Week 4 HW: Protein Design
Part A: Conceptual Questions
Why do beta-sheets tend to aggregate?
A beta-strand forms when a protein’s backbone (the repeating NH–Cα–CO chain shared by every amino acid) stretches into a nearly flat zigzag. When two or more strands line up next to each other and link through hydrogen bonds (an N–H on one strand pairs with a C=O on the neighbor), you get a beta-sheet.
The strands on the outer edges still have a full row of exposed N–H and C=O groups, which lets another strand dock and extend the sheet, and so on. That is the structural reason beta-sheets are prone to aggregation.
What forces pull sheets together?
- Hydrophobic effect (the biggest driver). In a beta-strand, side chains stick out alternately above and below the sheet. Many side chains are hydrophobic, so two sheets stack with their greasy faces inward.
- Hydrogen bonding (gives the structure regularity). Each strand that joins the sheet edge contributes roughly one H-bond per residue. Individually, H-bonds in water are not enormously strong (breaking one with a neighbor just lets you form one with water instead), but across a strand of ten or more residues they add up meaningfully.
- Van der Waals packing (stabilizes stacked sheets). These forces are weaker and shorter-range, arising from temporary, fluctuating dipoles.
Part B: Protein Analysis and Design
Briefly describe the protein you selected and why you selected it.
I chose a monoclonal antibody for several reasons:
- It can target specific proteins on cell surfaces with extreme precision, directly applicable to therapeutics.
- It can recruit the immune system (via its Fc region) to destroy tagged cells, combining specificity with immune effector functions.
- It can be engineered with ML and computational methods for improved binding affinity and reduced immunogenicity.
- It is highly specific to its target with fewer off-target effects compared to small-molecule drugs.
For this exercise I selected trastuzumab, famous for revolutionizing the treatment of HER2-positive breast cancer. It is a humanized IgG1 monoclonal antibody that binds the extracellular domain IV of HER2 (human epidermal growth factor receptor 2), blocking receptor dimerization and the downstream signaling that drives tumor growth.
How long is it? What is the most frequent amino acid?
The full trastuzumab IgG has two heavy chains (449 aa each) and two light chains (214 aa each), for a total of ~1,326 amino acids and ~148 kDa.
The crystal structure (PDB: 1N8Z) contains only the Fab fragment (the antigen-binding portion):
Heavy chain Fab (chain B, 220 aa):
Light chain (chain A, 214 aa):
| Property | Value |
|---|---|
| Combined Fab length | 434 amino acids |
| Most common amino acid | Serine (S), appearing 60 times |
How many protein sequence homologs are there for your protein?
Because trastuzumab is a humanized antibody with conserved IgG1 framework regions, BLAST returns a very large number of homologs (antibodies share roughly 70–90% identity in their framework regions). A BLAST search of the heavy chain Fab against UniProt returns over 250 homologs. The variable CDR (complementarity-determining region) loops are what give trastuzumab its HER2 specificity.
When was the structure solved? Is it a good quality structure?
Good quality means good resolution. Smaller is better; the benchmark is 2.70 Å.
| Field | Value |
|---|---|
| Deposited | 2002-11-21 |
| Released | 2003-02-18 |
| Published | Cho et al., Nature (2003) 421: 756–760 |
| Link | rcsb.org/structure/1N8Z |
| Resolution | 2.52 Å (good quality, better than the 2.70 Å benchmark) |
Are there any other molecules in the solved structure apart from protein?
Yes. In addition to the three unique protein chains (light chain A, heavy chain B, HER2 extracellular domain C), the structure contains:
| Molecule | Description | Copies |
|---|---|---|
| NAG | 2-acetamido-2-deoxy-β-D-glucopyranose (N-linked glycosylation sugar attached to HER2) | 2 |
| SO4 | Sulfate ion | 1 |
Does your protein belong to any structure classification family?
Yes. The overall complex is classified in the PDB under TRANSFERASE. The trastuzumab Fab itself belongs to the Immunoglobulin superfamily.
Visualize the protein as cartoon, ribbon, and ball-and-stick.
Cartoon
Ribbon
Ball and stick
Color the protein by secondary structure. Does it have more helices or sheets?
The structure has more sheets than helices: 215 atoms in sheets vs. 30 atoms in helices.
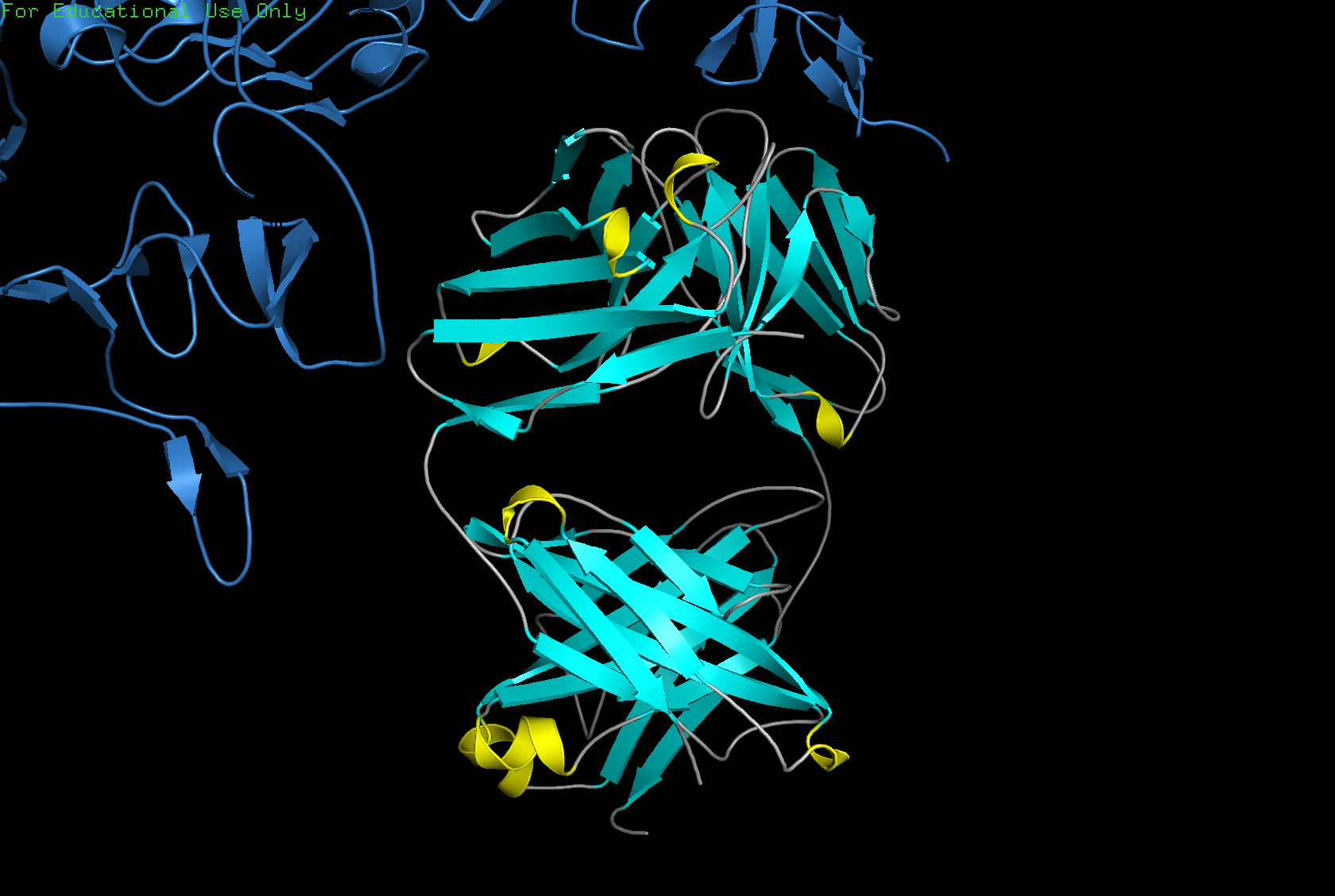
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs. hydrophilic residues?
Generally, proteins have a hydrophobic core and a hydrophilic surface, and trastuzumab follows this pattern. The immunoglobulin fold is a beta sandwich where:
- Hydrophobic residues (orange) point inward.
- Hydrophilic residues (blue) point outward.
This is hard to see in the visualization because the inward and outward surfaces are not so distinct. The CDR loops, which are the tips that contact HER2, are mixed: aromatic hydrophobics (Trp, Tyr) provide shape complementarity, while polar and charged residues form hydrogen bonds and salt bridges with the antigen.
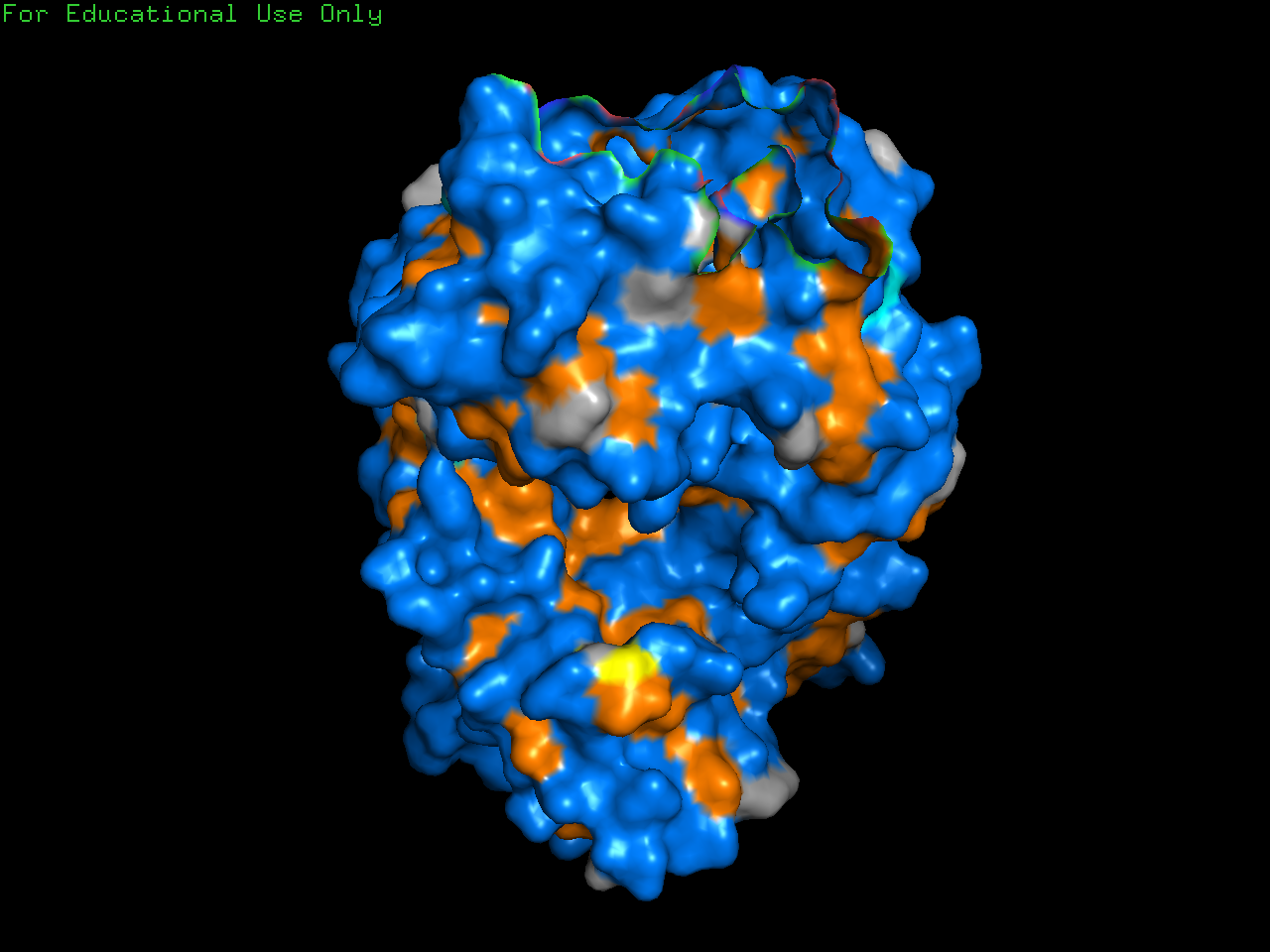
Visualize the surface of the protein. Does it have any holes (binding pockets)?
Yes, binding pockets are visible on the surface.
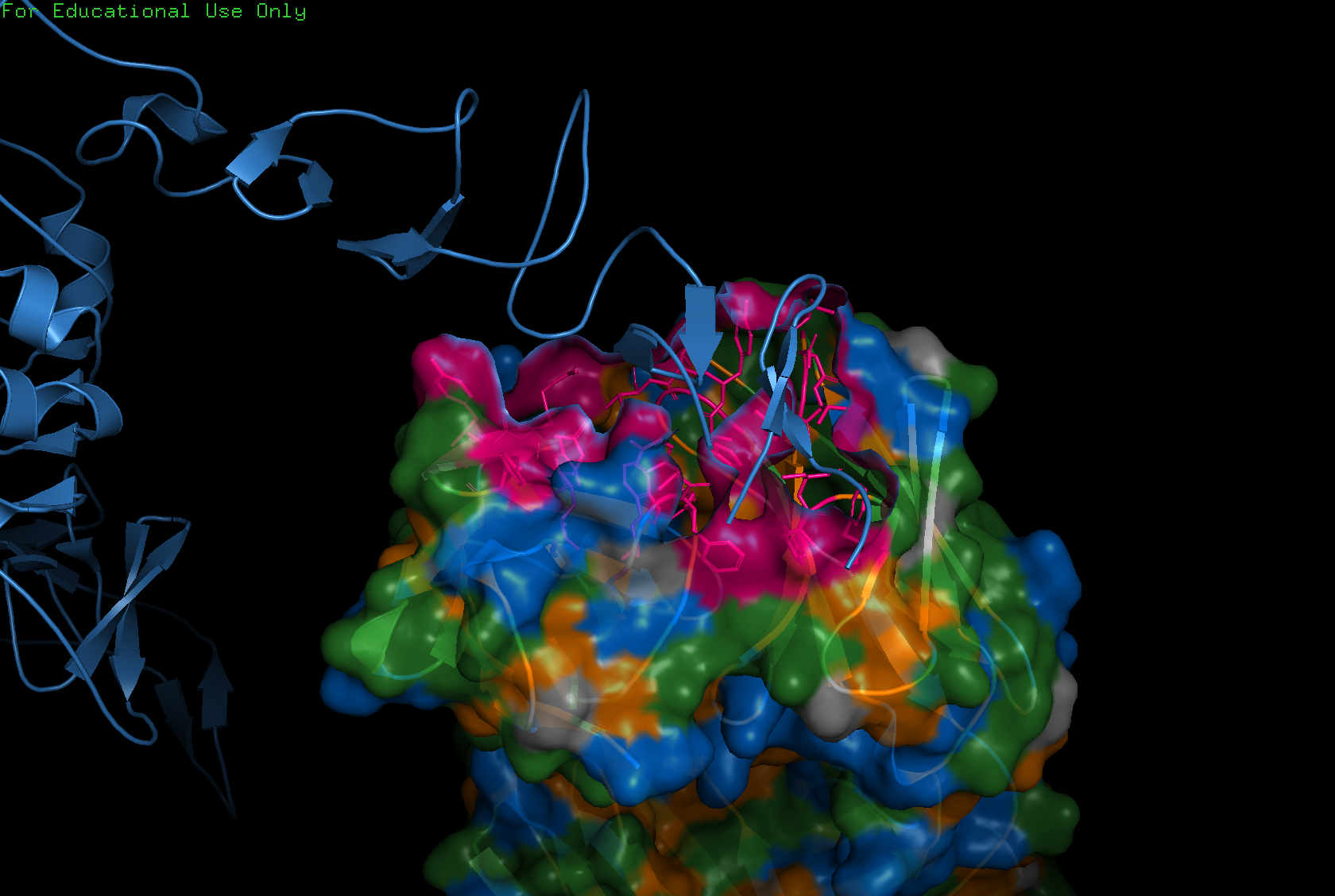
Part C: ML-Based Protein Design Tools
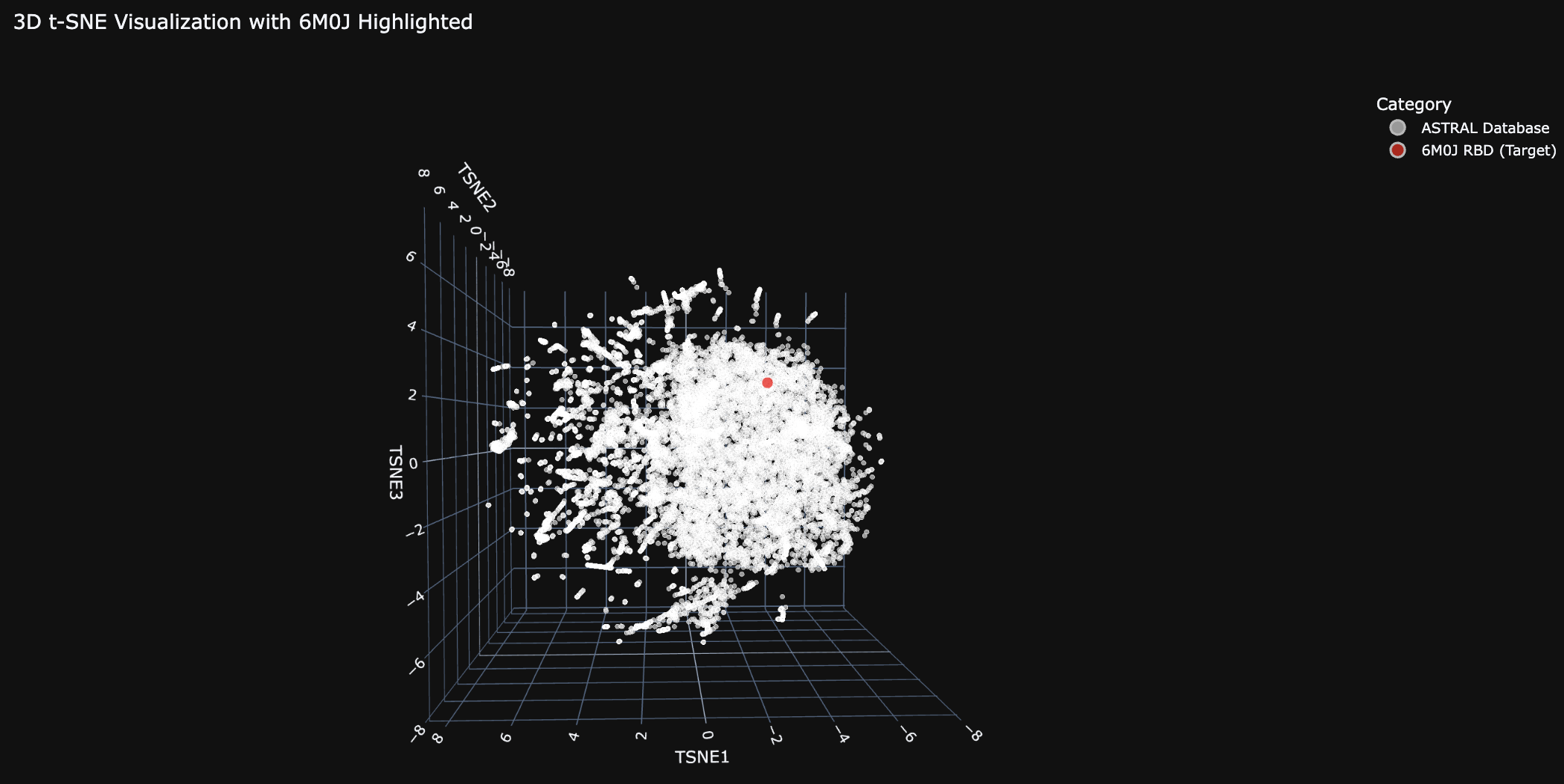
For this exercise I chose 6M0J, the SARS-CoV-2 Spike Receptor Binding Domain.
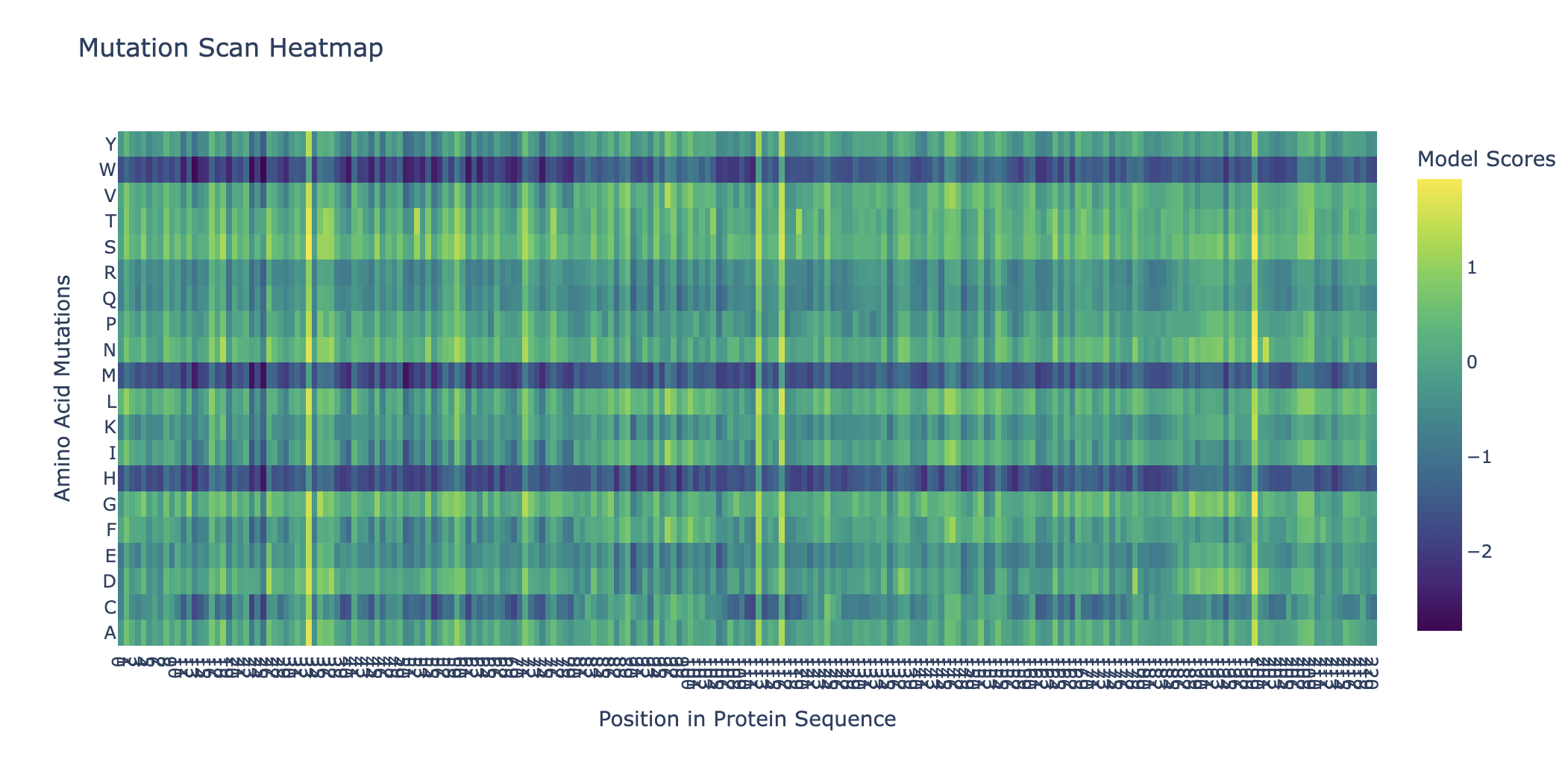
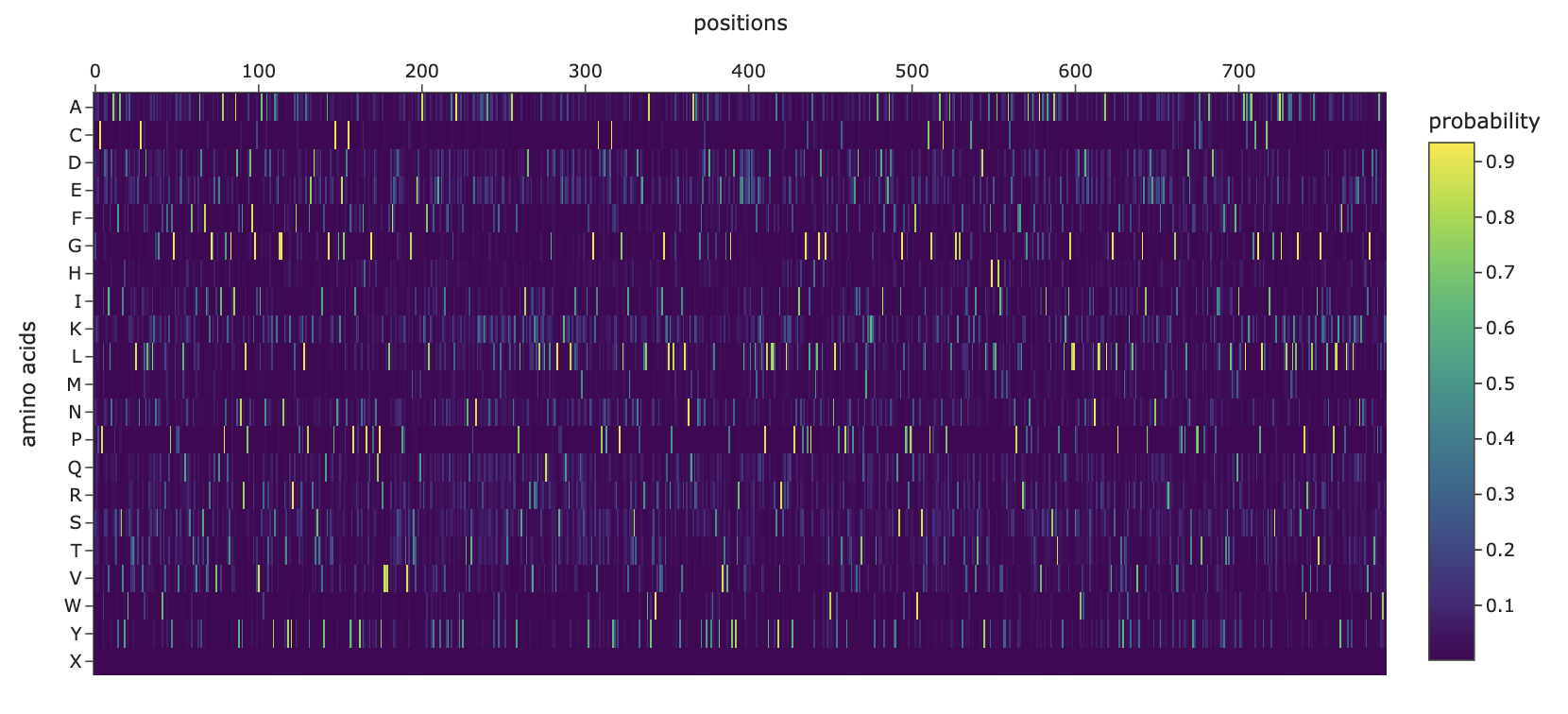
Deep Mutational Scans
Can you explain any particular pattern?
Horizontal patterns (rows): the rows for tryptophan (W), histidine (H), and methionine (M) are consistently darker across nearly all positions. These are large, bulky, or chemically complex amino acids that are difficult to accommodate at arbitrary positions without disrupting the fold. Small, simple amino acids like alanine or serine are more easily tolerated, so their rows appear lighter.
Vertical patterns (columns): the most striking pattern is the dark purple stripes at specific positions. These correspond to cysteine residues, which form the disulfide bonds that hold the shape together so it can bind the human ACE2 receptor. Because ESM2 learned from millions of sequences that these cysteines are almost never substituted in nature, it heavily penalizes any mutation at those positions. The darkest scores appear when cysteine is mutated to something like tryptophan or proline, which would not only break the disulfide bond but also create additional structural problems.
Latent Space Analysis
Do the formed neighborhoods approximate similar proteins?
Generally the proteins are clustered tightly. There are a few distinct clusters on the edges, which likely share a common evolutionary ancestor.
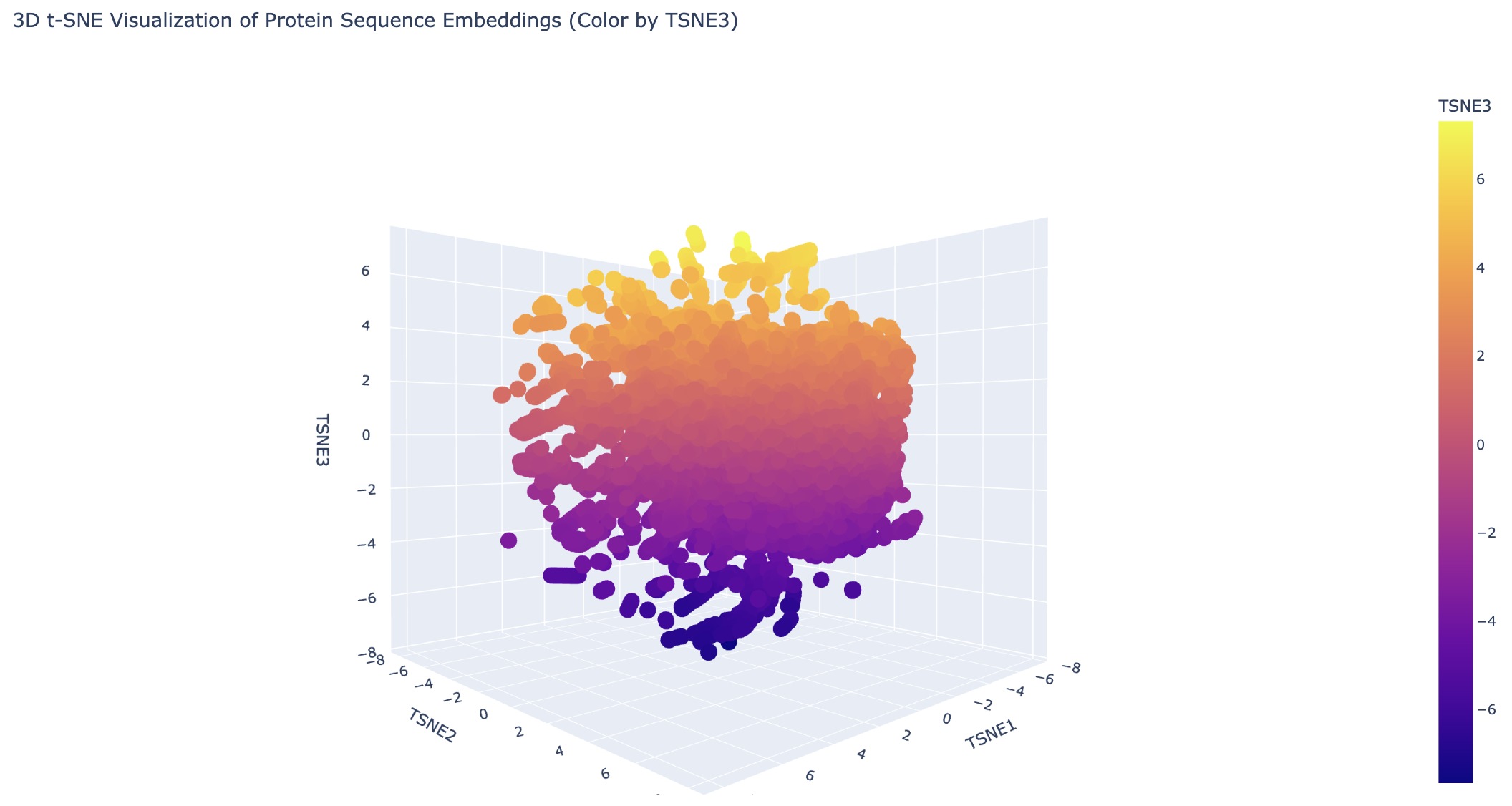
Place your protein in the resulting map.
The 6M0J protein falls within the main cluster.
Folding a Protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
| Metric | Score | Interpretation |
|---|---|---|
| pLDDT (local confidence, 0–100) | 25.516 | Low. Local structure unlikely to match the true structure. |
| pTM (global fold confidence, 0–1) | 0.129 | Low. Global topology prediction unreliable. |
This is likely because the 6M0J viral protein is normally part of a much larger Spike protein complex. The SARS-CoV-2 Spike RBD is unstable on its own.
Try changing the sequence. Is your protein structure resilient to mutations?
The original protein is not very resilient given its poor pLDDT and pTM scores. After redesign with ProteinMPNN, the structure became much more stable:
| Metric | Original | After ProteinMPNN |
|---|---|---|
| pLDDT | 25.516 | 92.095 |
| pTM | 0.129 | 0.881 |
Note: while the structural metrics improved dramatically, the redesigned protein could be functionally incorrect. Stability does not guarantee biological activity.
Inverse Folding
Compare the predicted sequence vs. the original.
Roughly half of the original amino acids were preserved. This is typical for ProteinMPNN, which optimizes the sequence for the target backbone rather than mimicking the native sequence.
| Metric | Original | ProteinMPNN |
|---|---|---|
| Energy score | 1.3747 | 0.8107 |
In ProteinMPNN, a lower score suggests the new sequence is potentially more stable or a better fit for the target backbone. This matches the pLDDT and pTM improvements above.
Input this sequence into ESMFold and compare to your original.
As noted above, the predicted structure after ProteinMPNN has higher pLDDT and pTM than the original.
Bacteriophage Engineering
For this exercise I worked with Alayah Hines and Terry Luo.
Computational Engineering of the MS2 Lysis Protein (L)
The MS2 L protein is a 75-amino-acid polypeptide that lyses E. coli by an incompletely understood mechanism. Its C-terminal transmembrane (TM) domain inserts into the cytoplasmic membrane and oligomerizes, depolarizing the membrane and triggering host autolytic enzymes to degrade the murein layer. Recessive, conservative missense mutations clustered around a conserved LS dipeptide strongly imply that L engages an unidentified host protein target rather than simply disrupting the bilayer. The dispensable N-terminal domain binds the chaperone DnaJ (with solved PDB structures), modulating lysis timing; removing it causes lysis ~20 minutes earlier. No experimental structure of L exists.
Goals:
- Stabilize L for more robust membrane accumulation.
- Accelerate lysis by bypassing DnaJ-dependent regulatory timing and improving delivery of functional L to the membrane.
Because the downstream lytic target is unknown, we do not attempt to enhance per-molecule toxicity at the point of target engagement. We focus on removing regulatory brakes and increasing the supply of functional protein.
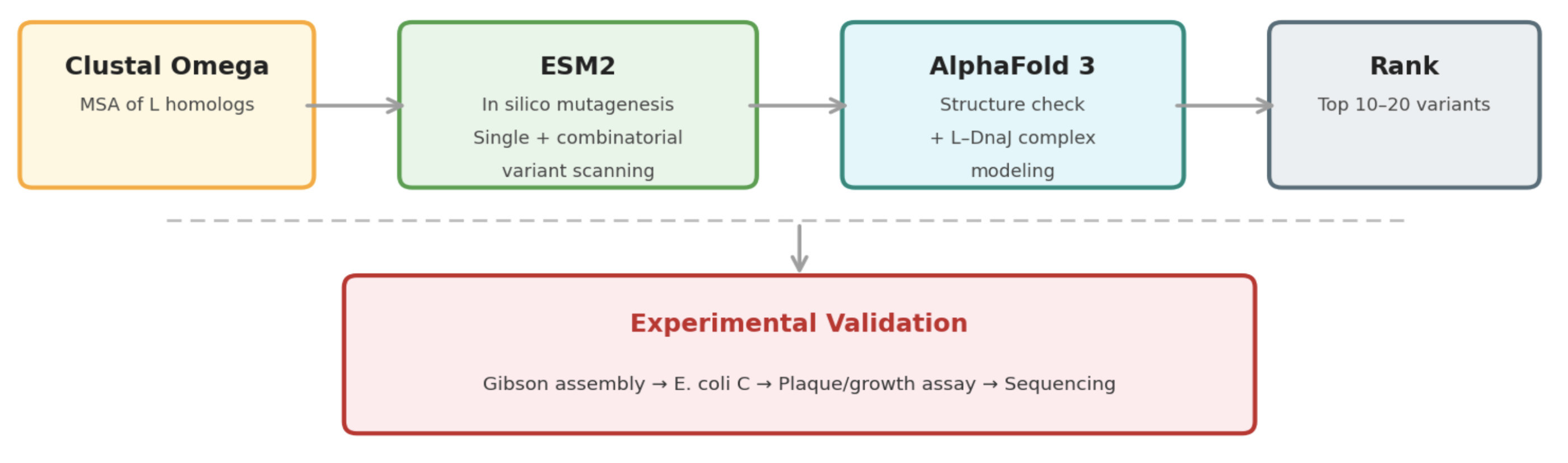
Pipeline: Three Tools, Each Non-Redundant
- Clustal Omega (Conservation Map). Align L homologs across Leviviridae (MS2, f2, R17, GA, PP7, AP205, PRR1, M12, KU1, JP34). Conserved C-terminal residues (especially the LS motif) are presumed to mediate the unknown heterotypic interaction and are excluded from mutation. This map constrains all downstream design.
- ESM2 + Deep Combinatorial Scanning (Fitness Oracle). Score every single-point mutation by log-likelihood change. Increases at mutable positions indicate stabilizing substitutions (Goal 1). N-terminal scanning identifies mutations that disrupt DnaJ binding (Goal 2). A strict preservation rule applies near the LS motif: mutations are evaluated for maintenance of wild-type fitness, not improvement. The genetics show that even conservative changes there cause recessive loss of function. Pairwise combinatorial scanning (~2M pairs) captures epistatic synergies at mutable positions.
- AlphaFold 3 (Structural Filter + Complex Model). Predicts variant structures as a sanity check (does the TM helix survive?) and models the L–DnaJ complex to verify that N-terminal truncations and mutations disrupt the regulatory interface. Used as a filter, not a design engine. The PAE matrix identifies confident interface contacts.
Ranking
Composite score: ESM2 log-likelihood gain (stability) + conservation preservation (all essential residues intact) + AF3-predicted DnaJ-binding disruption (for timing bypass). The top 10 to 20 variants advance to experimental validation.
Why Not More Tools?
ProteinMPNN is excluded because it is trained on crystallized globular PDB proteins, not predicted structures of disordered membrane peptides. Compute is instead invested in combinatorial ESM2 depth.
Pitfalls
- No experimental structure. All structural reasoning rests on AF3 predictions for a challenging target. Mitigated by treating AF3 as a filter and cross-referencing against the conservation map.
- Unknown lytic target. The central limitation. We cannot optimize target-binding affinity for an unidentified partner; engineering is restricted to upstream properties (stability, membrane delivery, DnaJ bypass).
- Autolysin bottleneck. If the lysis rate is limited by host autolytic enzyme activity rather than L accumulation, stabilization gains may show diminishing returns. The plaque assay will reveal this.
Pipeline Schematic