluorescence develops gradually, which can limit early signal detection in short reactions.
mKO2: Sensitive to pH changes (acid sensitivity), so fluorescence intensity can decrease if the reaction environment becomes slightly acidic over time.
mTurquoise2: Requires proper protein folding for high quantum yield, and misfolding can significantly reduce fluorescence output.
mScarlet_I: Optimized for fast maturation and high brightness, making it well-suited for strong fluorescence but still dependent on proper folding conditions.
Electra2: A newer fluorescent protein with enhanced brightness but potential sensitivity to oxygen and folding conditions, which can impact fluorescence consistency.
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Hypothesis: Increasing magnesium glutamate concentration and optimizing energy regeneration (e.g., glucose-based system) will improve folding efficiency and sustained expression of mRFP1, leading to higher fluorescence over 36 hours.
Protein: mRFP1
Reagents: Increased magnesium glutamate concentration and a glucose-based energy regeneration system
I don’t think these applies to me, as I don’t have the lab equipment nor received an email instruction.
Expected Effect: Enhancing magnesium levels will improve ribosome activity and protein folding, while sustained energy supply from glucose will extend protein synthesis time, allowing more mRFP1 to properly fold and mature. This will result in higher fluorescence intensity over a 36-hour incubation.