Week 2 HW: DNA Read, Write, & Edit
Gel Electrophoresis Designs
A pattern in the style of Paul Vanouse’s Latent Figure Protocol
I created an image of Mount Fuji with clouds in the sky. The image is inverted to make the figure easier to see.
Note: We worked in groups during lab this week, so the design we ran on the gel was different from the one above.
DNA Design Challenge
The protein
RES-701-3 is a tiny natural protein made by soil bacteria (Streptomyces). It belongs to a family called lasso peptides, named because the structure looks like a lasso or slipknot: the tail of the protein threads through a loop, creating a knot that is extremely hard to unravel.
This knotted shape makes lasso peptides unusually tough. They resist digestive enzymes, heat, and harsh chemical environments. These are properties that most proteins lack, and that make lasso peptides attractive as potential drugs.
RES-701-3 blocks a receptor on the surface of blood vessel cells called the endothelin type B receptor (ETB). The endothelin system controls blood vessel tightening and relaxation, and it becomes dysregulated with age, contributing to high blood pressure and vascular disease. RES-701-3 acts as an inverse agonist: it blocks the receptor and pushes it toward a state less active than its resting baseline.
In nature, the bacterium makes this peptide in two parts:
| Section | Sequence |
|---|---|
| Leader | MSDITLTPMDLLDLDELAAGGGRSTARE |
| Core | GNWHEPEIDGWNPHGW |
An enzyme cleaves the core away from the leader to produce the active peptide.
Reverse translation: protein to DNA
| Section | Nucleotide sequence |
|---|---|
| Leader | ATGAGCGATATTACCCTGACCCCGATGGATCTGCTGGATCTGGATGAACTGGCTGCTGGTGGTGGTCGTAGCACCGCTCGTGAA |
| Core | GGTAACTGGCATGAACCGGAAATTGATGGTTGGAACCCGCATGGTTGGTAA |
Codon optimization
Different species use different codons preferentially, and have abundant matching tRNAs for those codons. RES-701-3 comes from Streptomyces, which strongly prefers codons rich in G and C. Twist offers a Streptomyces coelicolor codon optimization profile.
It’s also worth noting that Shihoya et al. (2025) used Streptomyces venezuelae and reported the highest yields to date. In a real drug development setting, that organism might be a better choice.
The codon-optimized variant for the leader and core together:
Expression cassette parts
Promoter
The ermE*p promoter is the most widely used for gene expression in Streptomyces.
Ribosome binding site
A Shine–Dalgarno (SD) sequence, AAGGAG, is a good RBS for Streptomyces with leader peptides. It is positioned 6 to 10 nucleotides upstream of the start codon, so we use a 7-nucleotide spacer downstream and add CGACG upstream:
Start codon
The standard ATG.
Coding sequence
Both the leader and core peptide sequences in tandem.
His tag
A short stretch of six histidines that lets you fish the protein out of a mixture using a nickel column. In practice, a His tag is not a great choice for RES-701-3, because it would interfere with binding to the ETB receptor.
Stop codon
TGA is the preferred stop codon in Streptomyces because it is GC-rich, matching the organism’s overall DNA composition. (For comparison, the typical stop codon in many organisms is TAA.)
Terminator
The fd terminator from a bacteriophage, commonly used in Streptomyces expression vectors.
Reagents
To produce the mature peptide we also need three biosynthetic enzymes: LasB1, LasB2, and LasC. LasB1 binds the leader and delivers the precursor to LasB2, which cuts the leader off; LasC then closes the lasso ring on the core. These enzymes are not easy to order, and the highest-yield expression host (S. venezuelae) is also less common, so RES-701-3 is probably not the most practical target for class.
Twist DNA Synthesis Order
The lasso peptide order has been prepared. Below is the expression cassette in Benchling.
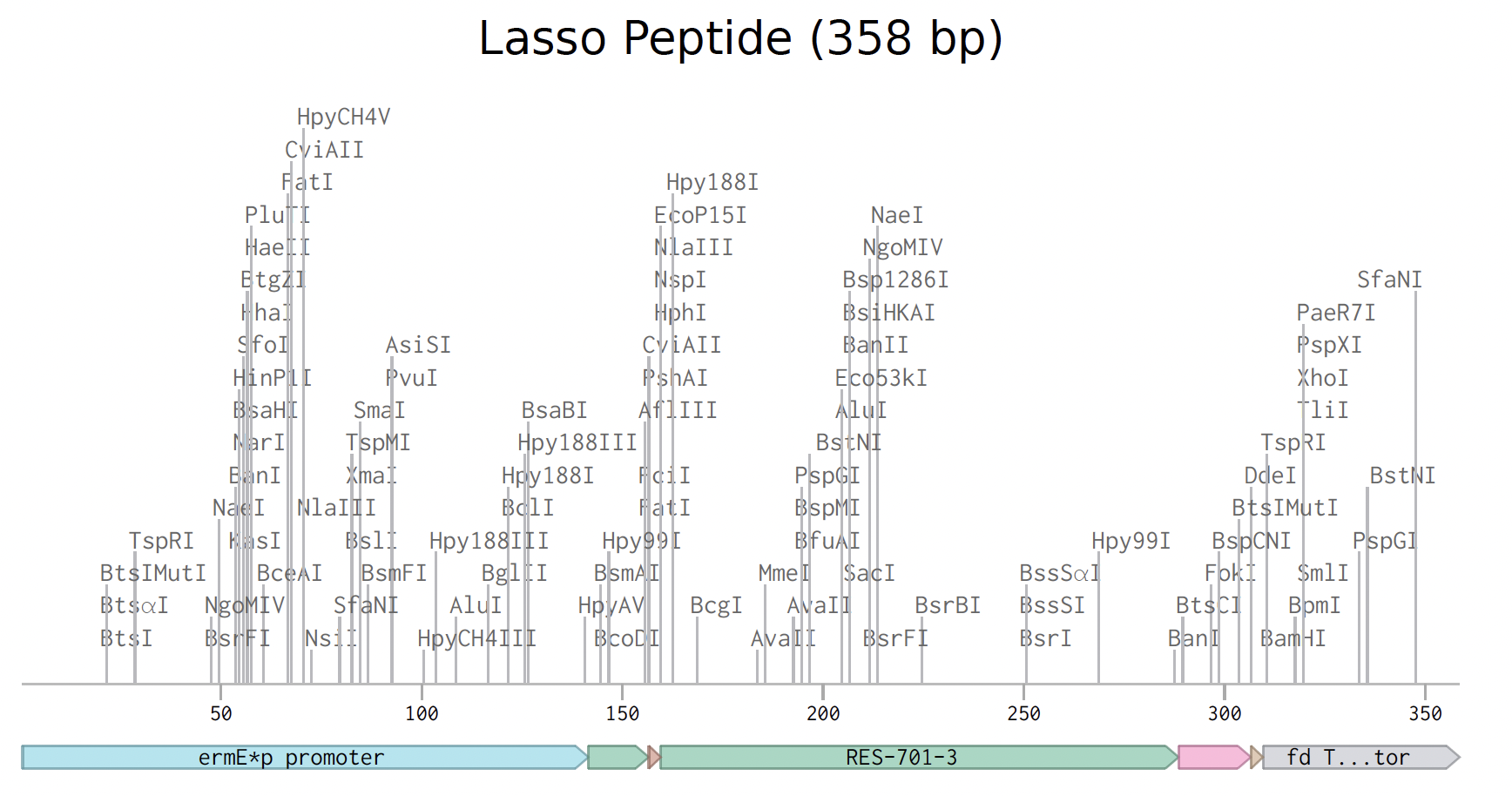
I used gene fragments rather than a clonal gene because the standard cloning vectors are designed for E. coli, not Streptomyces.
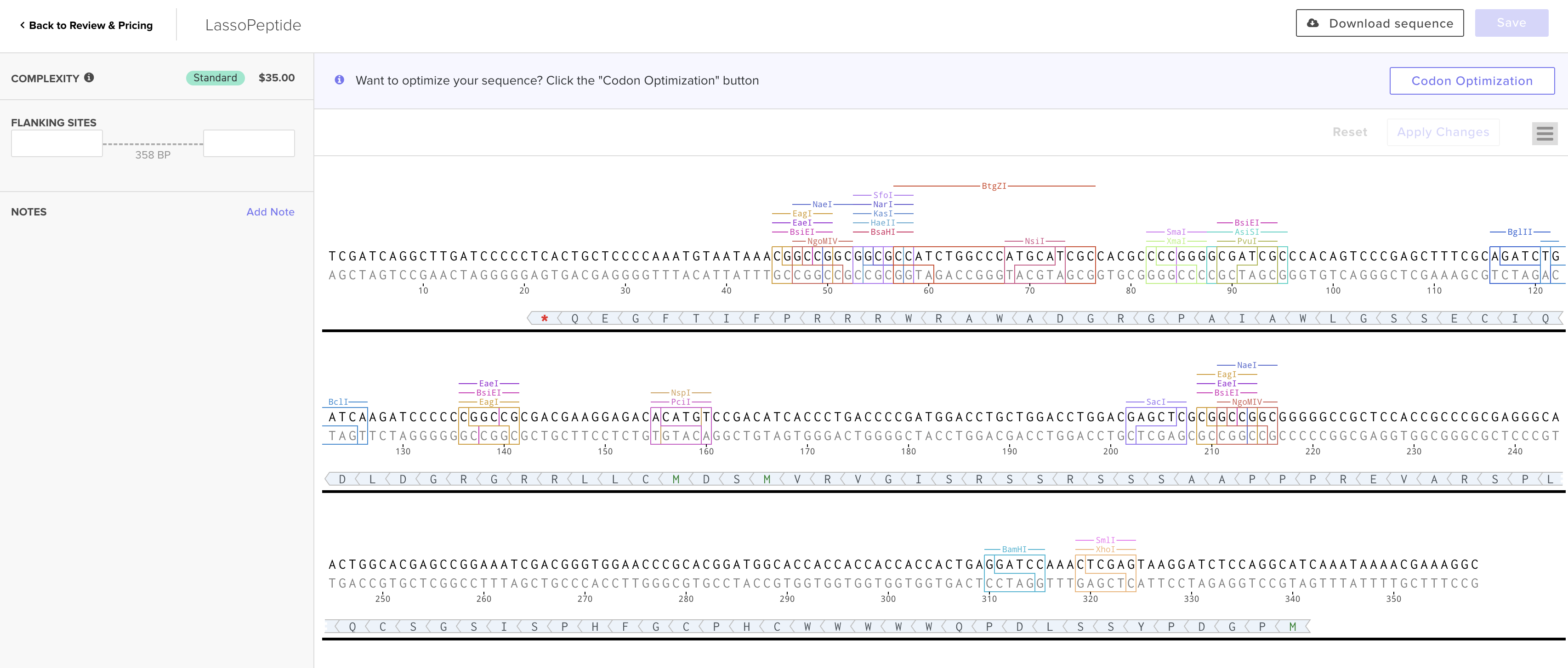
DNA Read / Write / Edit
5.1 DNA Read
What DNA would you sequence, and why?
I would sequence the whole genomes of all ~6,000 mammalian species. The largest current collection is the Zoonomia project, with around 250 whole genomes plus maximum-lifespan data for most of those species. Expanding this to cover all mammals, paired with their lifespan records, would let us train models that identify DNA patterns predictive of how long a species can live. More genomes means better predictions about which parts of DNA are linked to longevity.
What sequencing technology, and why?
Illumina short-read sequencing (second generation). It produces highly accurate short reads (~150–300 bp) and is well suited to spotting small genetic differences between species.
Generation
Second generation. First-generation Sanger sequencing reads one fragment at a time and is too slow and expensive for whole genomes. Second-generation methods sequence millions of short fragments in parallel, which is fast and cheap.
Input and preparation
The input is genomic DNA extracted from tissue or blood samples. Essential preparation steps:
- DNA extraction. Isolate high-quality DNA from the biological sample.
- Fragmentation. Break the DNA into smaller pieces.
- Adapter ligation. Attach short known sequences to the ends of each fragment so the sequencer can recognize and process them.
- PCR amplification. Make many copies of each fragment to boost the signal.
- Quality check. Verify the library is the right size and concentration before loading onto the sequencer.
Essential steps and base calling
Fragmented DNA is bound to a glass flow cell, amplified into clusters, and sequenced one base at a time. In each cycle, a fluorescently labeled nucleotide is added; a camera captures which color fires at each cluster (each of the four bases has a different color), and the machine records the base. The process repeats hundreds of times to read out each fragment.
Output
Digital sequence files (typically FASTQ) containing millions of reads of A, T, C, and G along with quality scores indicating the confidence of each base call. These reads are assembled and aligned computationally to reconstruct each species’ genome.
5.2 DNA Write
What would you synthesize, and why?
I would use the trained models from the sequencing analysis to predict specific DNA sequences associated with high maximum lifespan, then synthesize those predicted longevity-linked sequences (specific gene variants or regulatory elements found in long-lived species like bowhead whales or naked mole-rats) so they can be tested in cell cultures or animal models. The goal is to move from computational prediction to experimental validation: do these DNA sequences actually promote cellular health and longevity?
Technology choices
- Oligonucleotide synthesis (Twist Bioscience): for short to medium DNA fragments (up to a few thousand base pairs). Chemical synthesis on microchips runs many sequences in parallel, making it fast and affordable.
- Gibson Assembly or Golden Gate Assembly: for stitching shorter synthesized fragments into larger constructs using enzymes that join DNA pieces seamlessly.
Essential steps
- Sequence design. Use computational models to design target sequences, optimizing codon usage for the target organism and avoiding problematic features (long repeats, extreme GC content).
- Oligonucleotide synthesis. Short single-stranded DNA pieces (oligos, ~50–200 bases) are built base by base on a solid support. Each cycle adds one nucleotide.
- Assembly. Overlapping oligos are combined and joined enzymatically into longer double-stranded fragments (a few hundred to a few thousand bp).
- Cloning. The assembled fragments are inserted into a circular DNA carrier (plasmid vector) and introduced into bacteria, which copy the DNA as they grow.
- Verification. The final constructs are sequenced to confirm they are correct.
- Large construct assembly. Verified fragments are stitched together using Gibson or Golden Gate assembly to create larger constructs.
Limitations
- Speed. Synthesizing and assembling long constructs (>10 kb) can take weeks.
- Accuracy. Chemical synthesis introduces errors at roughly 1 in 200 bases per oligo. Errors are corrected through screening and verification, but this adds time and cost.
- Scalability. Very long or repetitive sequences are hard to synthesize; oligos may misassemble or fold in unwanted ways. Sequences with extreme GC content are also harder to build reliably.
5.3 DNA Edit
What would you edit, and why?
I would edit specific genes in model organisms (such as mice) to replace native sequences with the longevity-associated variants identified above. For example, if the model predicts that a particular DNA-repair gene variant is linked to longer lifespan in mammals, I would edit a mouse to carry that variant and test whether swapping in these “long-life” variants extends lifespan or improves age-related outcomes like cancer resistance or cellular repair.
Technology choice
CRISPR-Cas9. It is the most precise, versatile, and widely used genome-editing tool available. It works at specific genomic locations in living cells and organisms, including mammalian systems like mice.
Essential steps
- Target selection. Identify the exact genomic location to edit.
- Guide RNA design. Design a short RNA matching the target site.
- Cutting. Cas9, guided by the RNA, binds the matching DNA site and creates a double-strand break.
- Repair. The cell’s repair machinery fixes the break. If a DNA template carrying the desired sequence is provided, the cell can use it as a blueprint via homology-directed repair.
- Screening. Edited cells are sequenced to confirm the desired change.
Inputs
| Category | Components |
|---|---|
| Design | Target DNA sequence, custom guide RNA, donor template DNA flanked by sequences matching the cut site. |
| Molecular | Cas9 protein or mRNA, synthesized guide RNA, donor template DNA, delivery reagents. |
| Biological | Target mouse cells. |
Limitations
- Off-target edits. The guide RNA can bind similar sites elsewhere, causing unintended cuts.
- Low HDR efficiency. Only a fraction of edited cells carry the precise desired change, requiring extensive screening.
- Delivery. Getting CRISPR components into every target cell efficiently, especially in living animals, is still difficult, and some tissues are harder to reach than others.