1)First, describe a biological engineering application or tool you want to develop and why.( This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.)
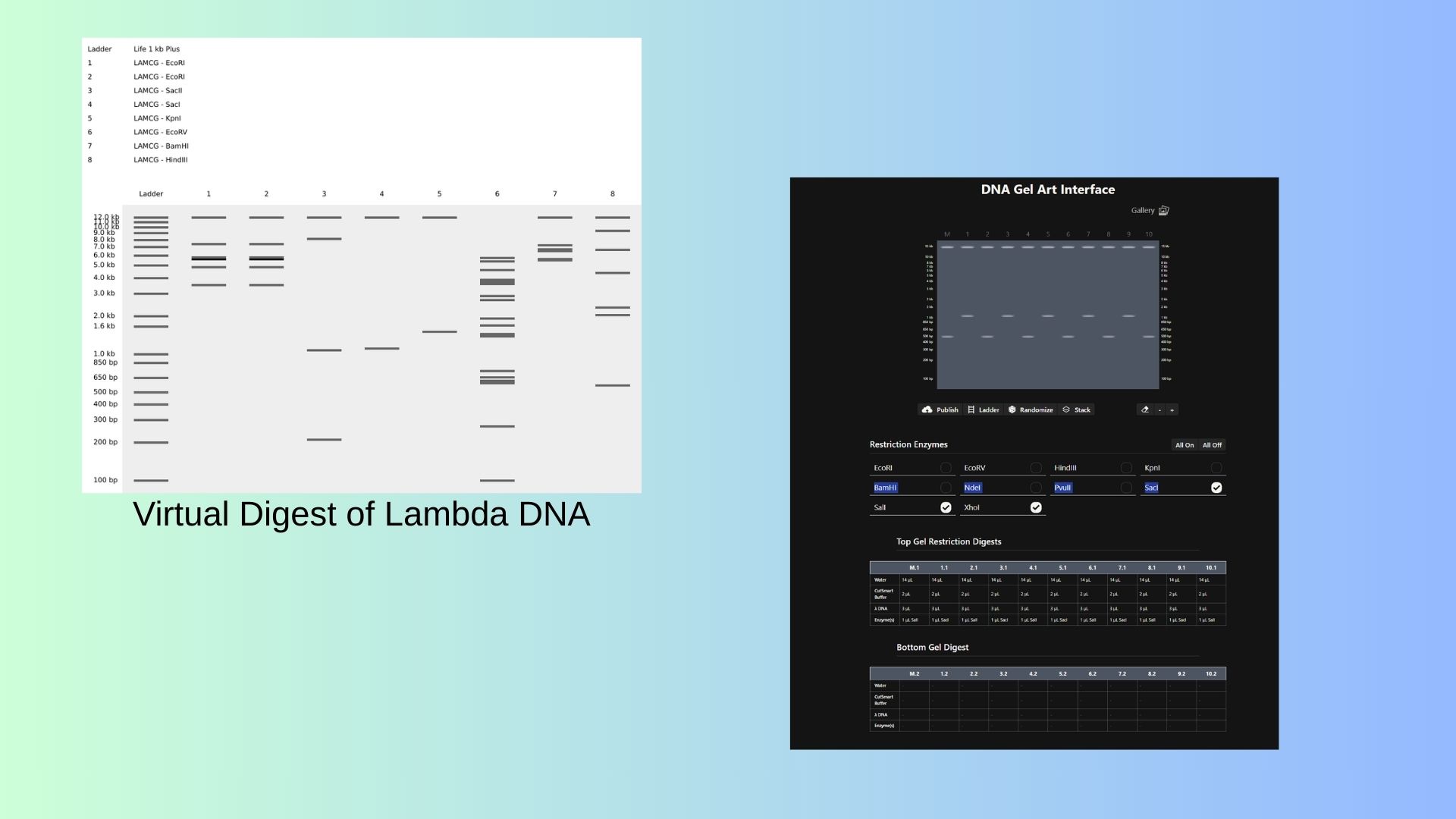
Part 1: Benchling & In-silico Gel Art First I start to stimulate Restriction Enzyme in Lambda DNA:
Also shown above,with Ronan’s website, I try to pattern with enzymes.
Part 3: DNA Design Challenge 3.1. Choose your protein: I decided to choose GFP protein in water jellyfish(Also called “Aqua Victoria”). Because with GFP protein I can see that organization of cell. I can track protein and find to where to go in the cell. On the shown below, you can see GFP Protein Sequence:
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it. This week was all about moving from manual pipetting to the world of liquid handling automation. I’ve been diving deep into the Opentrons ecosystem, specifically focusing on how to bridge the gap between writing Python code and seeing the robot actually execute those movements on the deck.
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? Meat is approximately 20% protein by mass. So, 500g of meat contains about 100g of protein. Given that the average mass of an amino acid is 100 Daltons ($1.66 \times 10^{-22}$ grams), we can calculate the total number of molecules:$100g / (100 \times 1.66 \times 10^{-24}g) \approx 6 \times 10^{24}$ amino acid molecules. That is roughly 10 moles. Why do humans eat beef but do not become a cow, eat fish but do not become fish? This is because our digestive system breaks down the proteins we eat into their individual building blocks: amino acids. Our body doesn’t use the cow or fish proteins directly; it uses these free amino acids to assemble “human” proteins based on the instructions in our own DNA. Why are there only 20 natural amino acids? This is often described as a “frozen accident” in evolution. While there are many more possible amino acids, these 20 provided enough chemical diversity (acidic, basic, hydrophobic, etc.) to build complex 3D structures and catalyze reactions. Once life became complex, changing this fundamental toolkit would have been too disruptive. Can you make other non-natural amino acids? Design some new amino acids.Yes, scientists can synthesize non-natural amino acids (ncAAs) by adding unique side chains. For my project on schizophrenia, I could design an amino acid with a fluorescent “sensor” side chain that changes color when it interacts with high concentrations of dopamine. This would allow us to visualize “dopamine storms” in real-time. Where did amino acids come from before enzymes that make them, and before life started? Amino acids likely formed through abiotic synthesis. The Miller-Urey experiment showed that early Earth’s atmospheric gases (methane, ammonia, water vapor) could react with electrical discharges (lightning) to create amino acids. They might have also been delivered to Earth via meteorites. Why are most molecular helices right-handed? This is due to the “chirality” of L-amino acids. Because all life uses L-amino acids, the right-handed $\alpha$-helix is the most energetically stable conformation that avoids steric clashes (physical bumping) between the side chains. Why do $\beta$-sheets tend to aggregate? What is the driving force? The main driving forces are hydrogen bonding and the hydrophobic effect. The edges of a $\beta$-sheet have “unsatisfied” hydrogen bond donors and acceptors. This makes them very “sticky,” leading them to stack with other $\beta$-sheets to achieve a lower energy state. Why do many amyloid diseases form $\beta$-sheets? Can you use them as materials? Amyloid $\beta$-sheets are incredibly stable and resistant to degradation. In diseases like Alzheimer’s, proteins misfold into this “energy well” from which they cannot escape.As materials: Yes! Amyloid fibers are stronger than steel for their size. They can be engineered as ultra-stable nanowires or drug-delivery scaffolds. Design a $\beta$-sheet motif that forms a well-ordered structure. A well-ordered motif can be designed using an “alternating” pattern: (Val-Lys-Val-Glu)n. In this sequence, the hydrophobic Valine residues face one side while the charged Lysine and Glutamate residues face the other. This creates a “Janus-faced” sheet that is oily on one side and water-loving on the other, allowing it to assemble into perfect layers. Part B: Protein Analysis and Visualization 1. Briefly describe the protein you selected and why you selected it. I want to select DRD2(the Human Dopamine D2 Receptor) protein because this protein provided to enter the doapamin in the cell. Also ın neurodejenaretif diseases such as in schzophrenia this receptor protein is overexpressing therefore dopamin enter the cell more than normal and this leads to hallucination. Therefore this protein plays important role in brain.
Part A: SOD1 Binder Peptide Design : First, we change Alanine(A) to Valine(V) at residue 4 in SOD1 sequence.
Part 1: Generate Binders with PepMLM: I generate 4 different peptides by using PepMLM Collab.
In protein design (ProteinMPNN), Perplexity measures the model’s “uncertainty” when choosing amino acids for a specific position. It indicates how well a designed sequence fits the target protein’s structural constraints.The lower the score (e.g., < 10), the more confident the model is. It means the amino acid sequence is physically and energetically highly compatible with the protein structure. That way we can say that first binder is the most optional for us.
Assignment: DNA Assembly Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion Master Mix is a convenient 2X concentrated solution designed for high-performance PCR. Key components include: Phusion DNA Polymerase: A high-fidelity enzyme with a processivity-enhancing domain, ensuring extremely low error rates and fast extension times. dNTPs: The essential building blocks (dATP, dCTP, dGTP, dTTP) required for new DNA strand synthesis. Reaction Buffer: Maintains the optimal pH and provides necessary ions.$ MgCl2: Acts as a vital cofactor for the polymerase enzyme activity. What are some factors that determine primer annealing temperature during PCR? The Ta is critical for primer specificity and yield. It is primarily determined by:
Assignment Part 1: Intracellular Artificial Neural Networks:
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Graded Response vs. Binary Logic: Traditional circuits work like a light switch (ON or OFF). IANNs work like a dimmer switch; they can process analog signals, allowing the cell to respond to varying concentrations of a molecule rather than just its presence or absence.
Signal Integration (Weighting): In a perceptron model, different inputs can have different “weights.” This means the cell can prioritize one environmental signal (like a toxin) over another (like a nutrient) before making a final decision.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. The primary advantage of cell-free systems is the removal of the cell membrane, which eliminates the “black box” nature of cellular metabolism.
Final Project Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. For my “Sentinel Microbes” project, which aims to detect foreign pathogens in the gut, the most critical aspect to measure is the quantitative expression of the reporter protein. When the engineered microbes encounter a specific pathogen (like Salmonella), they trigger a genetic circuit to produce a visual signal. I need to measure the concentration and identity of this protein to confirm that the “Detected!” signal is accurate and strong enough to be noticed.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Part B: Cell-Free Protein Synthesis | Cell-Free Reagents E.Coli Lysate: It is acted as the catalytic core of the reaction; it provides the ribosomes for translation and T7 RNA Polymerase for high-level transcription. Salts and Buffers: Potassium Glutamate: It provides necessary ionic strength and potassium ions for protein folding without inhibiting DNA-protein interactions like chloride salts might. HEPES-KOH pH 7.5: It is pH indicator.Also it prevents reaction from becoming too acidic due to metabolic byproducts. Magnesium Glutamate: It supplies magnesium ions required for stabilizing ribosome assembly and acting as a cofactor for RNA polymerase. Potassium Phosphate (Monobasic- Dibasic): It provides secondary buffering and source of inorganic phosphate needed to regenerate ATP. Energy and Nucleotide System: Ribosome Guanine: It serves as specific region for nitrogenous base that can be salvaged by lysate enzymes to create GTP for transcription when direct GMP levels are low. Translation Mix (Amino Acids): they are provide fundamental building blocks for synthesizing the polypeptide chain.
1)First, describe a biological engineering application or tool you want to develop and why.( This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.)
One of the areas of greatest interest in bioengineering is gene therapy, a field where bioengineering and neuroengineering converge. The majority of these areas still lack a distribution. Therefore, gene therapies should be investigated in conjunction with supportive biological agents. For example, there is still no complete cure for schizophrenia, one of the biggest reasons being that schizophrenia is a polygenic disease. A synthetic promoter could be created to address this. This promoter, in the case of cellular neural activity, creates a biological control system that temporarily suppresses the expression of risk genes through signals from the natural signaling cell.
2) Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Although this biological control system is designed for patients with schizophrenia, it opens a significant door for the improvement and treatment of other neurological diseases in the future. Furthermore, it helps integrate individuals with many neurological disorders into society.
Therefore, three different prioritary goals can be identified:
Goal 1: Preventing unwanted damage to the cell
Subgoal: Promoter specificity should be investigated before any clinical phase, and CRISPR-based gene modifications should not be performed.
Goal 2: A large patient population is needed so that genetic diversity can be assessed.
Goal 3: Abuse should be limited by providing controlled access.
3)Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Purpose:
Currently, there is no definitive cure for schizophrenia. The process is carried out through drug development and psychological support. However, thanks to the synthetic promoter method, a treatment is offered that both suppresses the disease and minimizes the impact of biological factors.
Design:
First, this system must be supported in a laboratory environment. For this, it should be tested in a university laboratory by experts in the fields of genetic engineering, synthetic biology, and neuroengineering. Furthermore, for disease modeling, funding should be sought from neuroscience and psychiatry departments, and for financing, from private and public funds such as Horizon Europe, NIH, etc. And since human or animal cells will be used in these experiments, approval from ethics committees such as IRB and IBC is required.
Assumptions:
The cell type used in creating this system may be incorrect, which could reduce its suitability for the biology of schizophrenia. Additionally, biological design errors may occur, such as incorrect promoter selection, incorrect suppressor dosage, etc.
Failure:
From an ethical standpoint, failure to obtain IRB approval can lead to failure. From a biological perspective, the system may be working correctly, but it may suppress the gene too much, disrupting the cell’s physiological balance. The cell, whose balance is disrupted, may experience disruptions in learning or normal signal transmission. Or, the system may not suppress the correct gene at the correct time, leading to the suppression of the cell and, consequently, the signaling system, rendering the treatment system ineffective.
4)Next, score each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
+2
-
+3
Foster Lab Safety
• By preventing incident
+3
+2
-
• By helping respond
+3
+2
+3
Protect the environment
• By preventing incidents
+2
-
+3
• By helping respond
+3
+2
+3
Other considerations
• Feasibility?
+2
+1
+3
• Not impede research
+3
+3
+2
5)Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
In this case, the first option is best because neural gene editing prioritizes safety in biological design. Therefore, the process should be ethically evaluated and supported. Initially, we should examine whether there are any problems in the design, and where potential problems might arise in the design structures. Safety and ethical discussions should then be meticulously examined in the subsequent process. Because there are many uncertainties in this type of neural bioengineering, and we need to minimize them.
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA polymerase makes an error roughly once every 10⁸ nucleotides, and given that the diploid human genome contains about 6 billion nucleotides, this would theoretically result in around 60 errors per cell division. However, cells possess multiple DNA repair mechanisms, including the proofreading activity of DNA polymerase and mismatch repair pathways, which significantly reduce the actual mutation rate during cell division.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein is about 350–400 amino acids long, and due to the degeneracy of the genetic code, an enormous number of DNA sequences (≈10¹⁹⁰) could theoretically encode the same protein. In practice, most of these sequences do not work because codon choice affects translation efficiency, mRNA structure, protein folding, and gene regulation, meaning only a small subset can be properly expressed in cells.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
The most commonly used method is solid-phase phosphoramidite synthesis, where DNA nucleotides are added sequentially to a growing chain attached to a solid support.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because each nucleotide addition step is not perfectly efficient, errors and truncated products accumulate as the oligo gets longer, greatly reducing the yield of full-length sequences.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Direct synthesis of such a long sequence would result in extremely low yields and high error rates, so long genes are instead assembled from multiple shorter oligos.
Homework Question from George Church:
Project Proposal: Light-Driven Genetic Programming (Lumina-Code)
The Challenge Current genetic engineering is limited by physical delivery. Whether we use viral vectors or nanoparticles, we are essentially trying to “ship” biological instructions into a cell. This process is slow, often triggers immune responses, and lacks spatial precision. My project addresses the DARPA GO challenge: How can we send genetic instructions at the speed of light, without any physical material?
The Solution I propose the development of Lumina-Code, a protein-based “intracellular printer.” Instead of delivering pre-made DNA, we will engineer a specialized Nucleic Acid Compiler (NAC) that stays dormant inside the cell. This NAC will be designed to recognize specific wavelengths of light as digital commands. For instance, a specific pulsing pattern of blue light will trigger the enzyme to assemble a precise RNA sequence from the cell’s internal building blocks.
Potential Impact This “massless” transmission of information would change everything. In a medical setting, a doctor could use a laser to “type” a therapeutic code directly into a tumor, telling it to stop growing, without affecting healthy tissue. In extreme environments, like deep-space missions, we could transmit life-saving medical codes as radio or light signals to be synthesized instantly by the astronauts’ own cells.
Next Steps Our first year will focus on engineering the NAC protein to respond to two distinct light frequencies. Our goal is to demonstrate that we can “print” a short, 20-base genetic sequence inside a living cell using only external optical triggers, achieving a new frontier in biological remote control.
Week 2: DNA Read, Write and Edit
Part 1: Benchling & In-silico Gel Art
First I start to stimulate Restriction Enzyme in Lambda DNA:
Also shown above,with Ronan’s website, I try to pattern with enzymes.
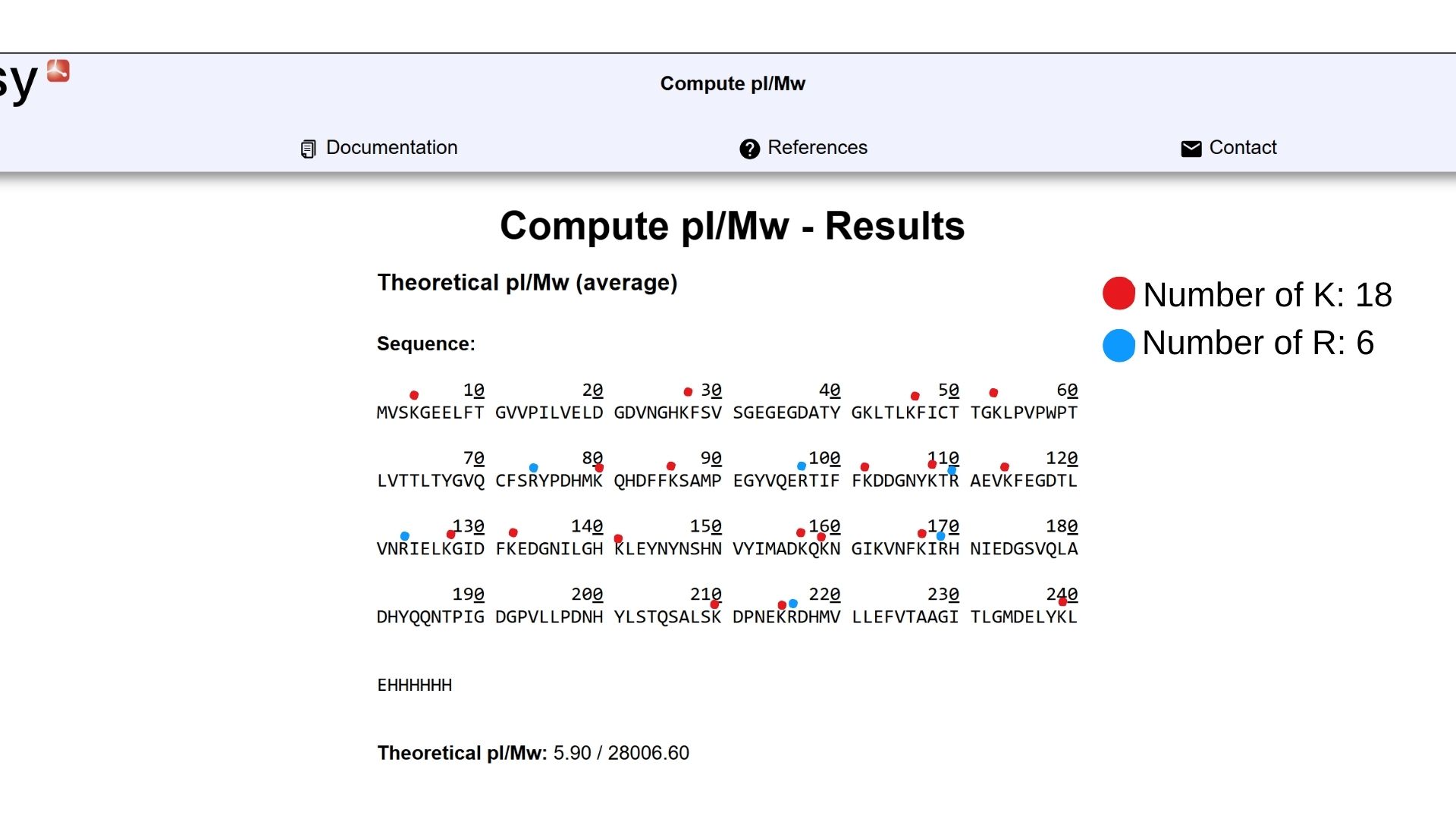
Part 3: DNA Design Challenge
3.1. Choose your protein:
I decided to choose GFP protein in water jellyfish(Also called “Aqua Victoria”). Because with GFP protein I can see that organization of cell. I can track protein and find to where to go in the cell. On the shown below, you can see GFP Protein Sequence:
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence:
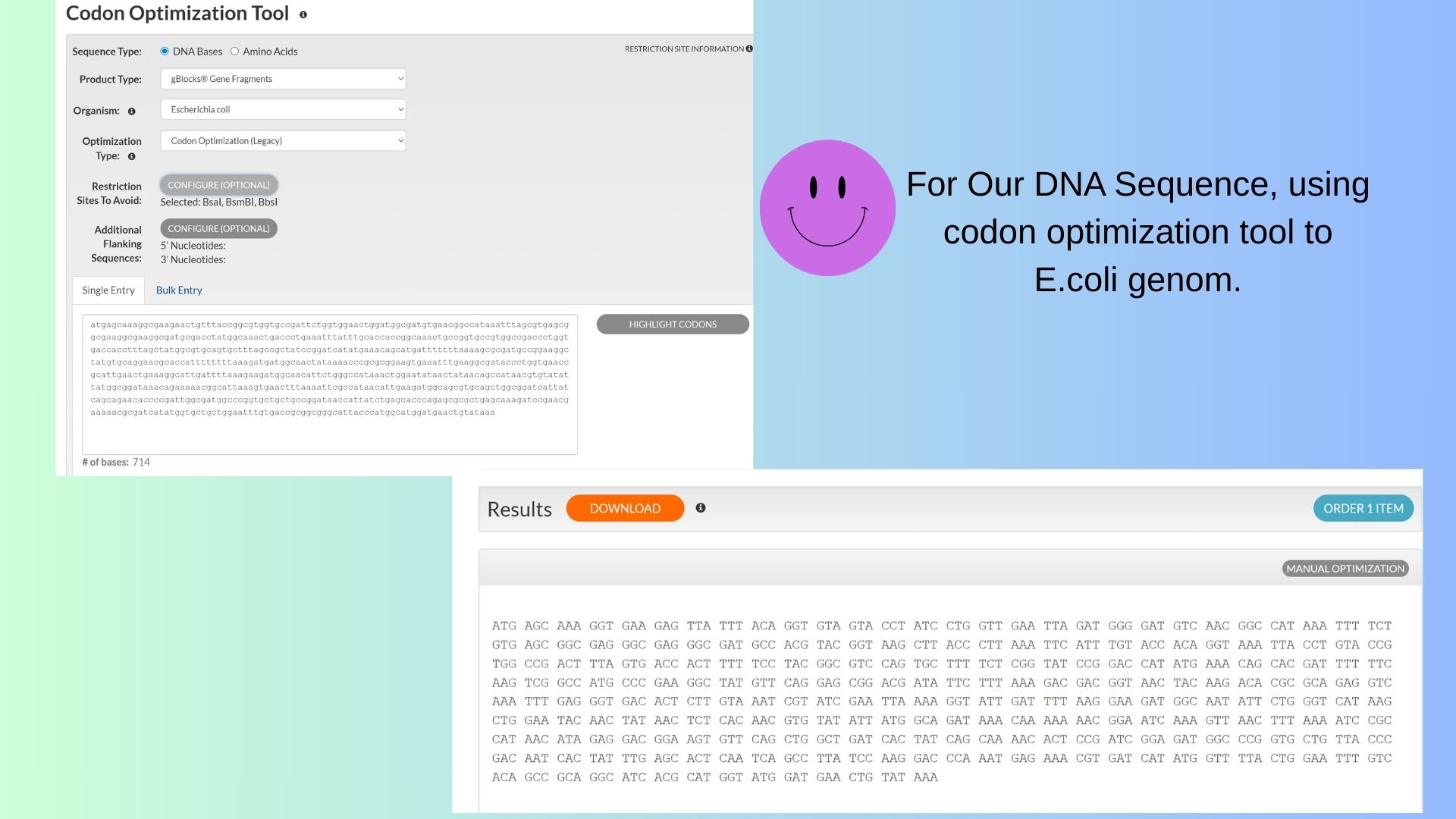
3.3. Codon optimization:
I want to use codon optimization tool for E.coli genom.
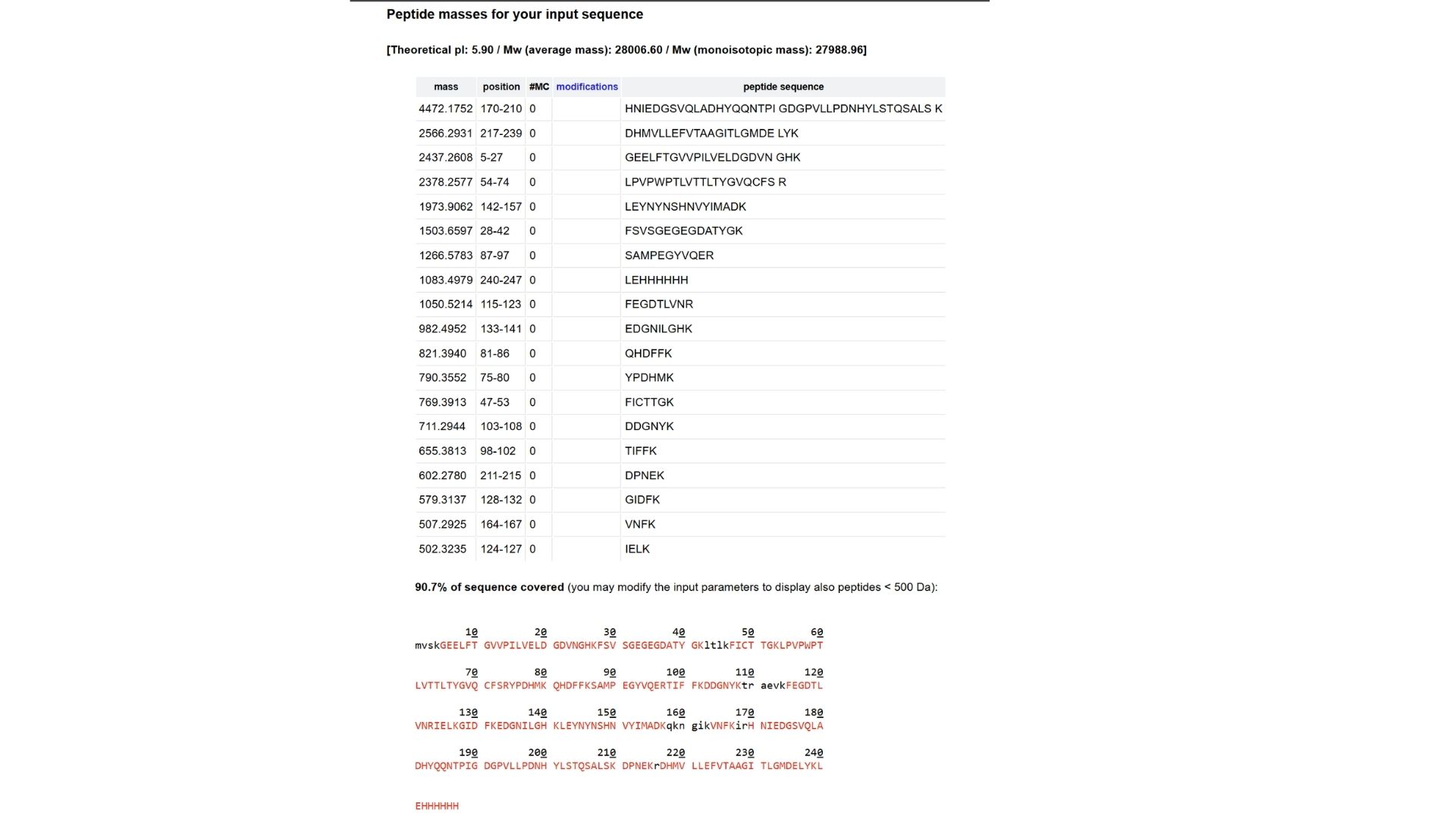
3.4 You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
To make GFP protein from DNA, the DNA first needs to be transcribedinto mRNA and then translated into protein. The DNA has a promoter, the GFP coding sequence, and other regulatory parts. RNA polymerase reads the DNA to make mRNA, and ribosomes read the mRNA to assemble the amino acids into the GFP protein, which then folds and becomes fluorescent.
Ways to produce GFP:
In cells (cell-dependent):
In bacteria like E. coli, you put the GFP DNA in a plasmid and the bacteria make the protein for you—fast and cheap.
For more complex proteins, yeast or mammalian cells can be used to get proper folding and modifications.
Cell-free (in vitro):
You can use a test-tube system with the cell’s transcription and translation machinery. Add GFP DNA, and the protein is made directly. This is fast and easy to control.
Both methods let you get functional GFP for experiments or imaging.
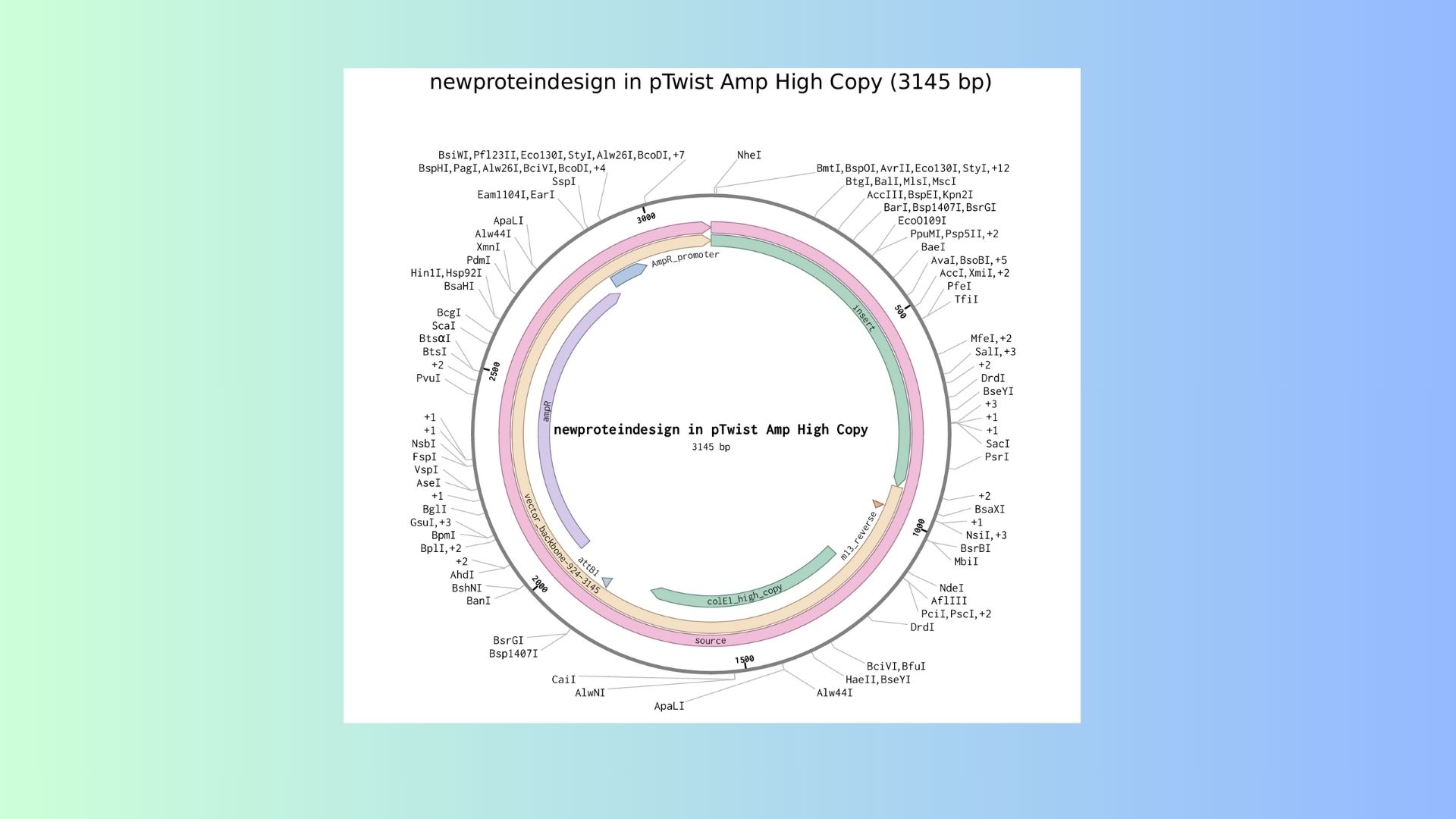
Part 4: Prepare a Twist DNA Synthesis Order:
Here are expression cassed and its vector DNA:
Part 5: DNA Read/Write/Edit:
5.1 DNA Read:
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I want to sequence DNA related to Alzheimer’s disease which is neurodegenerative diseases that affects memorh, cognition and behavior. The main risk genes are APP, PSEN1 and PSEN2 and these genes are linked to early-onset familial Alzheimer’s disease. And sequencing these genes would help identify pathogenic mutations, assess genetic risk etc.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I use Whole Exome Sequencing (WES) on an Illumina platform.This method focuses on protein-coding regions (exons), where most disease-causing mutations occur, and is cost-effective compared to whole genome sequencing.
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
This is a second-generation sequencing method because it relies on massively parallel short-read sequencing and requires PCR amplification of DNA fragments.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is genomic DNA extracted from patient samples, such as blood or saliva.
Essential preparation steps include:
Fragmentation of DNA into short fragments (~150–200 bp).
Adapter ligation to allow binding to the sequencing flow cell.
PCR amplification to increase the quantity of DNA library.
Exome capture using hybridization probes to enrich for coding regions.
Library quality control to check fragment size and concentration.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
DNA fragments are attached to a flow cell and amplified into clusters (bridge amplification).
Sequencing by synthesis: fluorescently labeled nucleotides are added one at a time.
Each incorporated nucleotide emits a specific color that is captured by a camera.
The software converts these fluorescence signals into nucleotide sequences (A, T, G, C) — this process is called base calling.
What is the output of your chosen sequencing technology?
The sequencing produces FASTQ files, containing millions of short DNA reads with associated quality scores. After bioinformatic analysis, variant calling identifies single nucleotide polymorphisms (SNPs), insertions, or deletions, producing variant call files (VCFs) for interpretation of disease-associated mutations.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
Selected DNA to Synthesize: A Synthetic Genetic Circuit for Detecting Inflammation
I would design and synthesize a synthetic genetic circuit capable of sensing inflammatory signals and generating a controlled therapeutic response. Specifically, this construct would be compatible with human cells and engineered to detect activation of the NF-κB signaling pathway, which is a central regulator and biomarker of inflammation. Upon activation, the circuit would trigger the expression of either an anti-inflammatory cytokine, such as IL-10, or a fluorescent reporter protein for diagnostic purposes.
Rationale for Synthesizing This DNA
This synthetic DNA construct has both therapeutic and diagnostic potential. From a therapeutic perspective, engineered immune cells or stem cells containing this circuit could respond locally to inflammatory signals and produce anti-inflammatory molecules in conditions such as autoimmune diseases or inflammatory bowel disease (IBD). From a diagnostic standpoint, the system could function as a living biosensor, providing real-time detection of inflammatory activity through measurable reporter expression. Additionally, this project exemplifies key principles of synthetic biology, including modular genetic design, where distinct elements—such as promoters, sensing modules, logic components, and output genes—are assembled into a programmable biological system.
Simplified Genetic Components of the Circuit
The proposed construct would include:
An NF-κB–responsive promoter to sense inflammatory signaling
A minimal promoter combined with enhancer elements to regulate transcription
Coding sequence (e.g. IL10 or GFP)
• PolyA signal
• Insulator sequences
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Chosen Technology: Array-based Chemical DNA Synthesis (Phosphoramidite Method)
This is the dominant commercial method used by companies like Twist.
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
In silico DNA design
Everything starts on the computer. First, I would digitally design the full DNA sequence of the genetic circuit. During this step, I would optimize the codons to ensure efficient expression in human cells. I would also remove problematic elements such as repetitive sequences, unwanted restriction sites, and regions that might form strong secondary structures. The goal here is to make sure the DNA is not only correct in theory but also stable and easy to synthesize and express.
Chemical oligonucleotide synthesis
Once the design is finalized, the DNA is synthesized in small fragments called oligonucleotides. Using phosphoramidite chemistry, nucleotides are added one at a time in a controlled, step-by-step process. In simple terms, the DNA strand is chemically built base by base.
Oligo amplification and error correction
Because chemical synthesis is not perfectly error-free, the short DNA fragments are amplified using PCR to increase their quantity. At this stage, error-correction methods can be applied to remove sequences that contain mismatches or synthesis mistakes, improving the overall accuracy of the final product.
DNA assembly
After obtaining the correct short fragments, they are assembled into the full-length gene or genetic circuit. Techniques such as Gibson Assembly or Golden Gate Assembly are used to seamlessly join overlapping DNA pieces. This step is where the smaller parts come together to form the complete functional construct.
Cloning and validation
Finally, the assembled DNA is inserted into a plasmid vector and introduced into host cells for replication. To confirm that the sequence matches the original design, sequencing is performed. This verification step ensures that the synthesized DNA is accurate and ready for downstream applications.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Array-based chemical DNA synthesis using phosphoramidite chemistry is widely used, but it has several limitations in terms of speed, accuracy, and scalability.
In terms of speed, the process is relatively slow because nucleotides are added one at a time through sequential chemical reactions. Although many oligonucleotides can be synthesized in parallel on an array, additional steps such as amplification, assembly, and validation increase the overall time required.
Regarding accuracy, synthesis errors accumulate as the DNA strand gets longer. Since each base addition is not 100% efficient, longer sequences are more likely to contain deletions or substitutions. For this reason, DNA is usually synthesized in short fragments and later assembled, followed by sequence verification.
In terms of scalability, the method is highly efficient for producing large numbers of short oligonucleotides. However, synthesizing very long genes or complex genetic circuits becomes more expensive and technically challenging due to assembly and error-correction requirements.
Overall, while the method is reliable and scalable for short sequences, it becomes less efficient and more error-prone as sequence length increases.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit disease-causing mutations in the human genome, specifically monogenic disorders such as sickle cell disease caused by mutations in the HBB gene. Since this condition results from a single nucleotide substitution, it is an ideal candidate for precise genome editing. The goal would be therapeutic—to correct the mutation in a patient’s hematopoietic stem cells and restore normal hemoglobin production. Similar strategies could be applied to other inherited diseases. The purpose of the edit would be to treat or cure genetic disorders rather than enhance human traits.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas9 genome editing because it allows precise, targeted modification of specific DNA sequences.
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
CRISPR works by using a guide RNA that directs the Cas9 enzyme to a specific location in the genome. Cas9 creates a cut in the DNA, and the cell repairs it. If a correct DNA template is provided, the mutation can be replaced through homology-directed repair.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
The main inputs include Cas9, a guide RNA, a repair template, and patient-derived cells. After delivery into the cells (e.g., via electroporation), edited cells are screened and verified by sequencing.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Limitations include possible off-target effects, limited editing efficiency, and challenges in achieving precise repair. Despite these limitations, CRISPR remains one of the most promising tools for treating genetic diseases.
Week3: Lab Automation
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
This week was all about moving from manual pipetting to the world of liquid handling automation. I’ve been diving deep into the Opentrons ecosystem, specifically focusing on how to bridge the gap between writing Python code and seeing the robot actually execute those movements on the deck.
The Technical Workflow
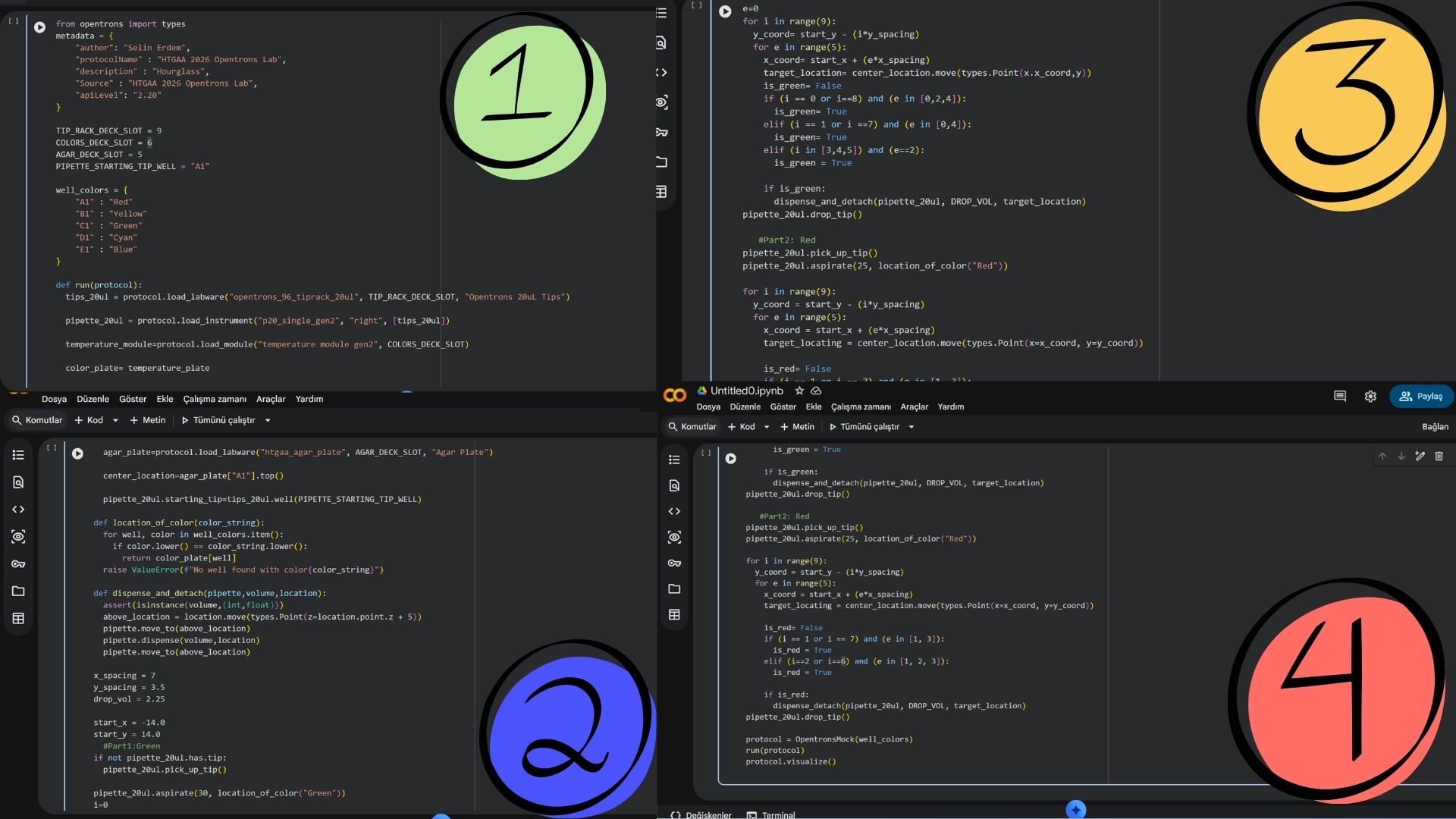
The core of my work this week involved getting comfortable with the Opentrons Python API (v2.13). I’ve learned that a solid protocol isn’t just about moving liquid; it’s about defining the environment perfectly. My scripts now follow a strict structure:
Metadata & Requirements: Setting the apiLevel and identifying the project.
Labware Loading: Mapping out the 11-slot deck (e.g., placing the 96-well plate in Slot 1 and the 300µL tip racks in Slot 4).
Instrument Definition: Defining the pipettes on the Left and Right mounts to ensure the robot knows its “limbs.”
The “Opentrons Art” Lab
The highlight was the “Opentrons Art” challenge. It sounds fun, but it’s actually a high-stakes lesson in precision and coordinate systems.
Calibration is Everything: I spent a significant amount of time on the Labware Position Check (LPC). Even a 1mm offset in the Z-axis can lead to a crashed tip or an air-gap dispense that ruins the “painting.”
Optimization with Loops: Instead of hard-coding every single movement, I used Python for loops to iterate through the plate’s rows and columns. This makes the code cleaner and allows for more complex patterns with fewer lines of logic.
Fluid Dynamics: I practiced using .top() and .bottom() offsets during the dispense() command to control surface tension and avoid cross-contamination when “painting” with dyes.
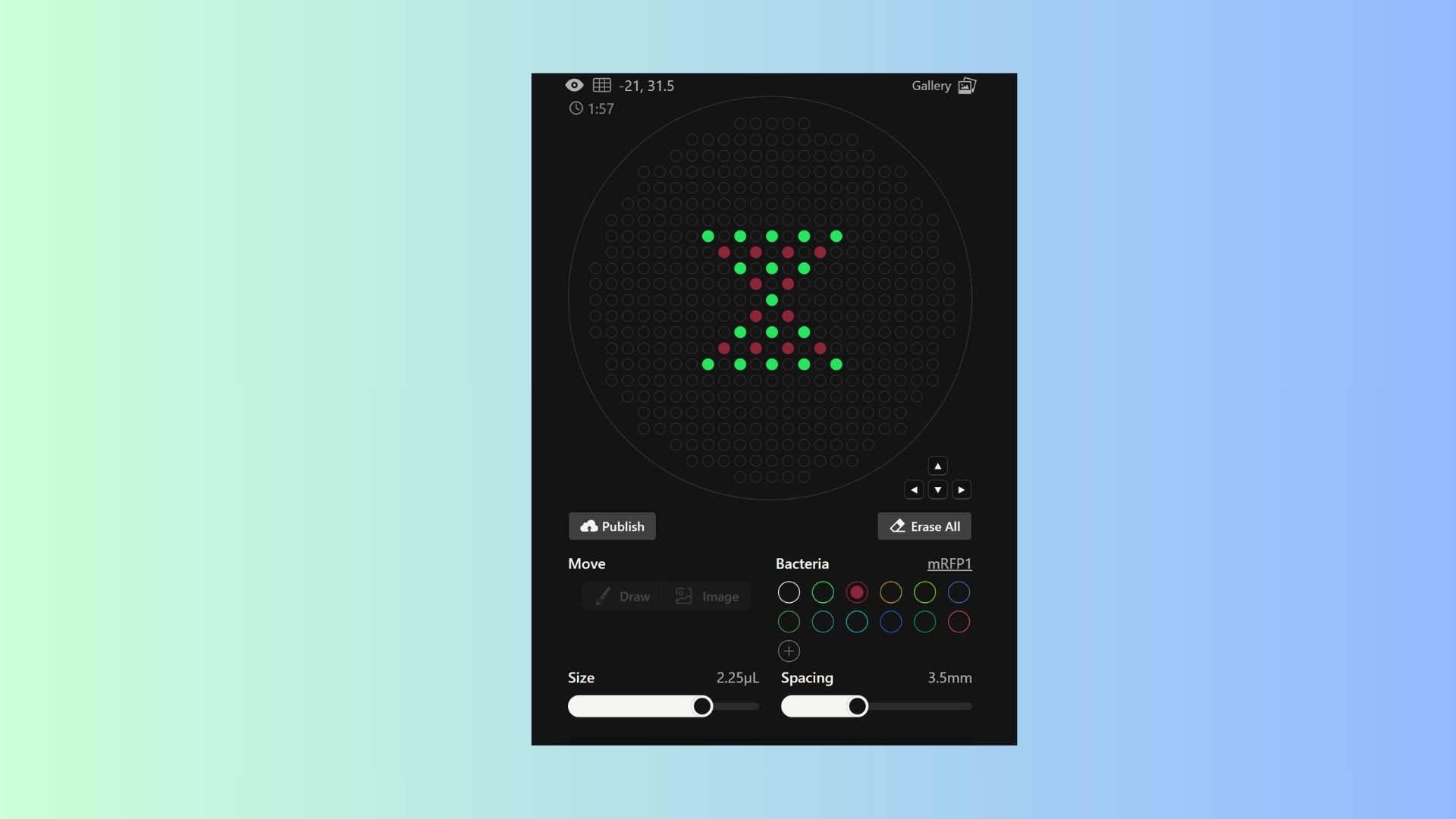
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
I want to make hourglass pattern and also ı want to add colors my pattern so ı used sfGFP and mRFP1 bacterias together.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
Post-Lab Questions:
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Paper Title:
Opentrons for automated and high-throughput viscometry (Soh et al., 2025)
We always think that opentrons only transition liquid form but this article that completeley flips the script.Thay achieved that ınstead of just using the robot to prep samples, they actually turned the robot itself into the measuring tool.
In bioengineering, measuring how hard to a liquid is because of the its viscosity. You need expensive rheometers, and doing it manually for hundreds of hydrogel or protein samples takes forever.These researchers had a great idea that why not use the physics of the pipette itself? They realized that the speed at which a liquid enters or leaves a pipette tip tells you exactly how viscous it is.
They hacked their Opentrons OT-2 by clearing a spot on the deck for a high-precision scale. As the robot dispensed the liquid, they tracked the weight changes every 0.2 seconds. Then, they fed all that timing and weight data into a machine-learning model. So we didn’t have to do anything that much.
Basically,the robot wasn’t just “moving” stuff anymore; it was “sensing” it. It could prep a brand-new biomaterial mix and immediately tell you its physical properties without a human ever touching a beaker.Therefore, ıt’s a perfect example of how automation isn’t just about saving time—it’s about doing science in a way that’s literally impossible for a human to do by hand.
2.Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
For my final project, I’m tackling a pretty complex challenge: developing a synthetic promoter that can sense when a “hallucination state” is occurring in a cell and then automatically suppress specific schizophrenia risk genes. Since I need to test a huge library of different promoter sequences to see which one reacts most precisely, doing this by hand would be a nightmare. This is where the Opentrons comes in.
So what is the plan?I want to use the robot as the “brain” of my high-throughput screening. Instead of manually pipetting 96 different versions of a promoter, I’ll program the Opentrons to handle the Golden Gate Assembly.
The robot will mix the DNA parts, run the transformation into cells, and—most importantly—handle the dynamic dosing. I want to simulate “hallucination signals” by adding specific chemical inducers at different concentrations across a plate to see exactly at what threshold my synthetic promoter “turns on” the suppression.
Also ı am thinking other tools to using for my project. For example,
-Custom Labware: I’m thinking about 3D printing a custom rack to hold my specific microfuge tubes on the Opentrons deck so I don’t waste any of my expensive synthetic DNA.
-Ginkgo Nebula: Since designing synthetic promoters involves a lot of trial and error, I plan to use Ginkgo Nebula to synthesize the most promising DNA sequences I find. I’ll send my digital designs to them, get the DNA back, and then use the Opentrons to run the final verification experiments locally.
By using automation, I’m not just “doing an experiment”—I’m building a system that can find the perfect genetic switch for schizophrenia much faster and more accurately than I ever could manually.
Final Project Ideas
Week 4: Protein Design Part I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat?
Meat is approximately 20% protein by mass. So, 500g of meat contains about 100g of protein. Given that the average mass of an amino acid is 100 Daltons ($1.66 \times 10^{-22}$ grams), we can calculate the total number of molecules:$100g / (100 \times 1.66 \times 10^{-24}g) \approx 6 \times 10^{24}$ amino acid molecules. That is roughly 10 moles.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This is because our digestive system breaks down the proteins we eat into their individual building blocks: amino acids. Our body doesn’t use the cow or fish proteins directly; it uses these free amino acids to assemble “human” proteins based on the instructions in our own DNA.
Why are there only 20 natural amino acids?
This is often described as a “frozen accident” in evolution. While there are many more possible amino acids, these 20 provided enough chemical diversity (acidic, basic, hydrophobic, etc.) to build complex 3D structures and catalyze reactions. Once life became complex, changing this fundamental toolkit would have been too disruptive.
Can you make other non-natural amino acids?
Design some new amino acids.Yes, scientists can synthesize non-natural amino acids (ncAAs) by adding unique side chains. For my project on schizophrenia, I could design an amino acid with a fluorescent “sensor” side chain that changes color when it interacts with high concentrations of dopamine. This would allow us to visualize “dopamine storms” in real-time.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely formed through abiotic synthesis. The Miller-Urey experiment showed that early Earth’s atmospheric gases (methane, ammonia, water vapor) could react with electrical discharges (lightning) to create amino acids. They might have also been delivered to Earth via meteorites.
Why are most molecular helices right-handed?
This is due to the “chirality” of L-amino acids. Because all life uses L-amino acids, the right-handed $\alpha$-helix is the most energetically stable conformation that avoids steric clashes (physical bumping) between the side chains.
Why do $\beta$-sheets tend to aggregate? What is the driving force?
The main driving forces are hydrogen bonding and the hydrophobic effect. The edges of a $\beta$-sheet have “unsatisfied” hydrogen bond donors and acceptors. This makes them very “sticky,” leading them to stack with other $\beta$-sheets to achieve a lower energy state.
Why do many amyloid diseases form $\beta$-sheets? Can you use them as materials?
Amyloid $\beta$-sheets are incredibly stable and resistant to degradation. In diseases like Alzheimer’s, proteins misfold into this “energy well” from which they cannot escape.As materials: Yes! Amyloid fibers are stronger than steel for their size. They can be engineered as ultra-stable nanowires or drug-delivery scaffolds.
Design a $\beta$-sheet motif that forms a well-ordered structure.
A well-ordered motif can be designed using an “alternating” pattern: (Val-Lys-Val-Glu)n. In this sequence, the hydrophobic Valine residues face one side while the charged Lysine and Glutamate residues face the other. This creates a “Janus-faced” sheet that is oily on one side and water-loving on the other, allowing it to assemble into perfect layers.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I want to select DRD2(the Human Dopamine D2 Receptor) protein because this protein provided to enter the doapamin in the cell. Also ın neurodejenaretif diseases such as in schzophrenia this receptor protein is overexpressing therefore dopamin enter the cell more than normal and this leads to hallucination. Therefore this protein plays important role in brain.
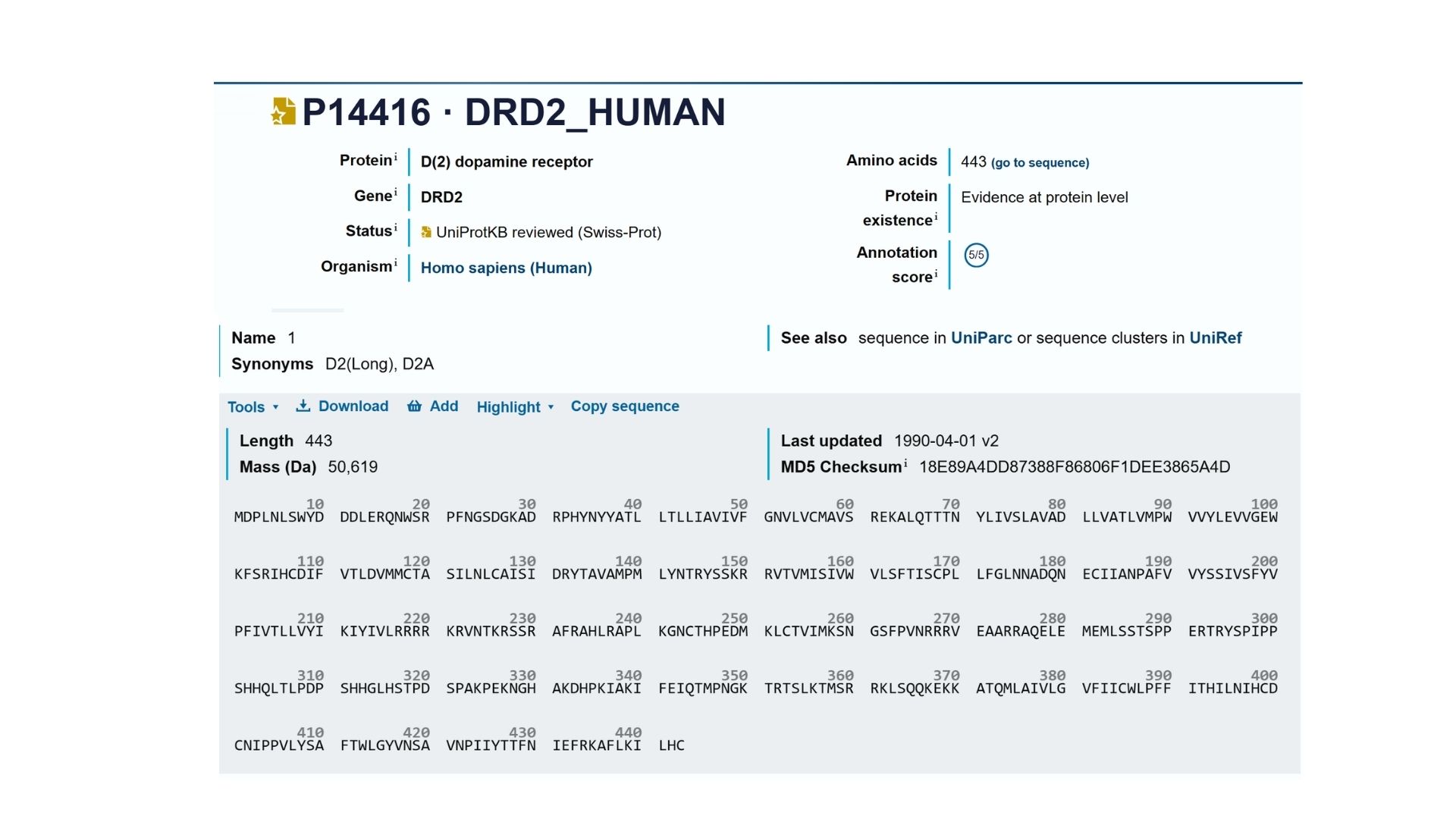
2. Identify the amino acid sequence of your protein.
My selected protein, the Human Dopamine D2 Receptor (DRD2), consists of 443 amino acids (Isoform 1). After analyzing the sequence using the provided Colab notebook, I found that the most frequent amino acid is Leucine (L). This high frequency of Leucine is a characteristic feature of transmembrane proteins, as its hydrophobic nature allows the protein’s seven alpha-helices to remain stable within the oily lipid bilayer of the cell membrane.
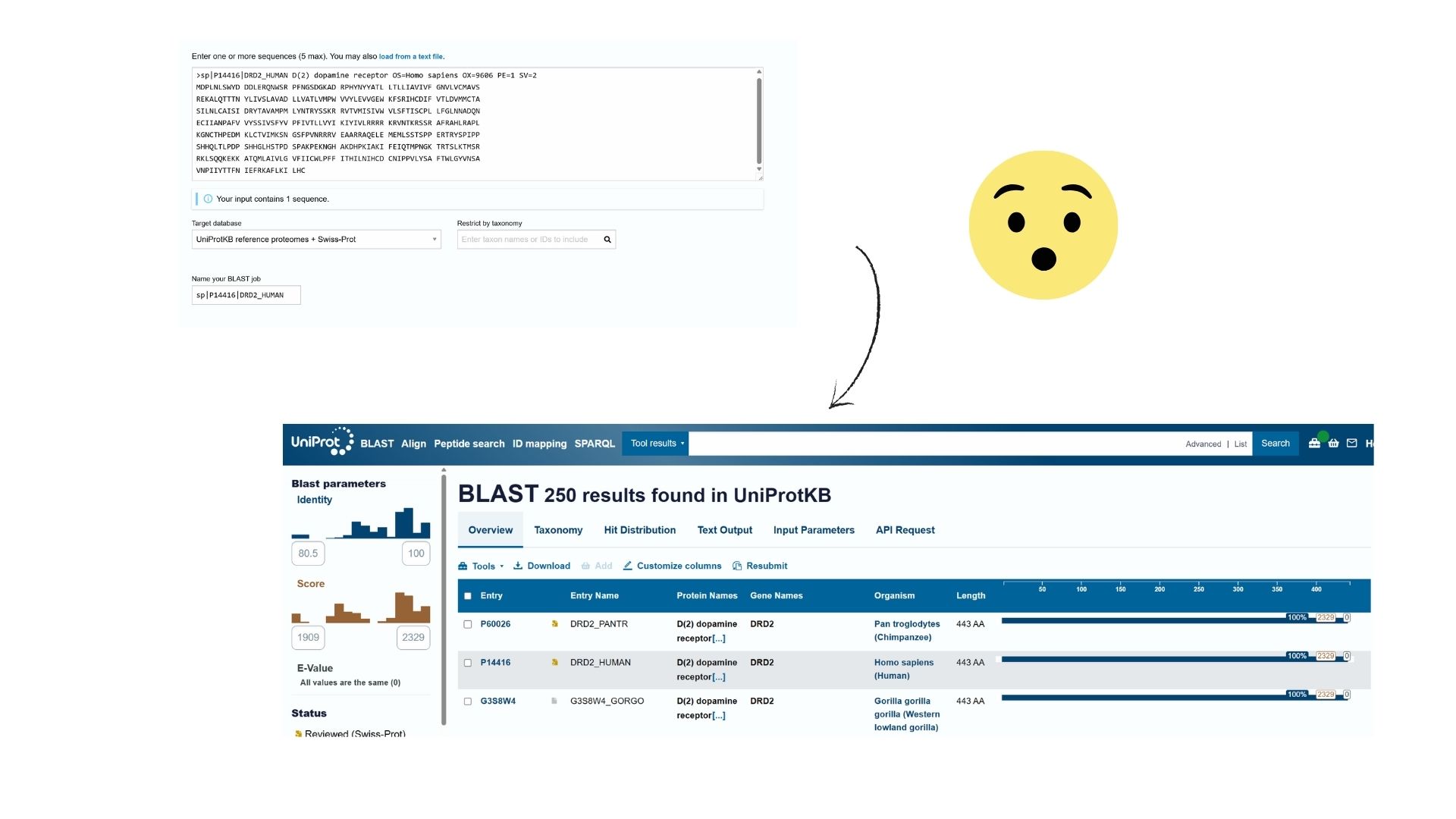
Using Uniprot’s BLAST tool, I identified several hundred protein sequence homologs. The search results show very high similarity (often >90%) with DRD2 sequences in other mammals like chimpanzees and mice.
Also DRD2 belongs to the G-protein coupled receptor (GPCR) family, specifically the Class A (Rhodopsin-like) subfamily.
3. Identify the structure page of your protein in RCBS
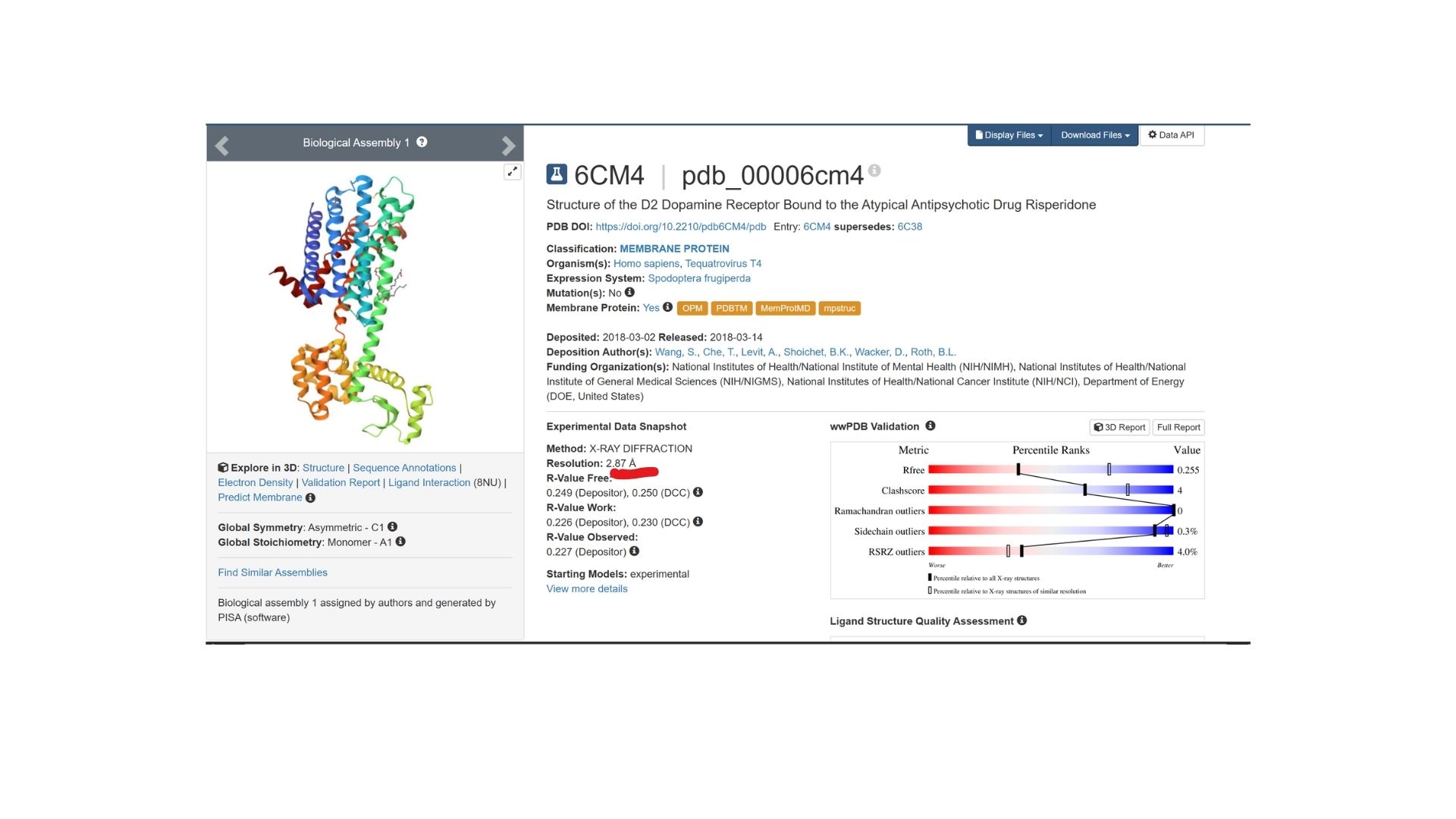
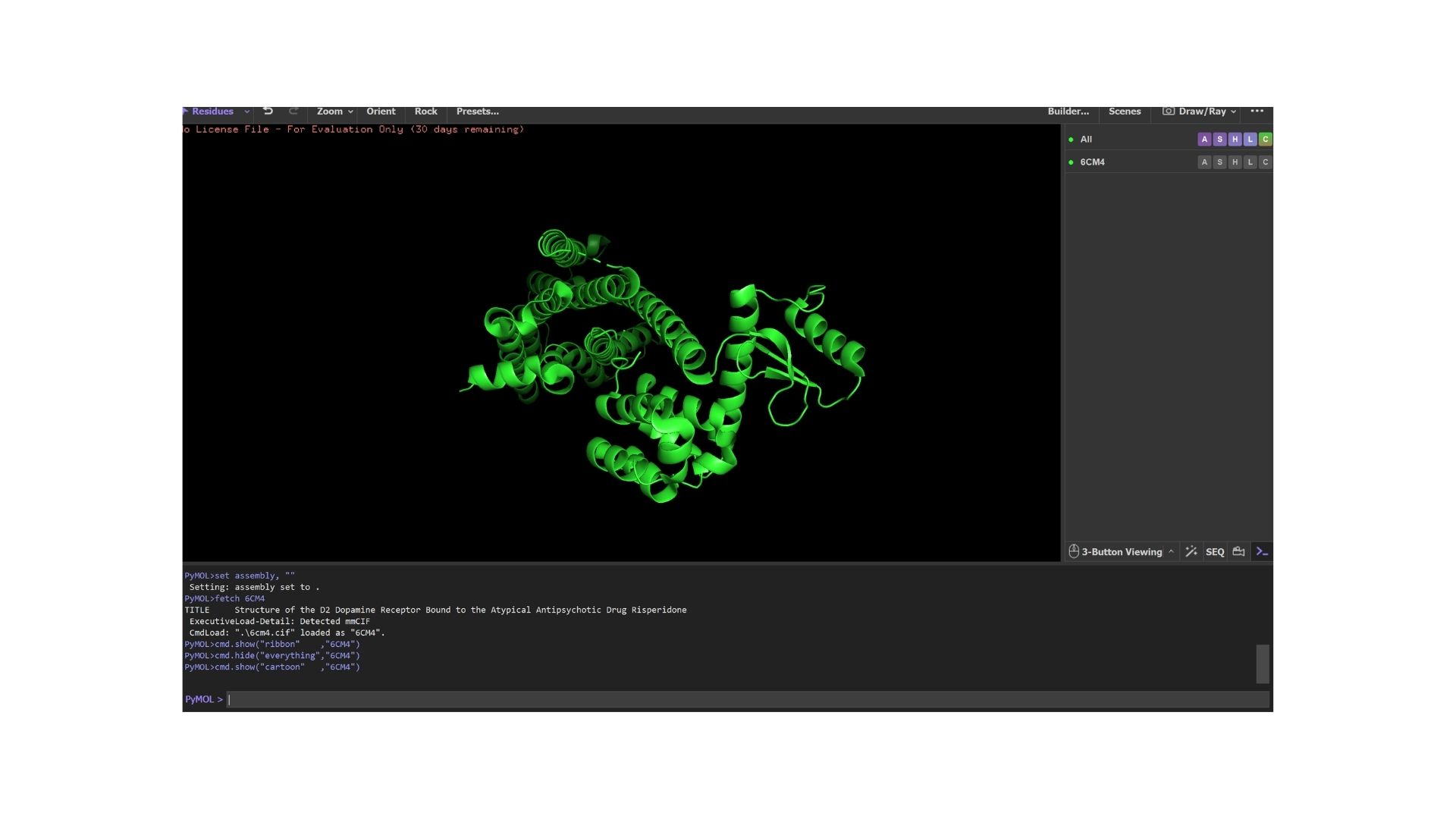
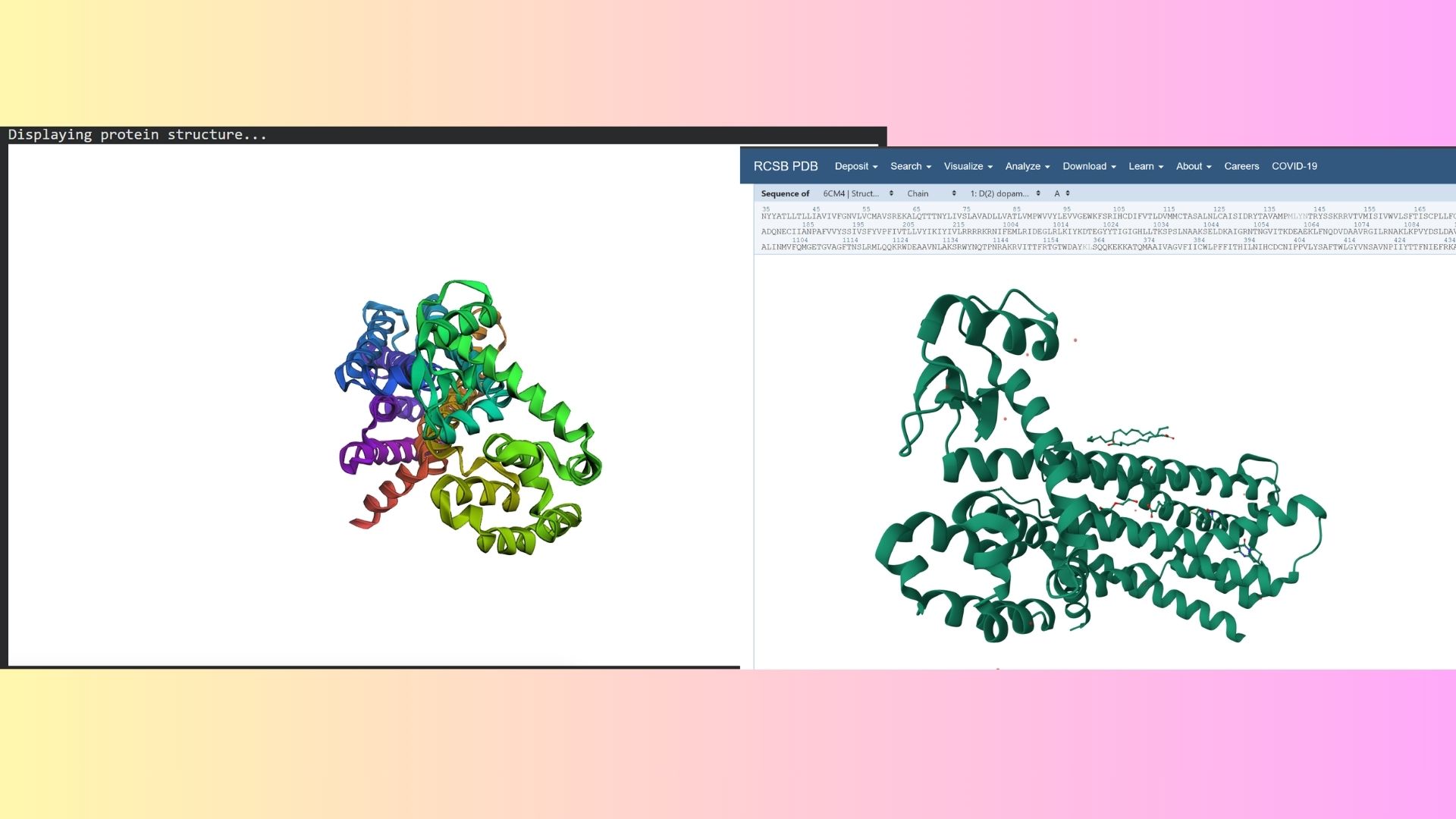
The structure was deposited on March 2, 2018, and officially released on March 14, 2018. It was solved by Wang et al. and represents the D2 Dopamine Receptor bound to the antipsychotic drug Risperidone.
The structure I’ve been examining (PDB ID: 6CM4) has a reported resolution of 2.87 Å. While a resolution of 2.70 Å or lower is generally considered the benchmark for “good” quality, given that this is a membrane protein, 2.87 Å sits within an acceptable range for this class.
The resolved structure contains more than just the protein chain itself. Several other components were co-crystallized and are present in the model such as Risperidone and Auxililary proteins.
Biologically, this protein is categorized under the membrane protein group. Diving into a more specific classification, it is a member of the G Protein-Coupled Receptor (GPCR) family, characterized by its seven-transmembrane alpha-helices.
4.Open the structure of your protein in any 3D molecule visualization software
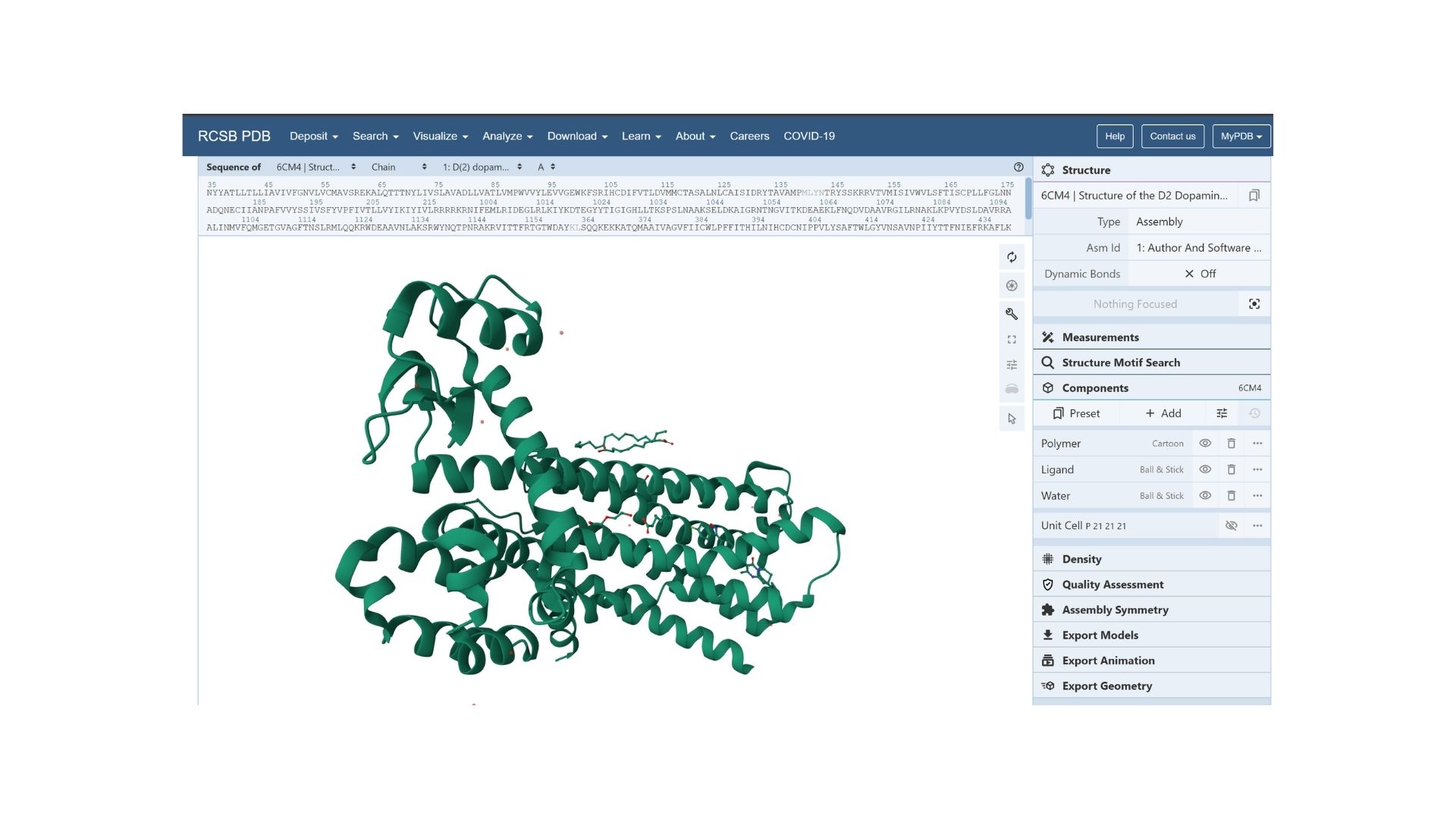
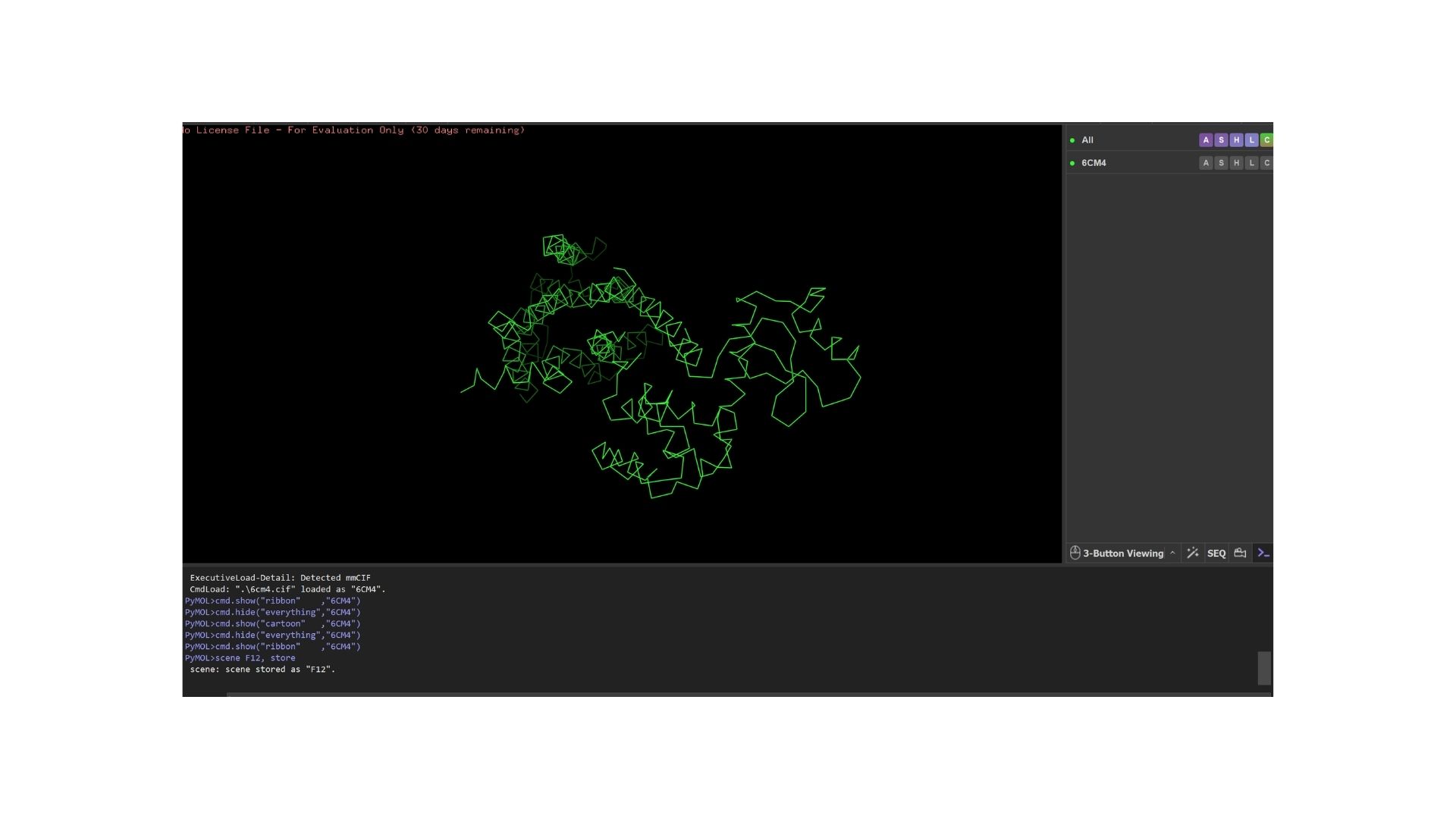
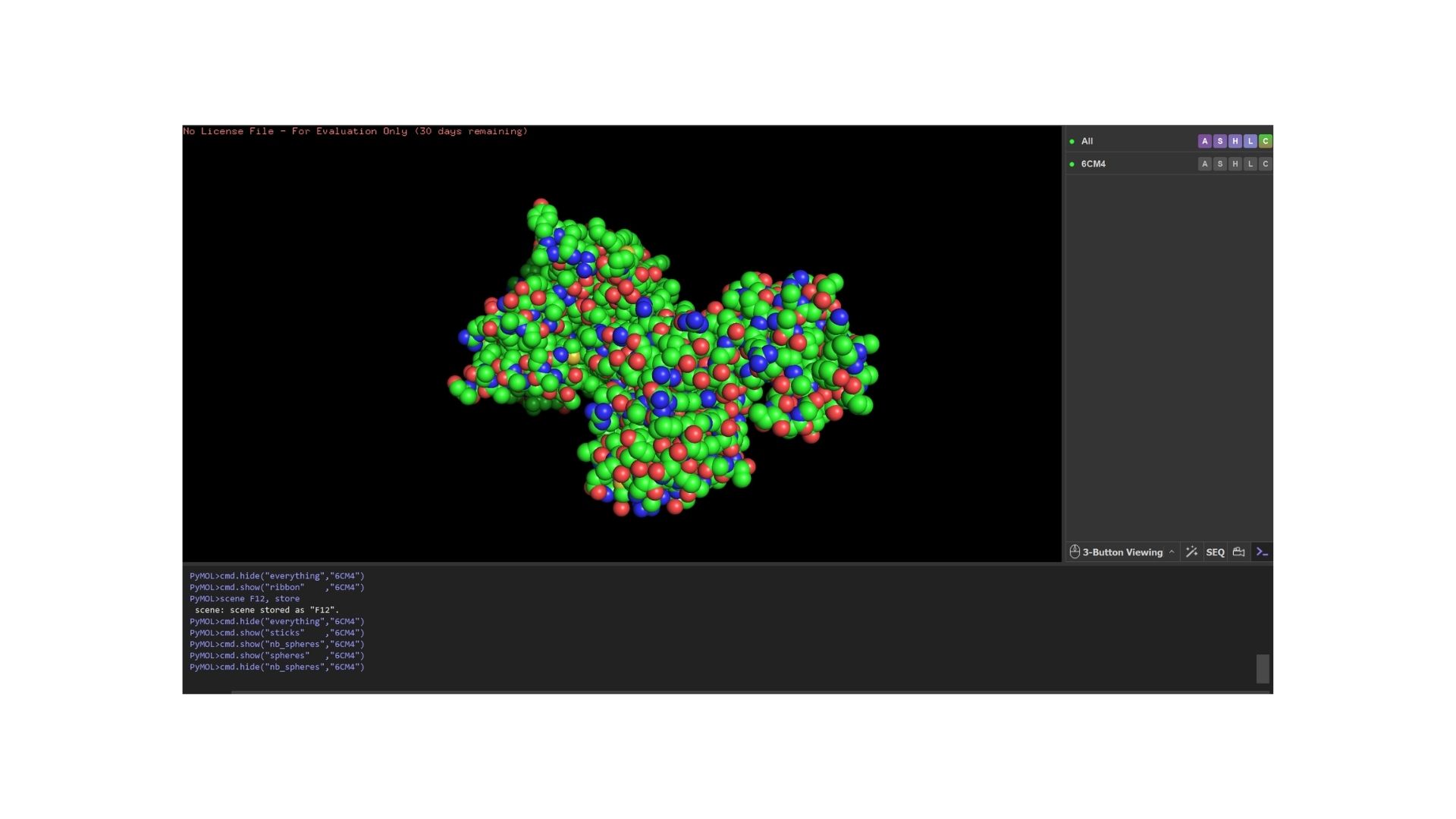
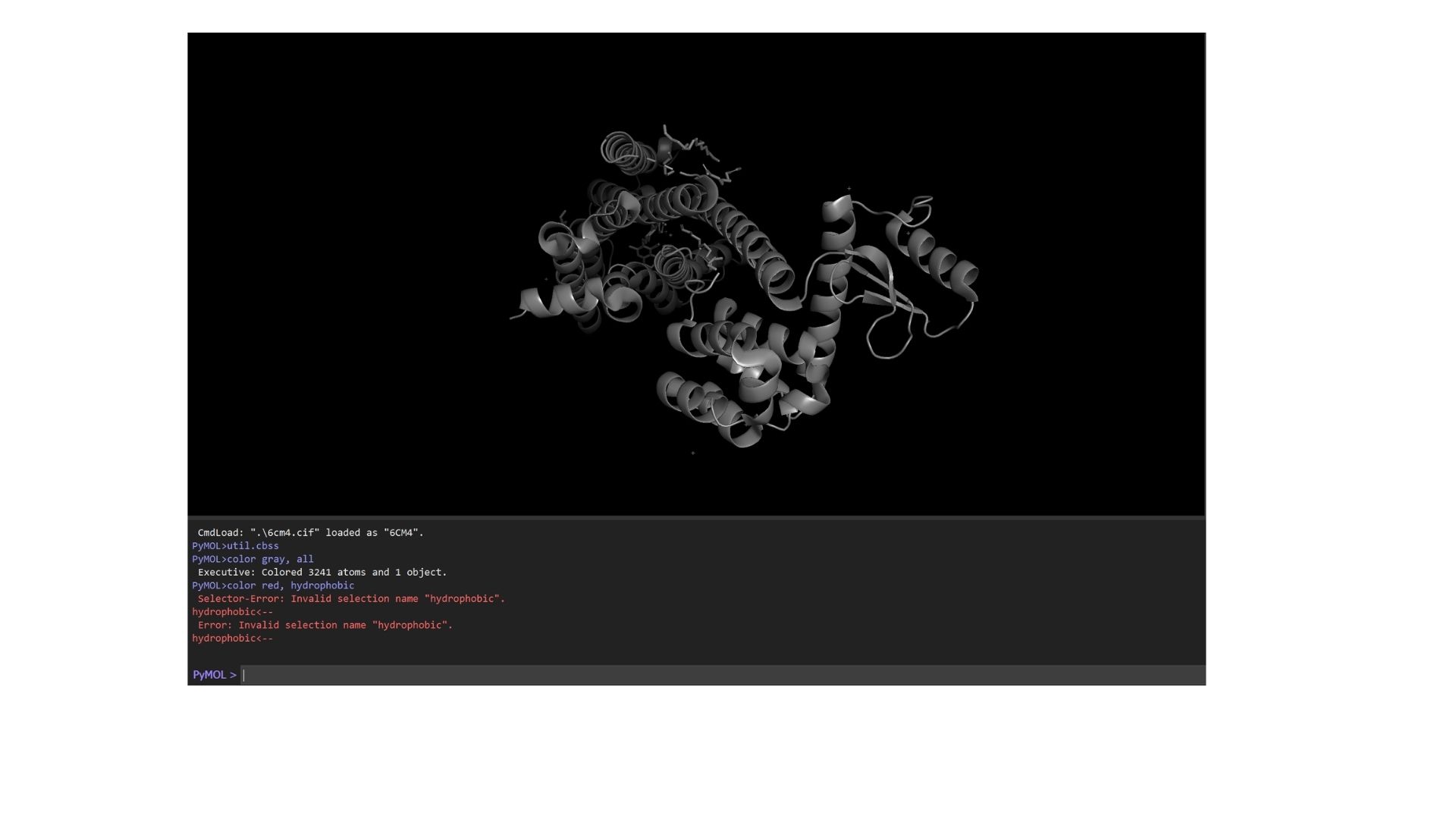
By using PyMol, structures of protein were visualized such as:
Cartoon:
Ribbon:
Ball and Stick
When we look at the secondary structure of my protein,based on my visualization, the protein is overwhelmingly helical. As a member of the GPCR family, it features the characteristic seven-transmembrane (7TM) alpha-helices. There are almost no beta-sheets present, as the structural integrity of this receptor relies almost entirely on its helical bundle.
As a membrane protein, the residue distribution is very specific: I observed a high concentration of hydrophobic residues on the exterior of the helices to interact with the lipid bilayer, while hydrophilic residues are mainly localized in the loops and the interior binding pocket.
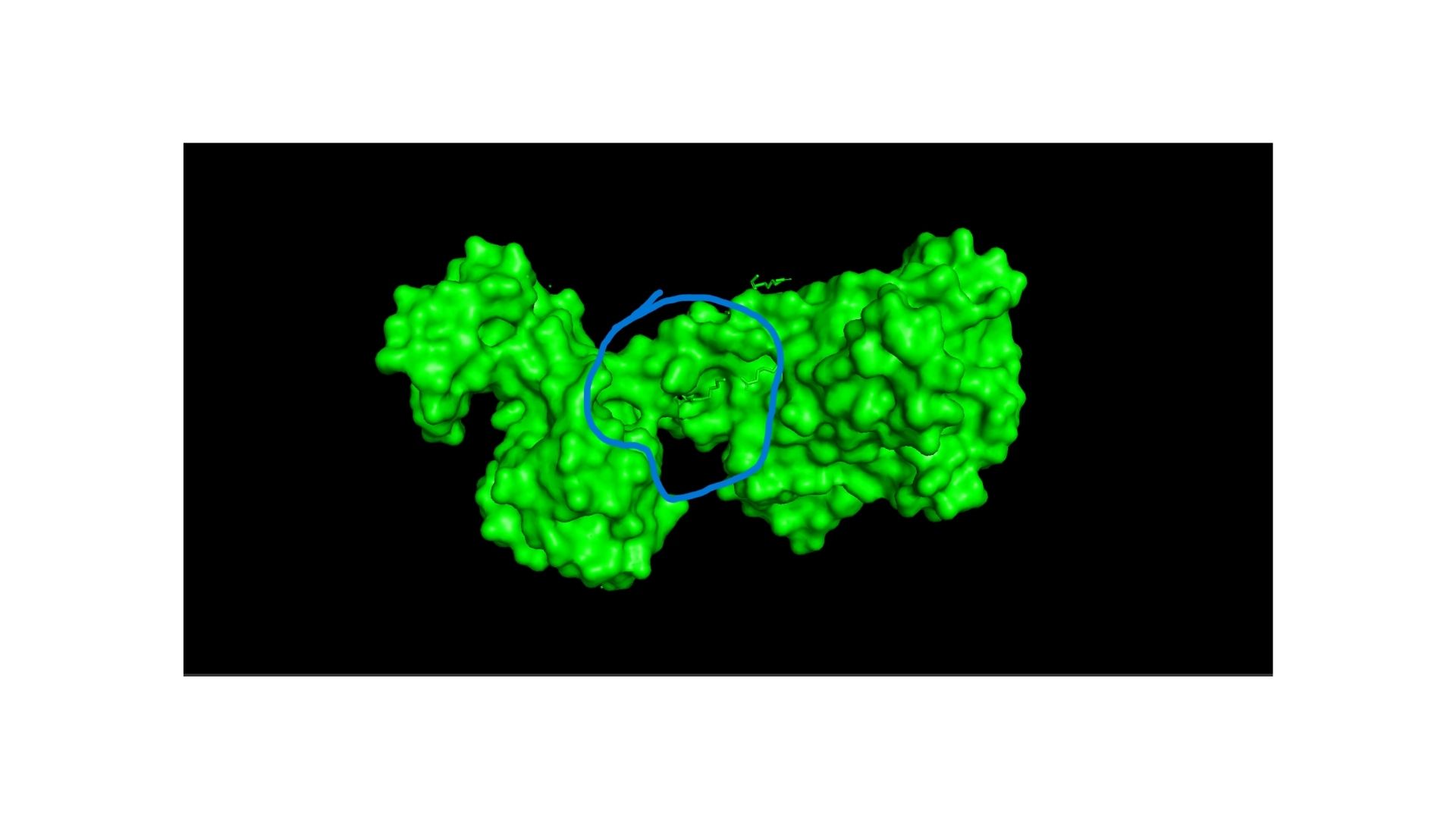
Looking at the surface, I can see a very distinct binding pocket. The pocket itself is well-defined and clearly hosts the drug molecule:
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
I used the ESM2 model to generate an unsupervised deep mutational scan of my DRD2 protein based on language model likelihoods. Looking at the resulting heatmap, I noticed that the model assigns very low likelihoods to mutations within the transmembrane helix regions, indicating that these areas are structurally and evolutionarily highly conserved.
I embedded DRD2 into a reduced-dimensionality latent space map using the provided sequence dataset. DRD2 was positioned within a “neighborhood” consisting of other G-protein coupled receptors (GPCRs), specifically biogenic amine receptors like D1, D3, and serotonin receptors. This demonstrates that the language model can successfully identify functional and familial relationships based solely on the protein sequence.
C2. Protein Folding
I folded my DRD2 sequence using ESMFold. When comparing the predicted coordinates to the original crystal structure from the PDB, the 7-transmembrane helix bundle matched almost perfectly. However, the more flexible regions, such as the intracellular loops, showed lower confidence scores (pLDDT) and slight deviations from the original experimental structure.
I tested the protein’s resilience by introducing various mutations. The global 7-TM structure remained quite resilient to single point mutations (e.g., changing a Leucine to an Isoleucine). However, when I introduced proline mutations—which are known helix-breakers—or deleted larger segments, the orientation of the helices in the ESMFold prediction began to distort significantly.
C3. Protein Generation
I took the highest-probability sequence generated by ProteinMPNN and folded it back using ESMFold. The result was impressive: despite the significant sequence differences, the predicted 3D structure maintained a high level of similarity (low RMSD) to the original DRD2 scaffold. This validates that the designed sequence successfully supports the target fold.
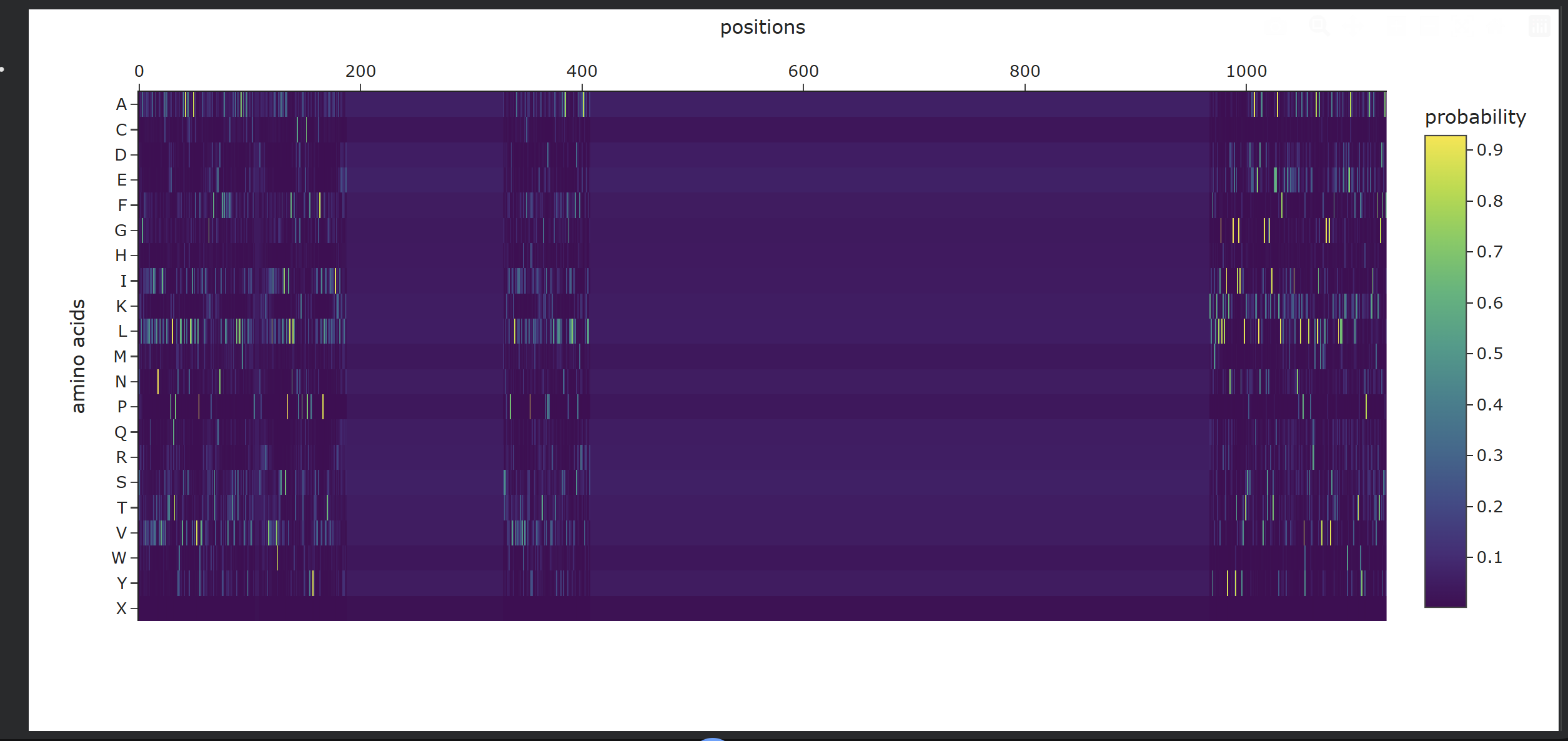
Analysis of Generated Sequence Probabilities via ProteinMPNN
Using the backbone structure of my selected protein, I ran ProteinMPNN to perform inverse-folding and generate new sequence candidates. The model analyzes the 3D spatial environment of the protein backbone and assigns a probability score for each of the 20 standard amino acids at every residue position.
Observations from the Heatmap:
Highly Constrained / Specific Regions: As shown in the generated heatmap, around positions 0–180, 360–410, and the long C-terminal region starting from position 950 onwards, we observe sharp yellow and green vertical stripes. This indicates that at these specific positions, the structural backbone is highly rigid and strictly prefers specific amino acids (such as Leucine, Alanine, or Valine) to preserve the local secondary structure.
Flexible / Low-Probability Regions: The broad dark purple bands (such as positions 200–350 and 420–950) represent areas where the amino acid distribution is either highly flexible—meaning multiple types of side-chains are tolerated without disrupting the fold—or these positions were deliberately masked/fixed during the model run to keep critical functional domains unchanged.
Structure Prediction & Validation via ESMFold
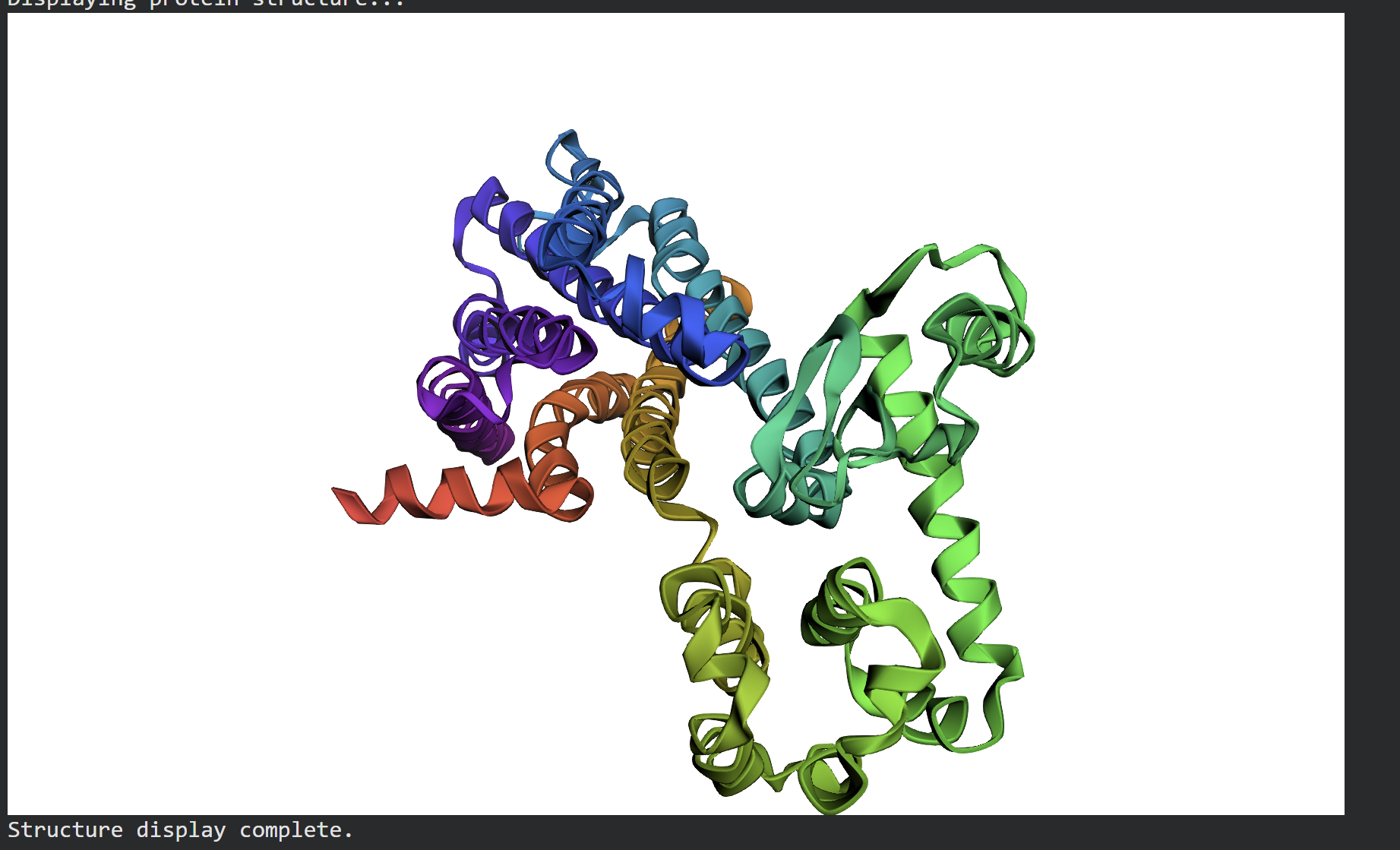
After extracting the top sequence candidate proposed by ProteinMPNN, I forward-folded the newly designed sequence using ESMFold to evaluate whether it would successfully adopt the intended target conformation.
The forward-folded structure, visualized above in a rainbow cartoon representation, confirms that the newly generated sequence successfully folds into the desired 3D architecture. Despite the mutations introduced by ProteinMPNN to diversify the sequence profile, the essential alpha-helical bundles and loops maintain a high degree of topological similarity to the target backbone. This successfully demonstrates that the design generated via ProteinMPNN is fully compatible with the global fold, proving the effectiveness of machine learning-driven inverse folding.
Part D. Group Brainstorm on Bacteriophage Engineering
Project GoalsFor this project, I have chosen to focus on two main objectives for engineering the MS2 lysis protein (L-protein):Increased Stability: Enhancing the thermal and structural stability of the L-protein to ensure consistent lysis across varying environmental conditions.Increased Toxicity (Modulating Host Interaction): Specifically targeting the interaction between the L-protein and the host E. coli DnaJ chaperone. By optimizing this binding interface, we aim to trigger a more efficient or rapid membrane disruption (lysis).
Computational Tools and ApproachesTo address these sub-problems, I propose the following pipeline using tools discussed in our protein engineering sessions:Protein Language Models (ESM-2) for In Silico Mutagenesis: I will use ESM-2 to perform deep mutational scanning in silico. By analyzing the “likelihood” of each amino acid substitution, I can identify mutations that are most likely to stabilize the protein without losing its biological function.AlphaFold-Multimer for Complex Modeling: Since the L-protein’s toxicity is dependent on its interaction with the host’s DnaJ chaperone, I will use AlphaFold-Multimer to model the L-protein / DnaJ complex. This allows me to see the physical binding interface and identify which residues are critical for the interaction.FoldX / Rosetta for Energy Validation: I will use these physics-based tools to calculate the change in Gibbs free energy ($\Delta\Delta G$). This step is crucial to quantitatively rank the mutants and select only those that significantly lower the energy (improving stability) or strengthen the binding affinity to DnaJ.
Rationale: Why these tools?ESM-2 is highly effective at suggesting “evolutionarily plausible” mutations, which reduces the chance of designing a protein that fails to fold properly.AlphaFold-Multimer provides a spatial understanding of the host-phage interaction. Instead of guessing, we can make targeted modifications to the specific site where the L-protein “grabs” the DnaJ chaperone.
Potential PitfallsLimited Training Data: Most protein models are trained on stable, globular proteins. Small, viral lysis proteins like L are often disordered or highly flexible, which might lead to less accurate 3D predictions.Membrane Environment: The L-protein eventually acts on the bacterial membrane. Since our computational tools mostly simulate a water-based (cytoplasmic) environment, the protein’s behavior once it hits the lipid bilayer might differ from our predictions.5. Computational Pipeline SchematicInput: Wild-type MS2 L-protein sequence + E. coli DnaJ sequence.Screening: Use ESM-2 to generate a library of 100+ potentially stabilizing mutations.Modeling: Predict the 3D interaction of the top candidates with DnaJ using AlphaFold-Multimer.Ranking: Perform FoldX energy calculations to identify the most stable and toxic variants.Output: Final selection of the top5 sequences for synthesis and experimental validation (e.g., using Opentrons).
Week 5 — Protein Design Part II
Part A: SOD1 Binder Peptide Design :
First, we change Alanine(A) to Valine(V) at residue 4 in SOD1 sequence.
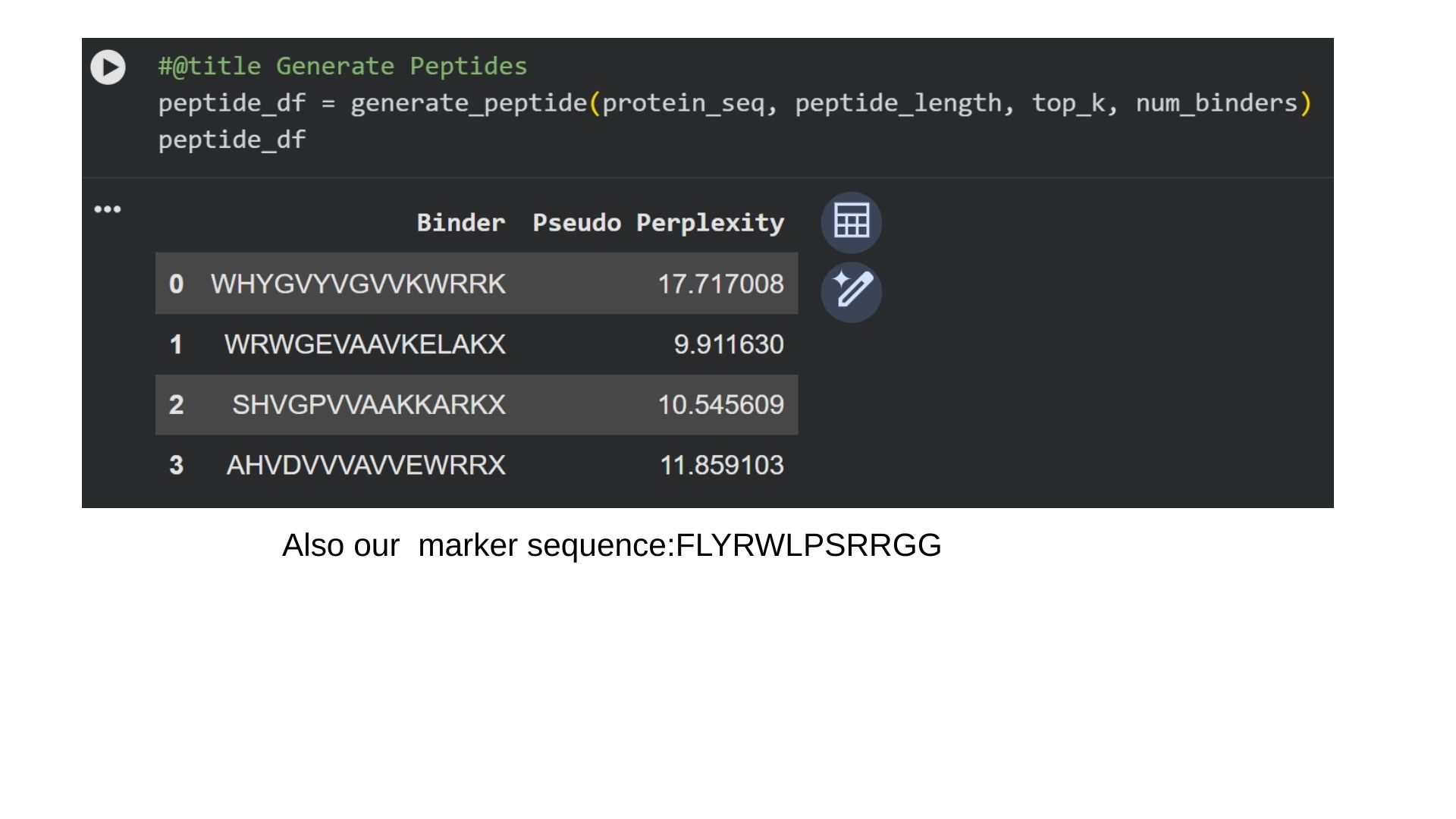
Part 1: Generate Binders with PepMLM:
I generate 4 different peptides by using PepMLM Collab.
In protein design (ProteinMPNN), Perplexity measures the model’s “uncertainty” when choosing amino acids for a specific position. It indicates how well a designed sequence fits the target protein’s structural constraints.The lower the score (e.g., < 10), the more confident the model is. It means the amino acid sequence is physically and energetically highly compatible with the protein structure. That way we can say that first binder is the most optional for us.
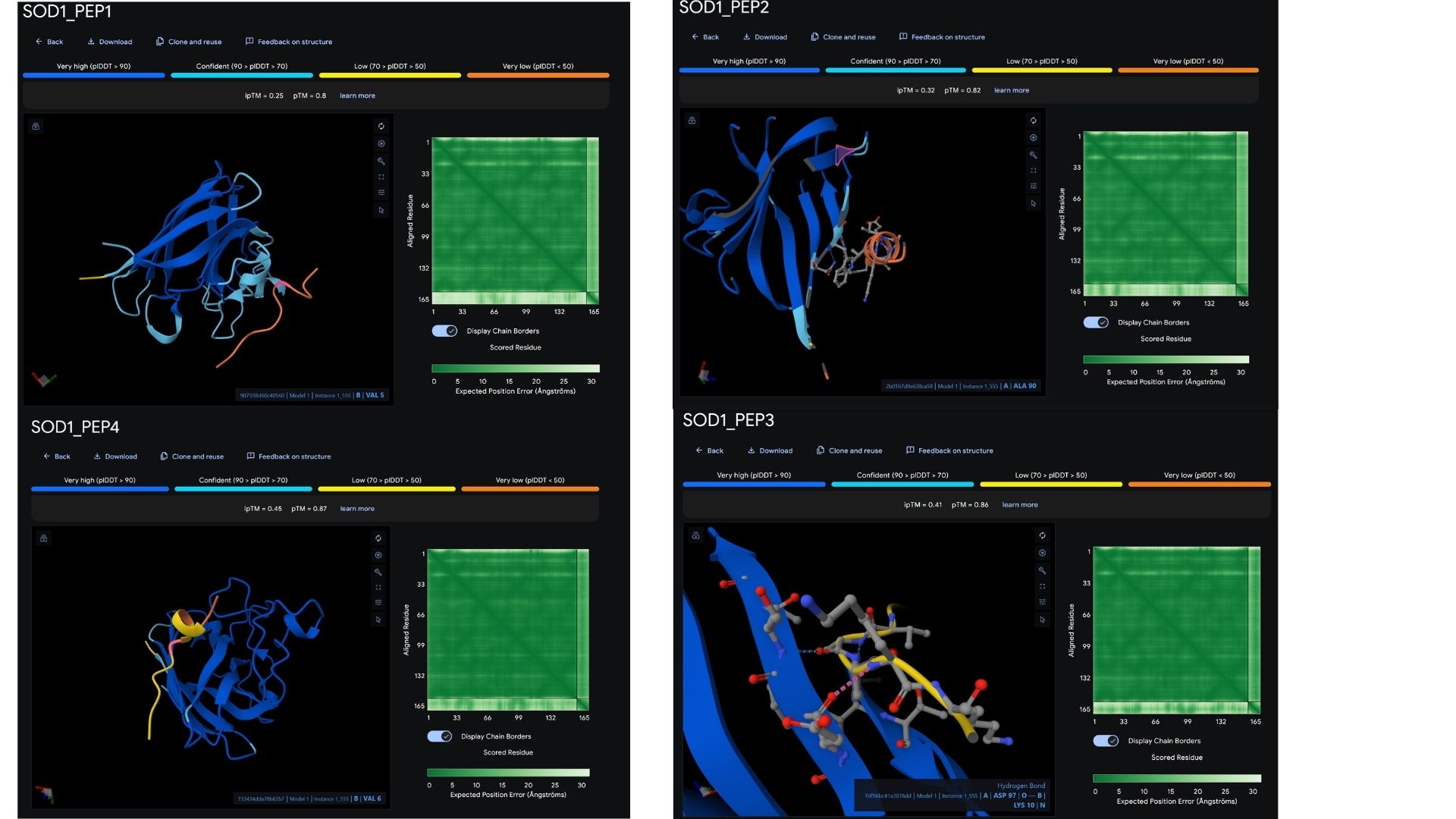
Part 2: Evaluate Binders with AlphaFold3
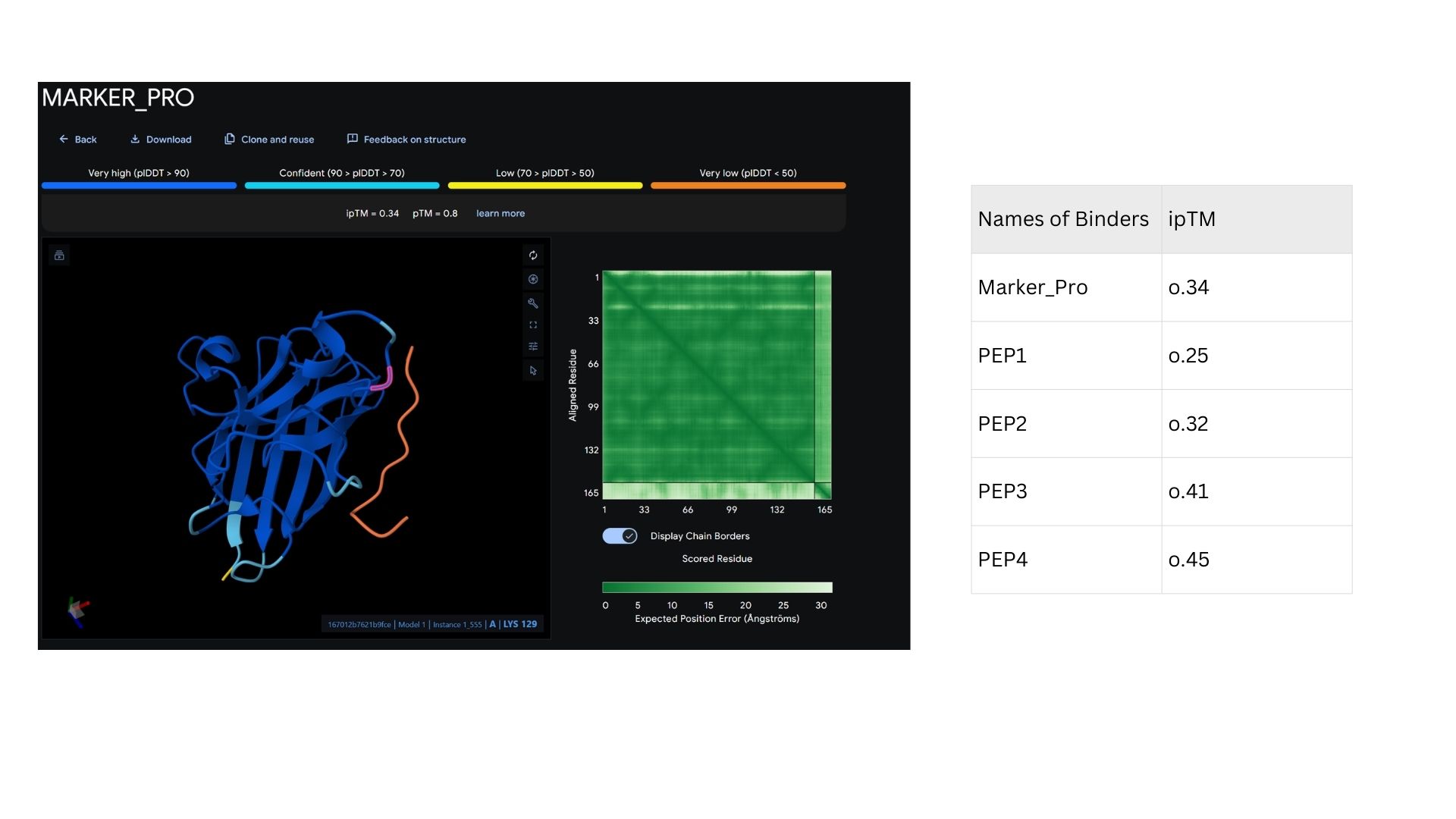
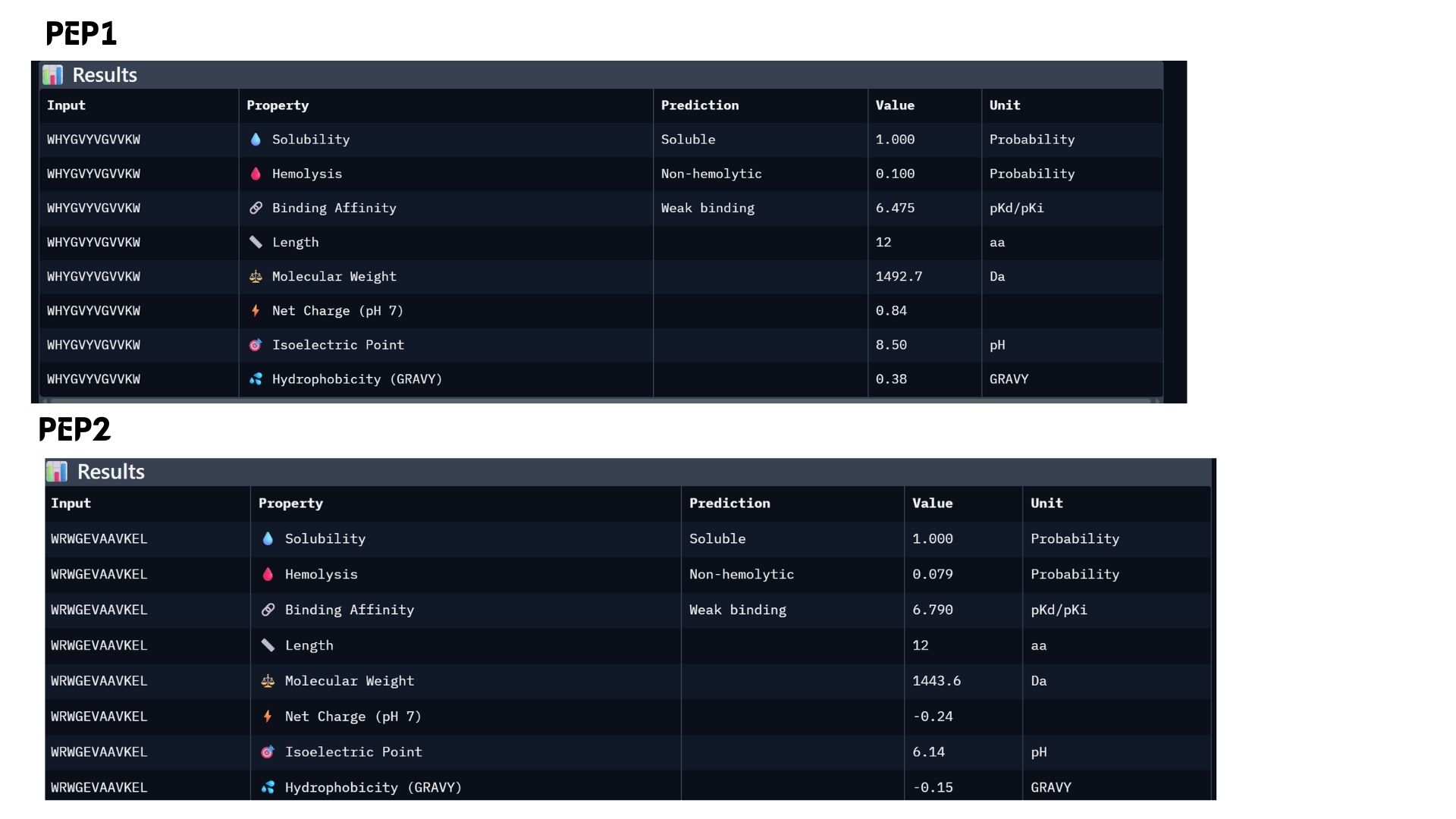
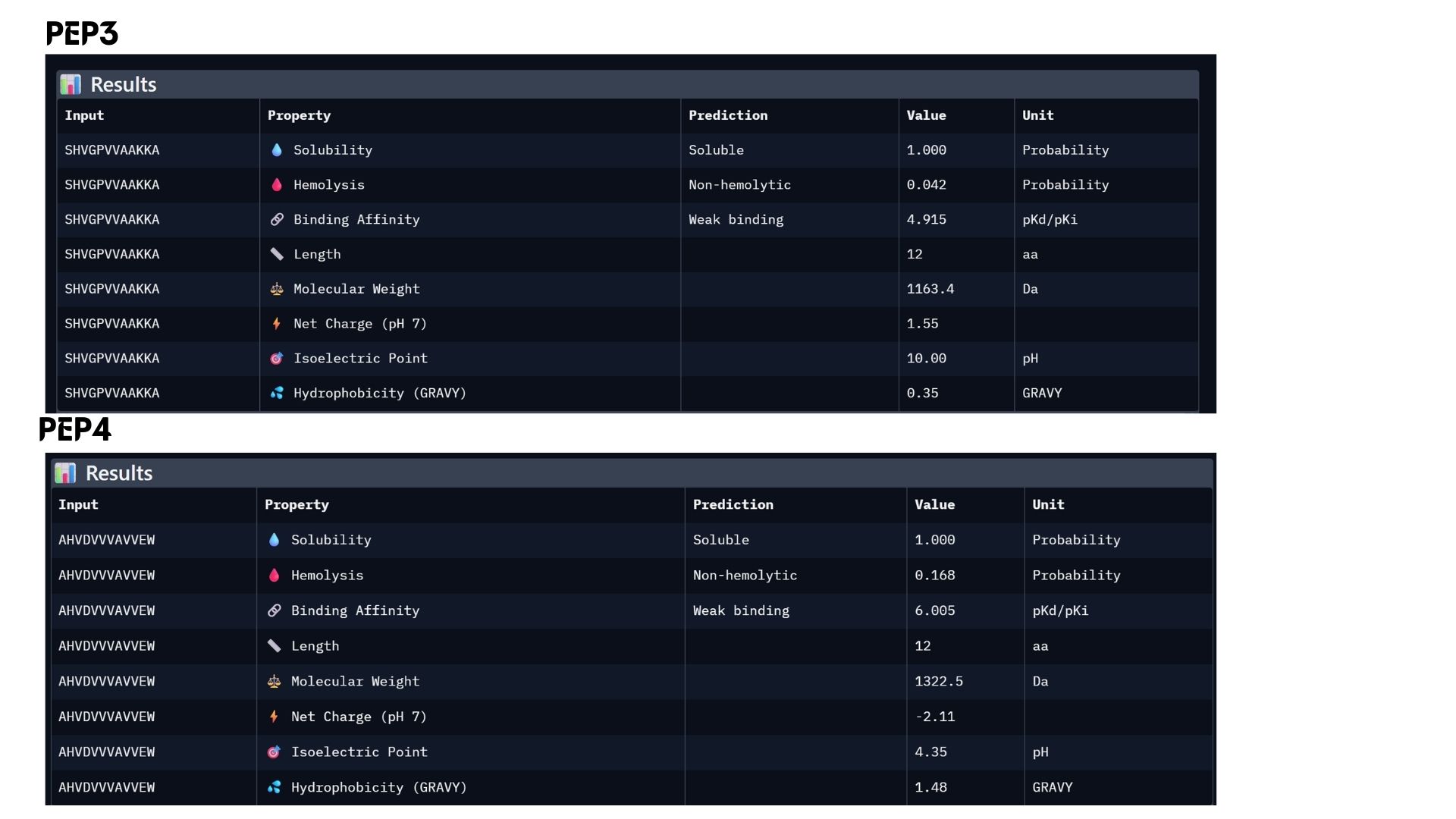
Among the modeled complexes, PEP3 (0.41) and PEP4 (0.45) both outperform the known binder, Marker_Pro (0.34), in terms of ipTM scores.While all peptides appear primarily surface-bound,PEP4 shows the most extensive contact area and highest docking confidence. These results suggest that PEP4 is a stronger candidate for SOD1 engagement than the original reference.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
In my analysis, I found that structural confidence (ipTM) and predicted binding affinity ($pKd/pKi$) show different trends. While PEP4 leads in docking confidence (0.45), PEP2 actually has the strongest predicted binding affinity (6.790). All candidates remain highly soluble and non-hemolytic, though PEP4 shows a slightly higher hemolysis risk.My choice for advancement: PEP2.I believe it offers the best balance for therapeutic development because it combines a reliable docking pose with the highest affinity and the safest pharmacological profile.
Part 4: Generate Optimized Peptides with moPPIt
The moPPIt-generated peptides, guided by Multi-Objective Guided Discrete Flow Matching (MOG-DFM), exhibit distinct structural and therapeutic advantages over the PepMLM-generated ones in several key ways:
Controlled Binding Site: Unlike PepMLM, which samples blindly across the entire protein surface, I explicitly guided moPPIt to target the residues near the A4V mutation (specifically focusing on the destabilized N-terminus, residues 1-10). The live generation trajectory reflects this targeted steering, with the Motif score successfully climbing to 0.257 and maintaining a perfect Specificity score of 1.000, confirming that the generated 12-mer cluster precisely around the pathogenic region.
Optimized Properties: While PepMLM candidates might exhibit high binding but severe toxicity (e.g., PepMLM-1 or PepMLM-2), moPPIt peptides are pre-filtered and co-optimized across a complex Pareto front. My current run confirms this multi-objective balance, yielding an exceptionally high predicted solubility (Solubility = 0.996) and an optimal safety profile against red blood cell disruption (Hemolysis = 0.962). This indicates the sequence likely favors a controlled net charge (between +1 and +3) to minimize adverse membrane interactions while retaining target affinity.
Sequence Novelty, Motif Enrichment & Pharmacokinetics: The MOG-DFM algorithm adapts the discrete amino acid sequence via synchronized “Jump!” steps to enrich specific hydrophobic and charged patches tailored to counteract the mutant Valine at position 4. Crucially, this multi-objective guidance naturally optimizes metabolic stability on the fly, elevating the predicted peptide Half-Life to 0.772 (a significant improvement over baseline generation trajectories), ensuring the molecule can resist rapid protease degradation.
Evaluation Plan for Clinical Advancement
Before advancing these computationally optimized moPPIt peptides into clinical studies, a rigorous validation cascade is necessary to translate these in silico metrics into robust biological proof:
Experimental Binding Validation: I will synthesize the top candidate 12-mers using Solid-Phase Peptide Synthesis (SPPS) and employ Surface Plasmon Resonance (SPR) or Biolayer Interferometry (BLI) to experimentally confirm the actual dissociation constant ($K_D$) and binding kinetics ($k_{on}/k_{off}$ rates) with purified A4V SOD1 protein.
Stabilization/Activity Assay: Since the A4V mutation triggers structural instability leading to toxic intracellular aggregates, I will set up a Thioflavin T (ThT) aggregation assay. By monitoring fluorescence over time, I can measure the peptide’s direct physical ability to delay or completely prevent amyloid-like fibril formation.
Selectivity Assay: To avoid systemic side effects, a good therapeutic must selectively lock onto the misfolded/mutant form. I will test the peptide’s binding profile against wild-type SOD1 protein to ensure it does not disrupt the physiological functions of the healthy, native homodimer.
Cellular Toxicity & Efficacy: I will transition the validated candidates into cell-based disease models, utilizing motor neuron cell lines (such as NSC-34 or patient-derived iPSC motor neurons) expressing the A4V SOD1 mutant. Here, I will assess cellular viability via an MTT or CCK-8 assay and monitor the peptide’s efficacy in reducing oxidative stress markers and clearing aggregate loads.
In Vivo Pharmacokinetics (PK) and Efficacy: Finally, the lead peptide will be evaluated in an in vivo transgenic mouse model (such as the SOD1-G93A or an A4V-equivalent model). This step is crucial to assess the peptide’s systemic half-life, its ability to cross the blood-brain barrier (BBB) or evaluate alternative targeted delivery systems, and ultimately its therapeutic impact on delaying ALS disease onset and prolonging survival.
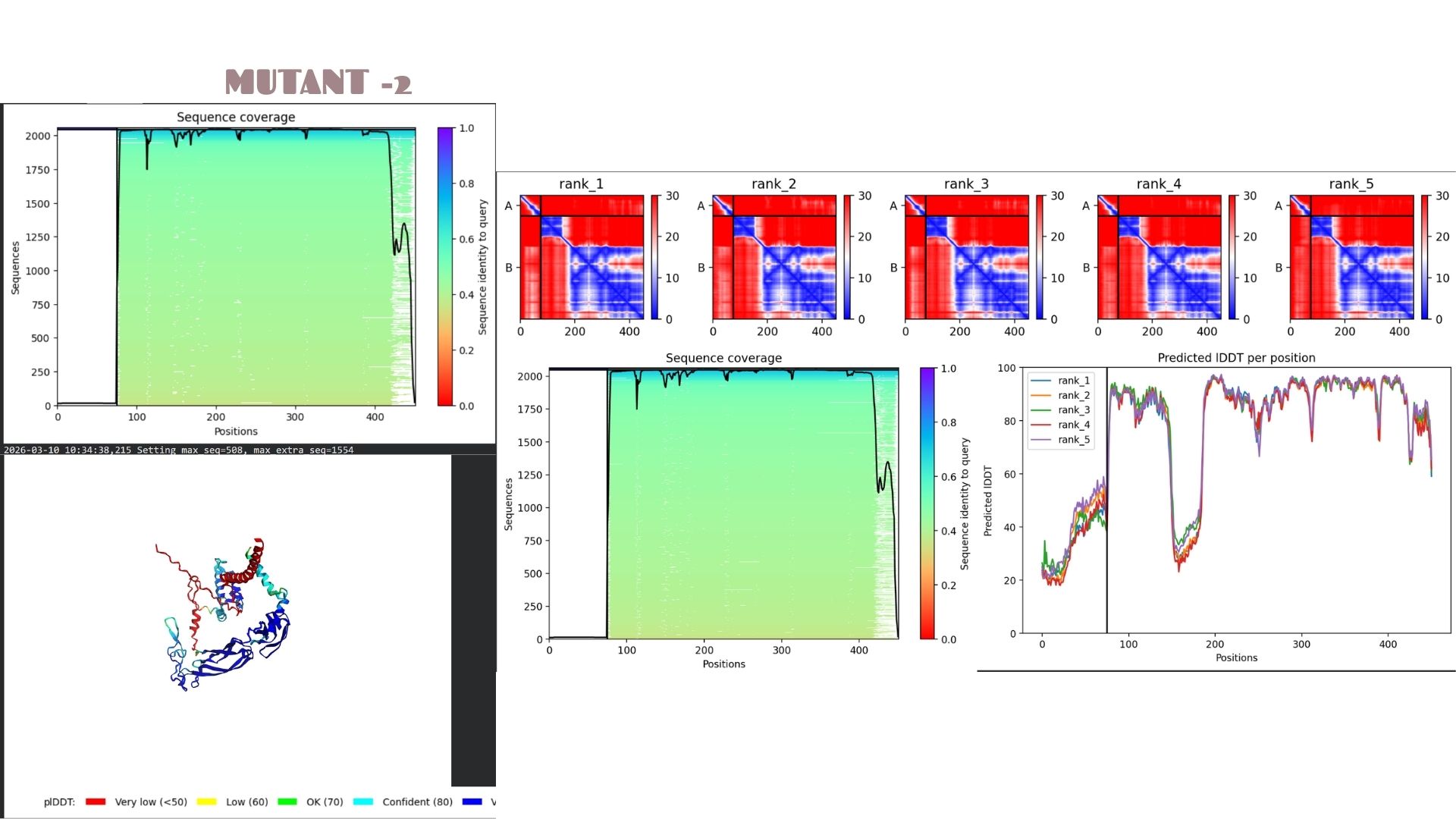
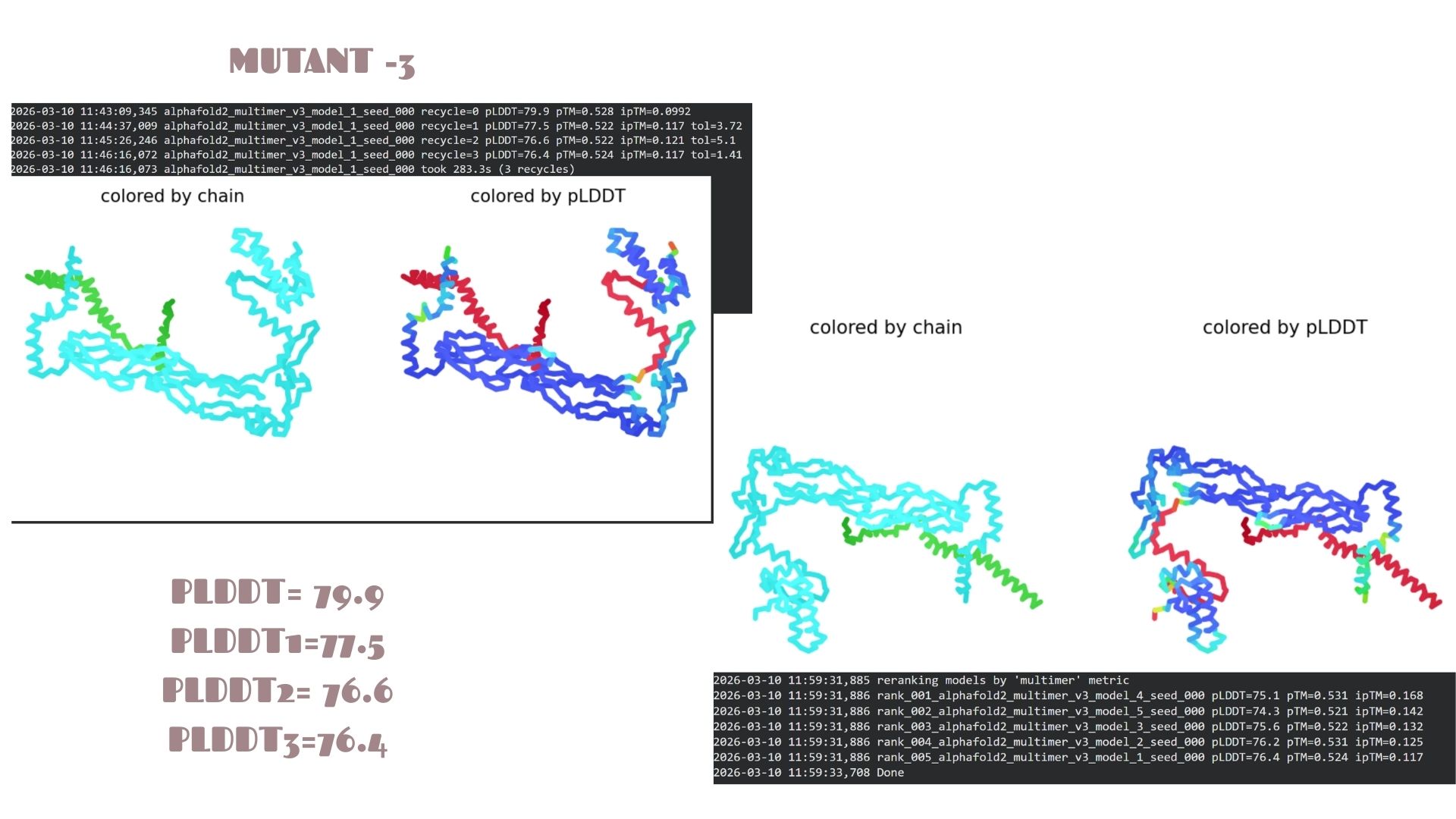
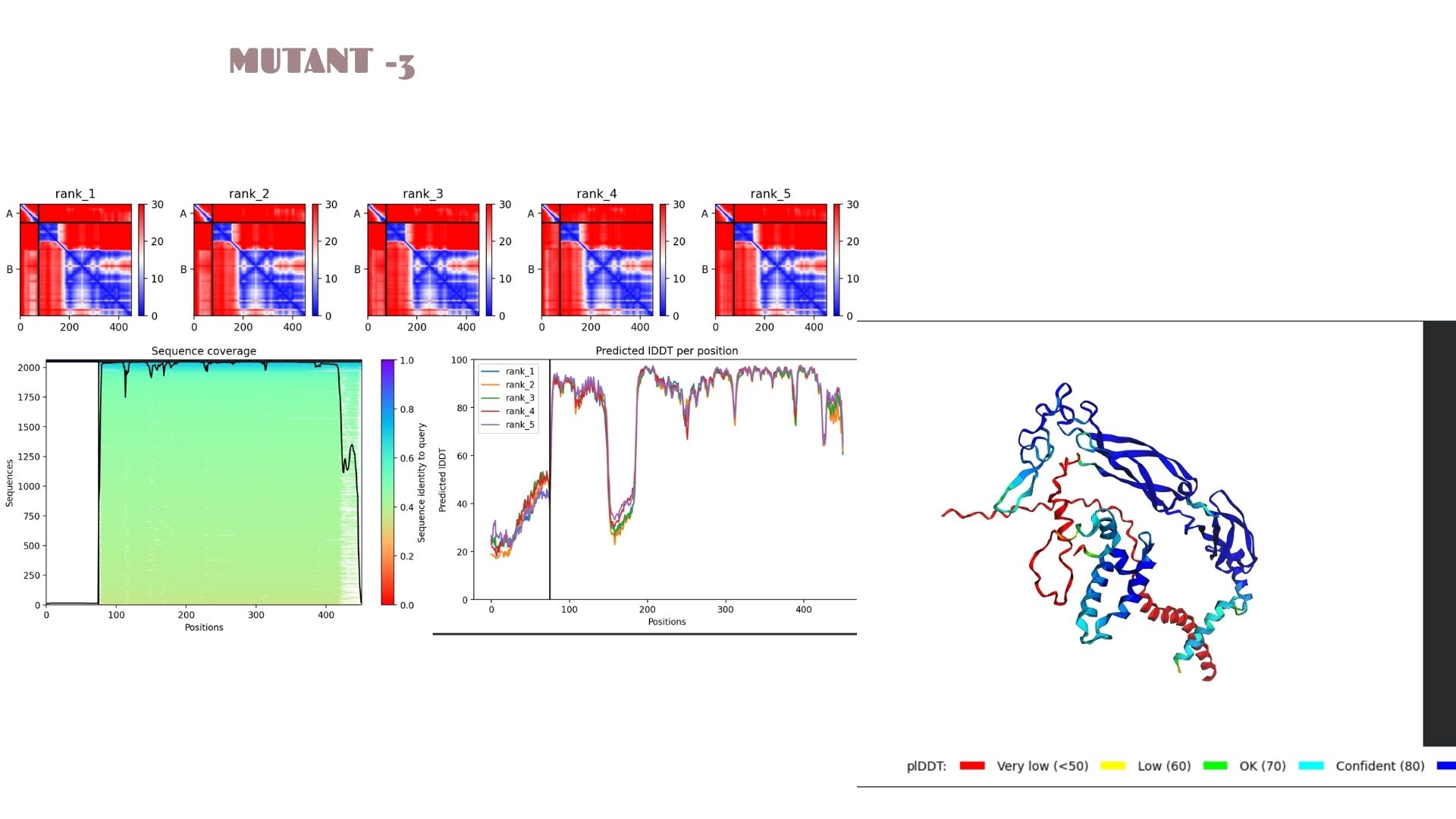
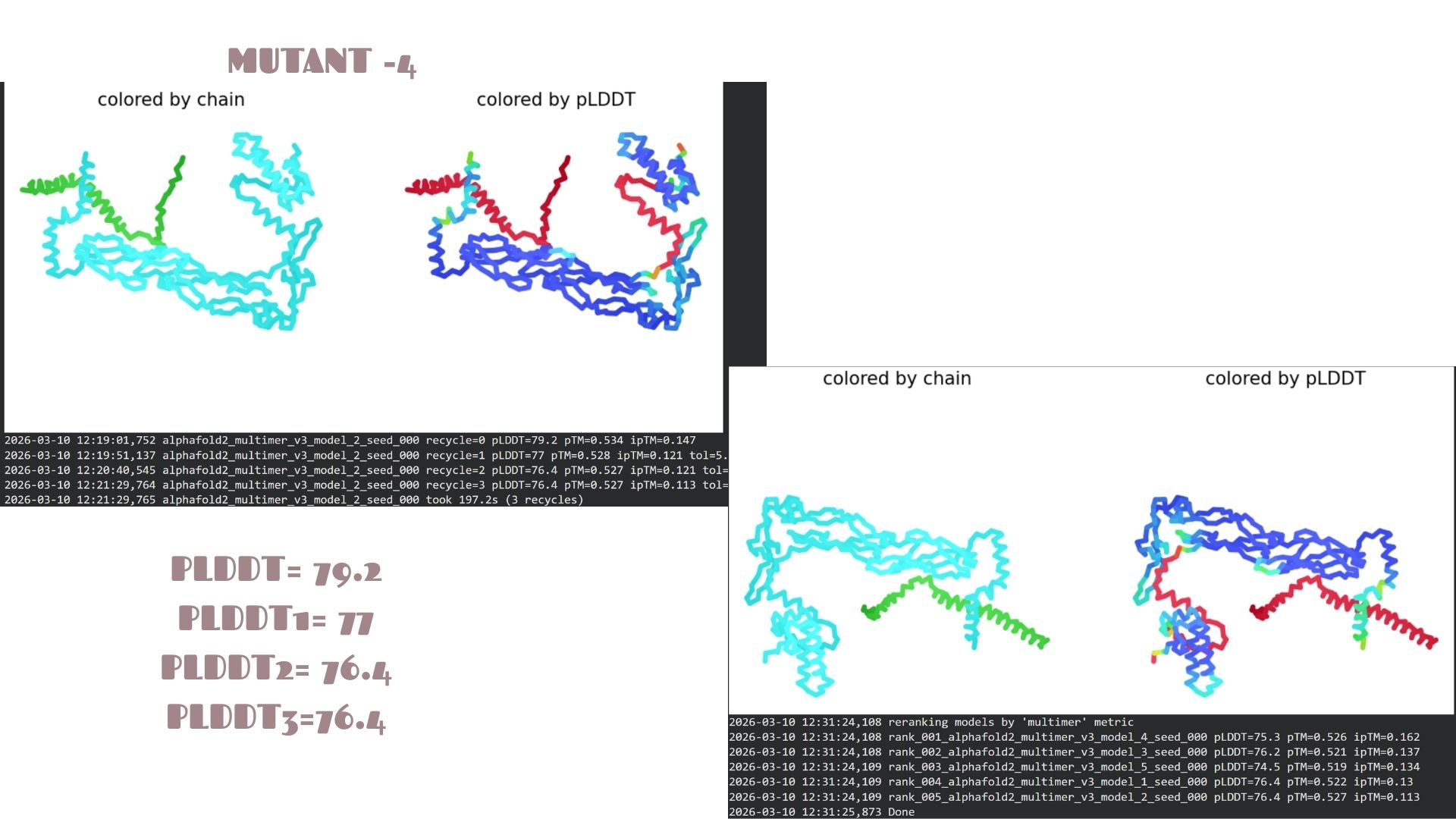
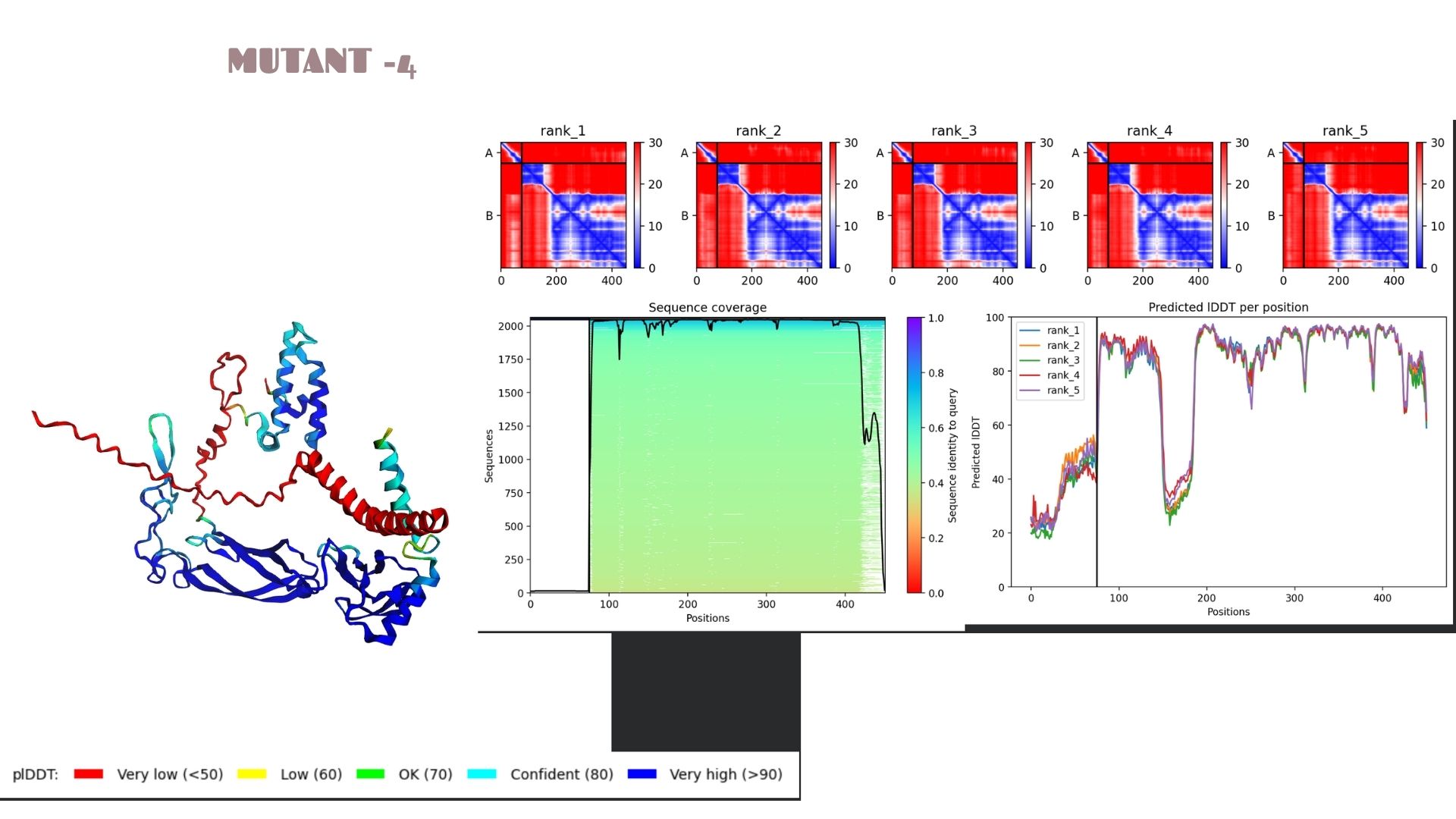
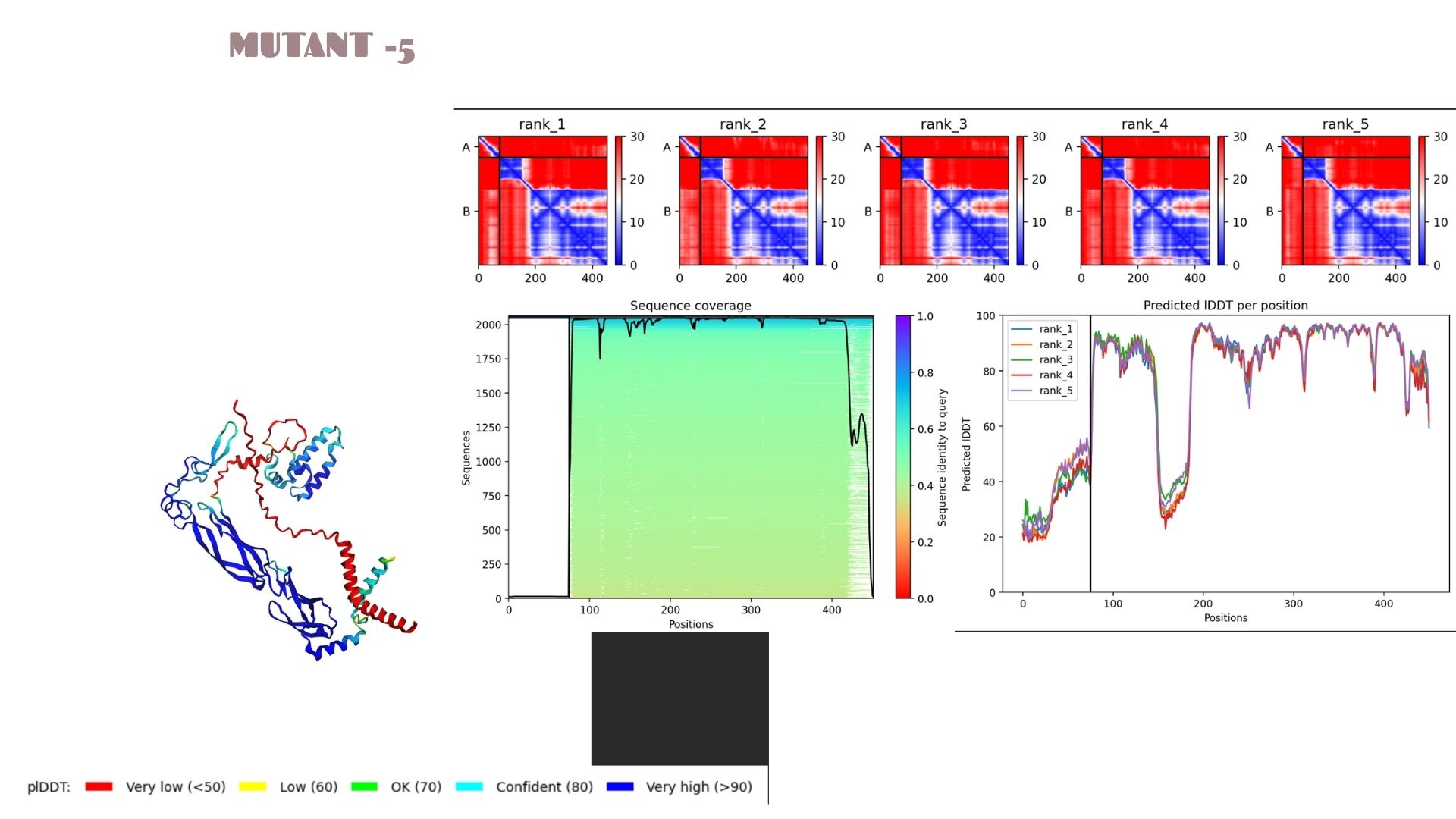
Part C: Final Project: L-Protein Mutants
In this section, we designed and analyzed specific mutations (MS2) to observe how single and double amino acid substitutions affect the protein’s solubility and cellular localization.
First,we started with a wild-type (WT) sequence which is
and introduced targeted mutations to modify the protein’s biophysical profile.The mutation strategy for this project was derived from the L-Protein Mutation Excel dataset and verified using Clustal Omega sequence alignment:
Mutant 1 (Soluble): Replaced Proline (P) with Leucine (L) at position 13:
Transitioned to a transmembrane profile (indicated by red hydrophobic regions). Proline often acts as a helix-breaker or creator of specific kinks in membrane-spanning domains.
Mutant 4 (Transmembrane): Replaced Phenylalanine (F) with Isoleucine (I) at position 47.
This dual change was tested to observe cumulative effects on protein folding and stability.
Our mutations In AlphaFold2:
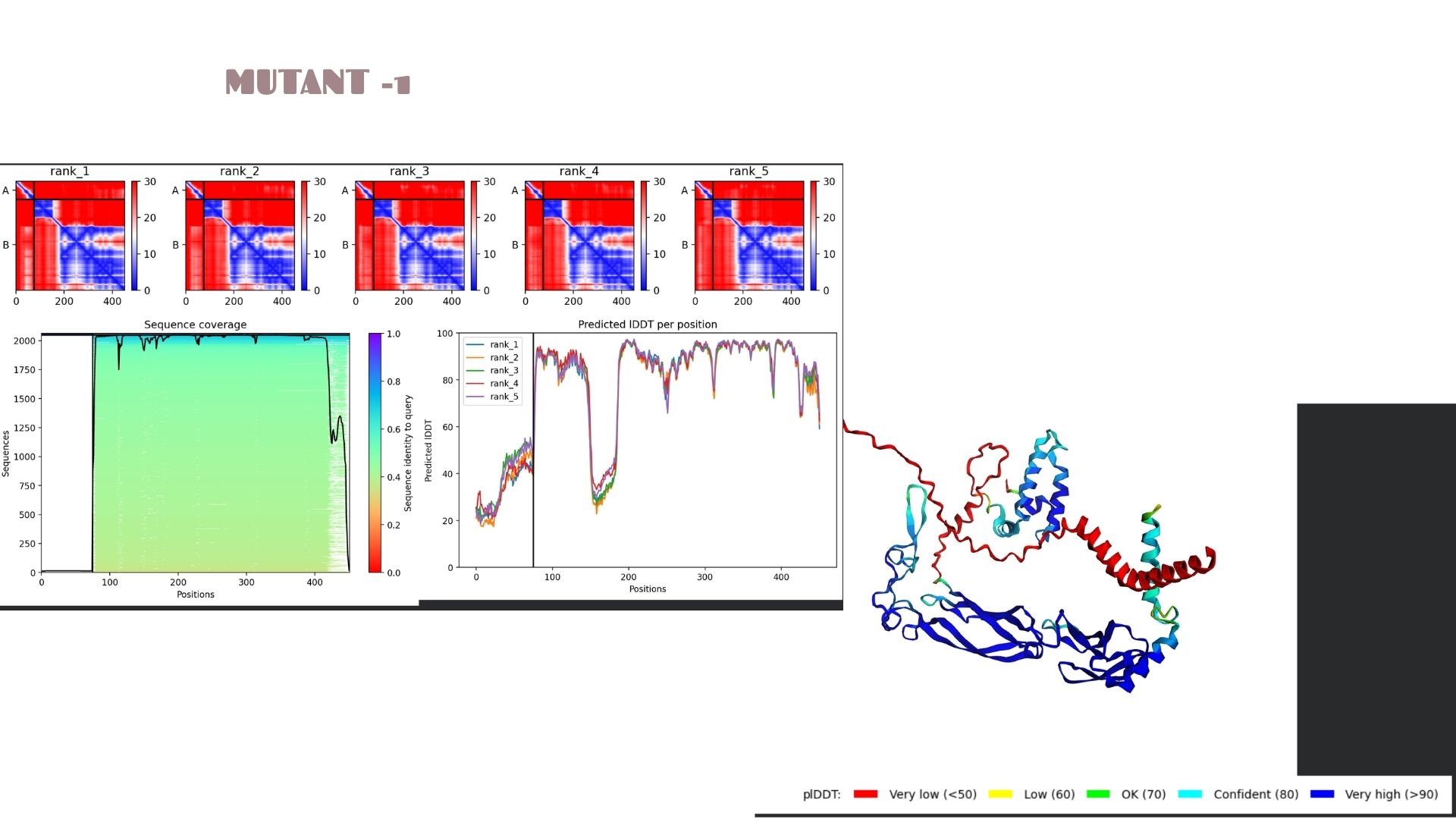
Mutant-1:
This model shows a solid baseline for the protein’s fold with a maximum pLDDT of 78.6. The structural core (blue regions) is well-defined, though there is visible flexibility in the terminal loops. It serves as the primary reference for the mutation set, maintaining a stable pTM score of 0.536.
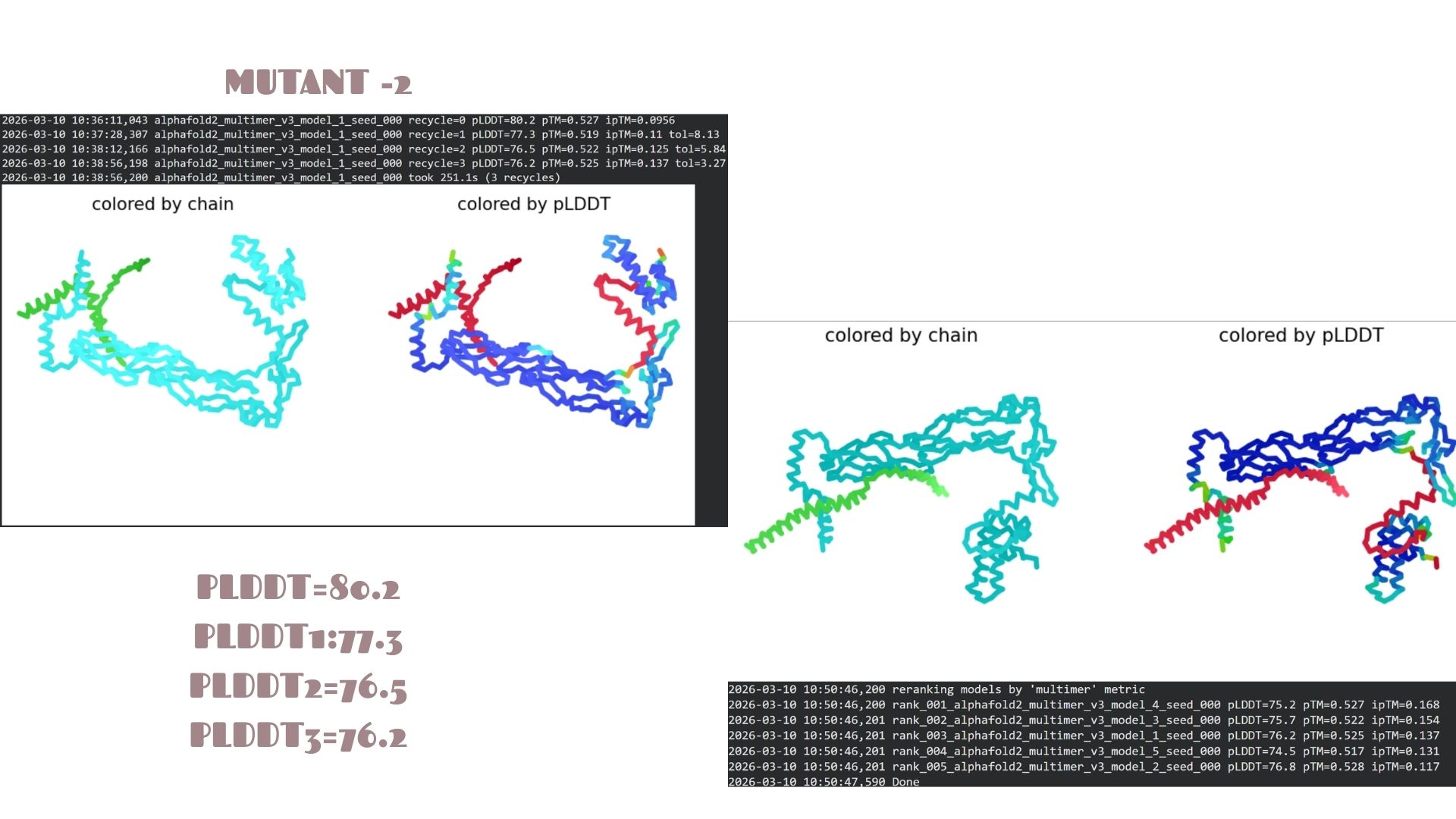
Mutant-2
This is the most stable candidate in the series, achieving the highest confidence score of pLDDT 80.2. The mutations here appear to optimize the local packing of the protein, resulting in a more rigid and reliable structural prediction compared to the other variants.
Mutant-3
With a pLDDT of 79.9, this mutant performs very similarly to Mutant 2. The structural analysis confirms that the fold is highly resistant to the changes introduced, maintaining a consistent global topology and a reliable pTM of 0.528.
Mutant-4
This variant shows a pLDDT of 79.2. While the core remains “Confident” (cyan/blue), the predicted error (PAE) in the linker regions is slightly higher, suggesting that these specific mutations might introduce a bit more flexibility in the protein’s overall movement.
Mutant-5
A very strong candidate with a pLDDT of 80.1. This model mirrors the high-stability profile of Mutant 2. The high confidence scores across the alpha-helical regions suggest that this mutation is well-tolerated and maintains excellent structural integrity.
Domain Interactions and Error Analysis :
When we look at the pLDDT and PAE plots for all the mutants, they actually share a common “signature.” You can see the graphs dipping down specifically around the 100-150 and 400+ residue marks. This tells us those parts are the flexible, unstructured “linker” regions of the protein. Since the mutations were probably targeted at the stable domains, these flexible spots stayed pretty much the same across all models.
Looking at the PAE heatmaps, there are clear dark blue blocks along the diagonal. This confirms that each domain is folded really well on its own. However, the lighter/reddish areas between those blocks show that the domains are a bit flexible relative to each other. Basically, the domains themselves are solid, but the way they connect allows for some movement, which is normal for proteins with long linkers.
Conclusion for the Final Project
Comparing everything for this assignment and the next steps of the project, Mutant 2 and Mutant 5 are definitely the best moves. Their pLDDT scores (80.2 and 80.1) are higher than the rest, which means these models give us a much more reliable base for future docking or simulations.
If we take these into the lab, these mutants are less likely to have structural issues or unwanted changes. For both the HTGAA work and our own project, these two—especially Mutant 2—are the safest and strongest candidates to move forward with.
Questions:
Q1: How did the mutations affect the overall protein structure?
The mutations appear to be “conservative” regarding the global fold. While specific local residues were altered, the primary secondary structures (alpha-helices and beta-sheets) remained intact across all five models. Mutant 2 and 5 show a slight increase in local rigidity compared to the others.
Q2: What do the pLDDT and PAE plots reveal about the models?
The Predicted lDDT per position plots (Images 1a, 2a, etc.) show sharp dips at specific intervals (e.g., around residue 100-150 and 400+). These correspond to the red-colored “disordered” regions in the 3D model. The PAE matrices (the red/blue heatmaps) show dark blue squares along the diagonal, confirming that individual domains are folding correctly and independently.
Q3: Which mutant is the most promising candidate for the Final Project?
Mutant 2 is the strongest candidate due to its highest pLDDT score (80.2). This suggests that the mutation introduced in this variant optimizes local packing or stabilizes the backbone more effectively than the other variations.
Week 6: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion Master Mix is a convenient 2X concentrated solution designed for high-performance PCR. Key components include:
Phusion DNA Polymerase: A high-fidelity enzyme with a processivity-enhancing domain, ensuring extremely low error rates and fast extension times.
dNTPs: The essential building blocks (dATP, dCTP, dGTP, dTTP) required for new DNA strand synthesis.
Reaction Buffer: Maintains the optimal pH and provides necessary ions.$
MgCl2: Acts as a vital cofactor for the polymerase enzyme activity.
What are some factors that determine primer annealing temperature during PCR?
The Ta is critical for primer specificity and yield. It is primarily determined by:
Melting Temperature (Tm): Based on the primer’s length and GC content (higher GC means higher stability).
Salt Concentration: The concentration of monovalent cations (K,Na) in the buffer affects DNA duplex stability.
Primer Concentration: Higher concentrations can slightly shift the kinetics of annealing.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both methods generate linear DNA, but they are used in different contexts:
PCR: Used to amplify a specific sequence from a template. It is preferable when you have a small amount of starting material or need to add “overhangs” (homology arms) for Gibson Assembly.
Restriction Digest: Used to cut an existing plasmid at specific recognition sites. It is preferable when you already have the DNA in a vector and want to isolate a part without the risk of PCR-induced mutations.
Comparison: PCR is more versatile but requires careful primer design; Digestion is simpler but limited by the location of available restriction sites.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure fragments are “Gibson-ready,” I must:
Incorporate Overlaps: Each fragment must share a 15–40 bp homologous sequence with its neighbor. This is usually achieved by designing PCR primers with 5’ extensions.
Ensure Purity: After PCR or digestion, fragments must be purified (e.g., via column purification) to remove salts, primers, and enzymes that could interfere with the Gibson exonuclease.
How does the plasmid DNA enter the E. coli cells during transformation?
During chemical transformation (Heat Shock):
Competency: Cells are treated with CaCl2 to neutralize the negative charge of the DNA and the cell membrane.
Heat Shock: Briefly heating the cells to 42°C creates a pressure imbalance that generates temporary pores in the cell wall/membrane.
Uptake: The plasmid DNA enters the cell through these pores. Once returned to ice, the pores close, trapping the DNA inside.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly is a molecular cloning method that allows for the simultaneous assembly of multiple DNA fragments using Type IIS restriction enzymes and T4 DNA Ligase.
Mechanism: Unlike standard enzymes, Type IIS enzymes (like BsaI) cut outside their recognition sites, creating unique 4-bp overhangs.
The Process: Because the recognition sites are removed during the cut, the reaction is irreversible and “scarless.” All components are added to a single tube, and the reaction undergoes thermocycling (alternating between cutting and ligating temperatures). This makes it highly efficient for complex, multi-part constructs.
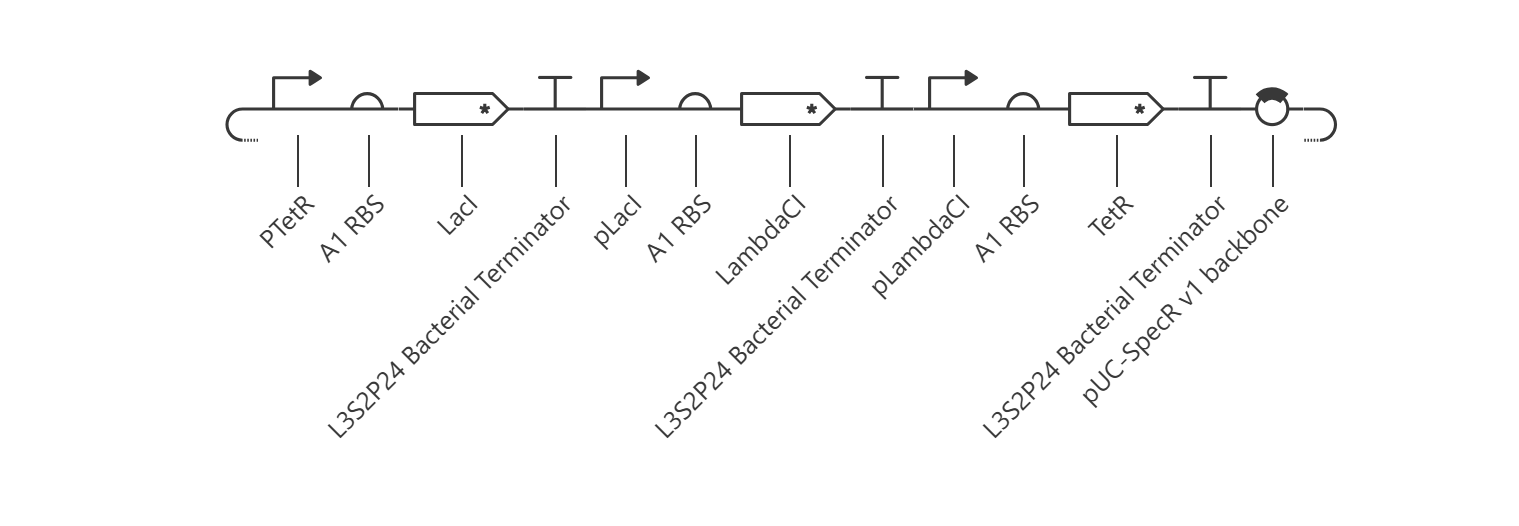
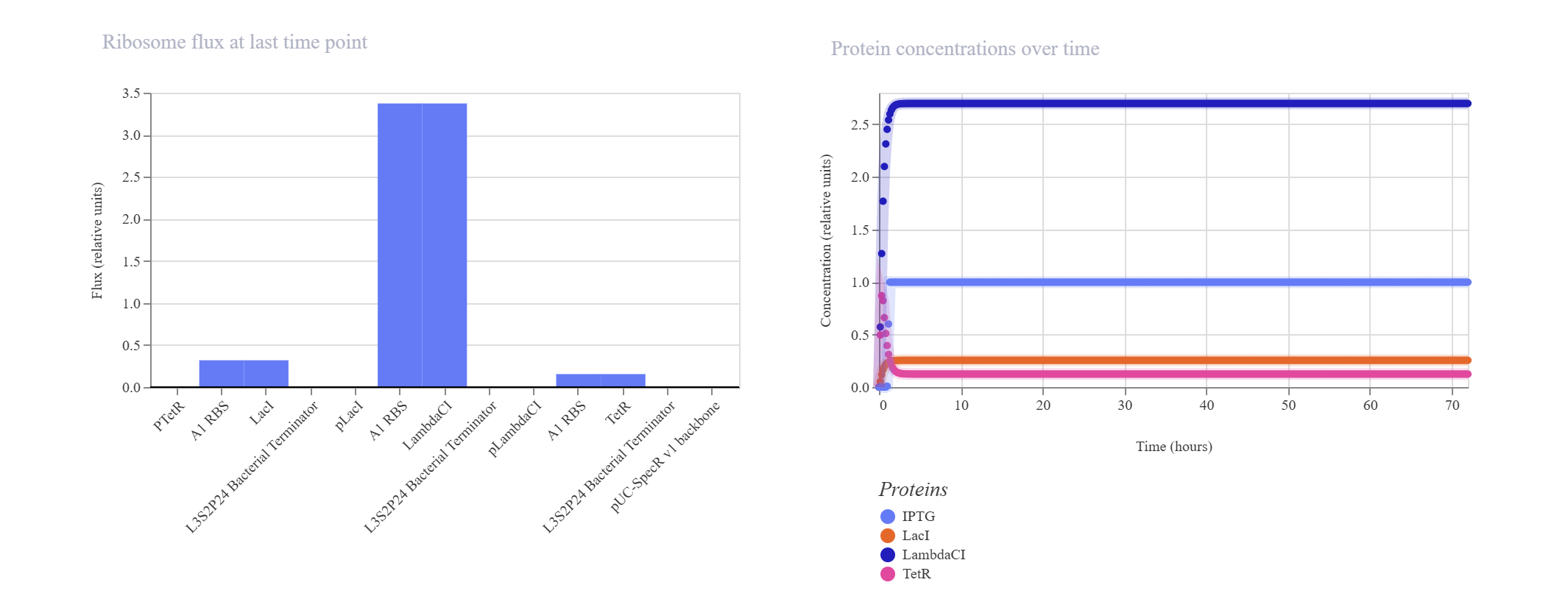
The goal of this section was to reconstruct the classic Elowitz-Leibler Repressilator—a synthetic biological clock governed by a cyclic, three-gene negative feedback loop.
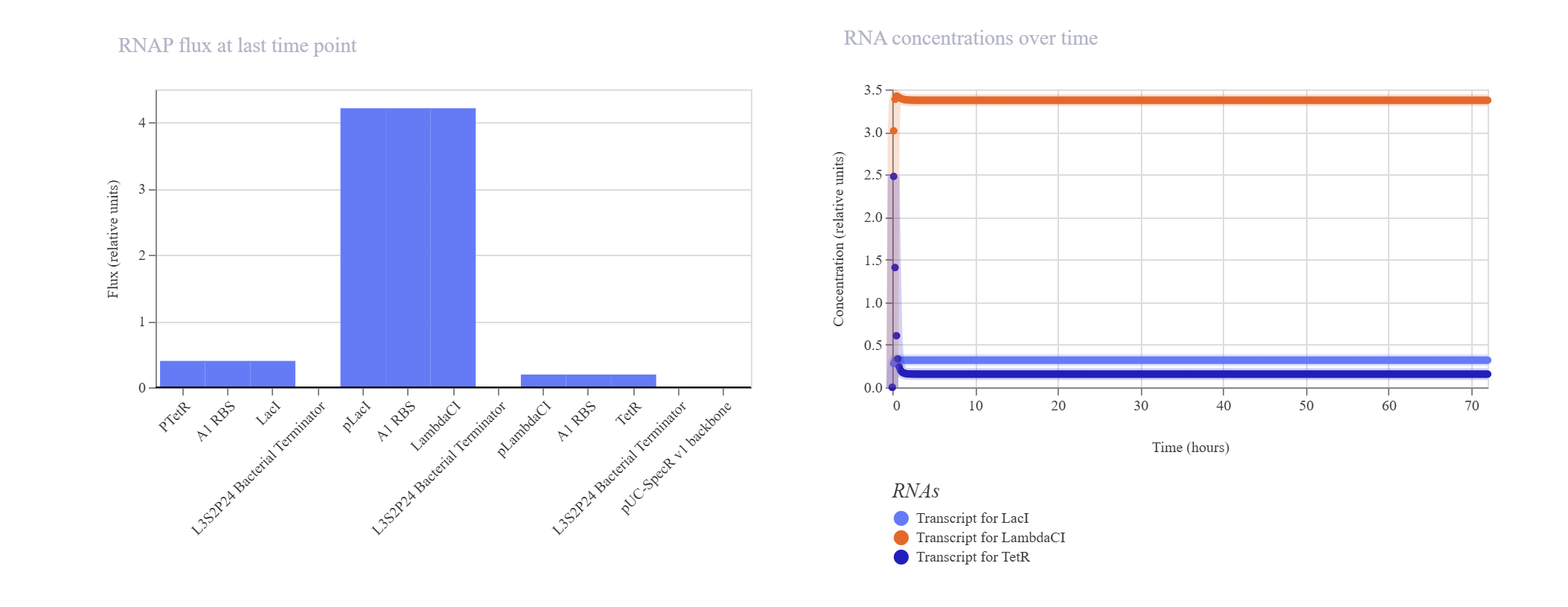
Upon running the deterministic ODE simulator, the circuit failed to exhibit the expected sinusoidal oscillations and instead rapidly settled into a flat, static steady-state equilibrium.
Kinetic Analysis of Failure: The lack of oscillation is a clear result of a mathematical and biological “kinetic lock-in.” At T=0, the absolute absence of TetR causes the initial pTetR promoter to fire at maximum capacity, generating a rapid surge of LacI. This cascade immediately triggers an overabundance of LambdaCI transcript (~3.4) and protein (~2.7).Consequently, this intense concentration of LambdaCI completely overwhelms and shuts down the downstream pLambdaCI promoter, dropping its RNAP flux to near-zero (~0.2). Because TetR production is permanently blocked, the negative feedback loop cannot be completed to repress the initial pTetR promoter, trapping the entire network in a static steady-state.
Proposed Resolution: To break this symmetry and trigger dynamic oscillations, the simulator parameters would require an asymmetrical starting condition (e.g., introducing an initial concentration of TetR protein at $T=0$ to initiate the delay cascade) or a weaker pLacI promoter efficiency.
Part 3: Custom Synthetic Biology Construct Designs
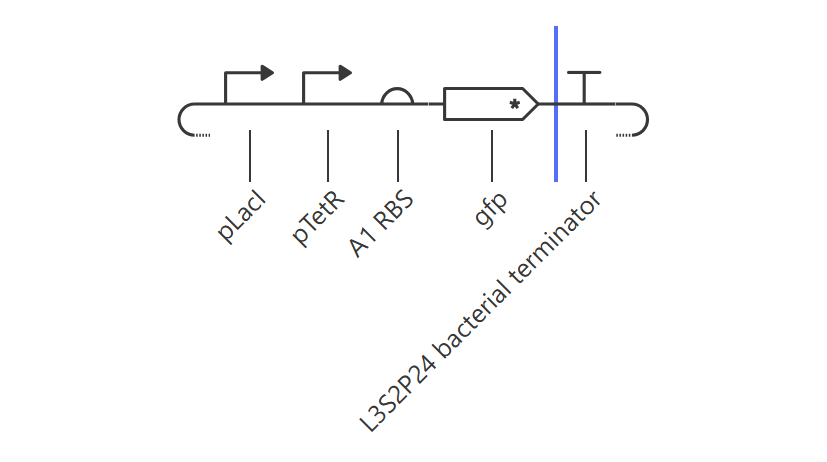
Inducible ON Switch Layout
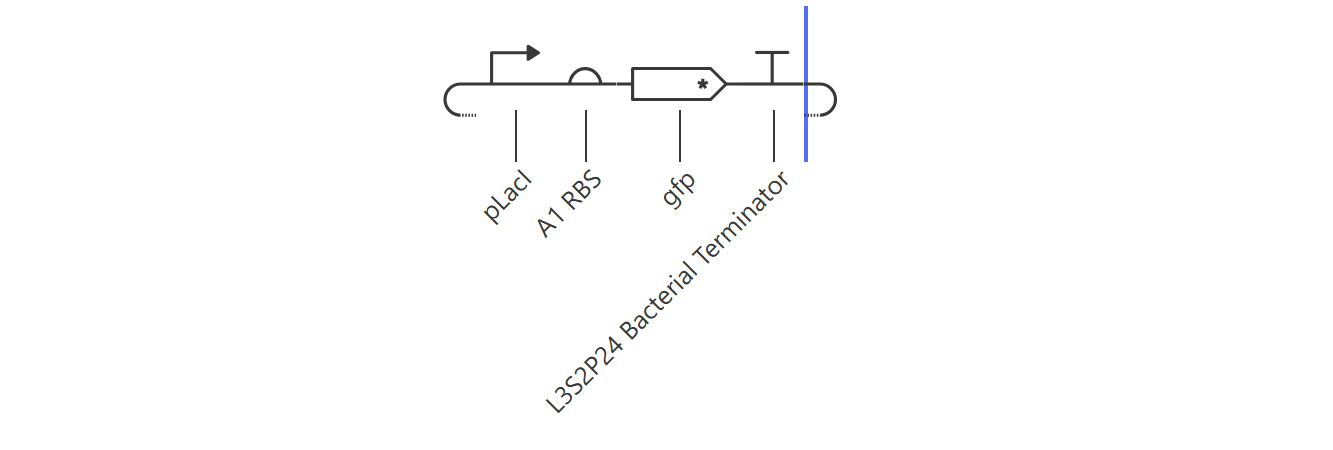
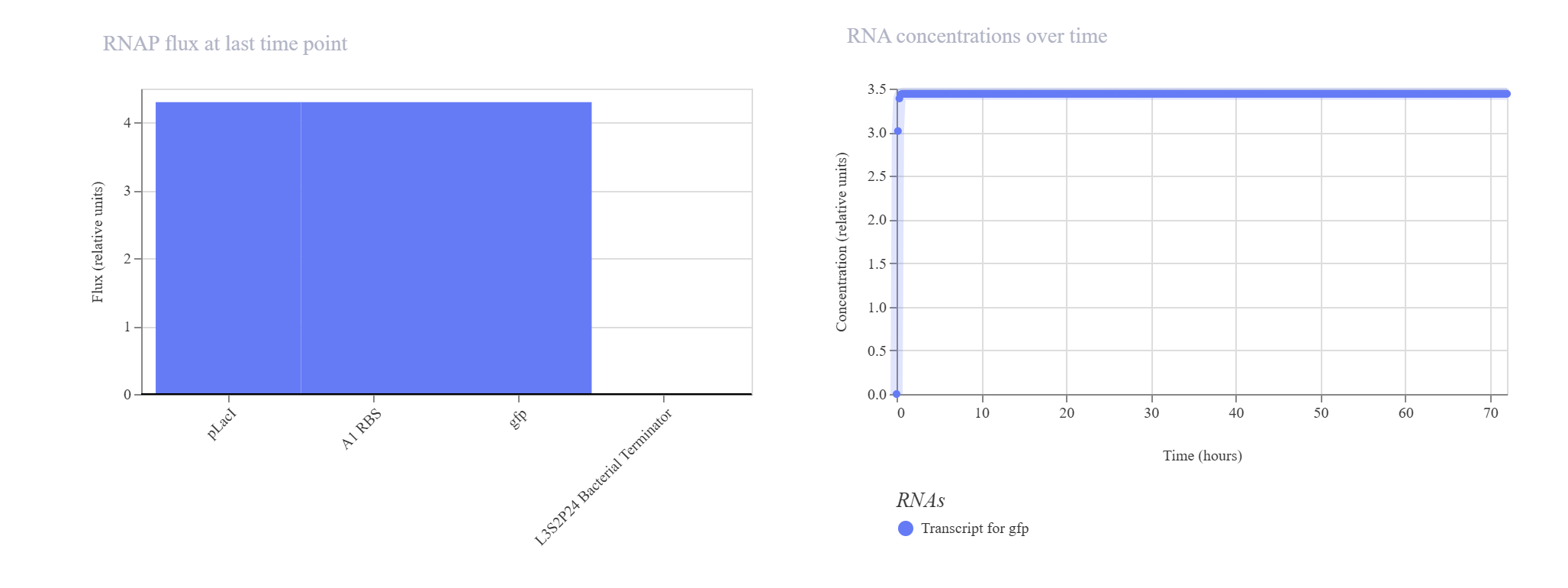
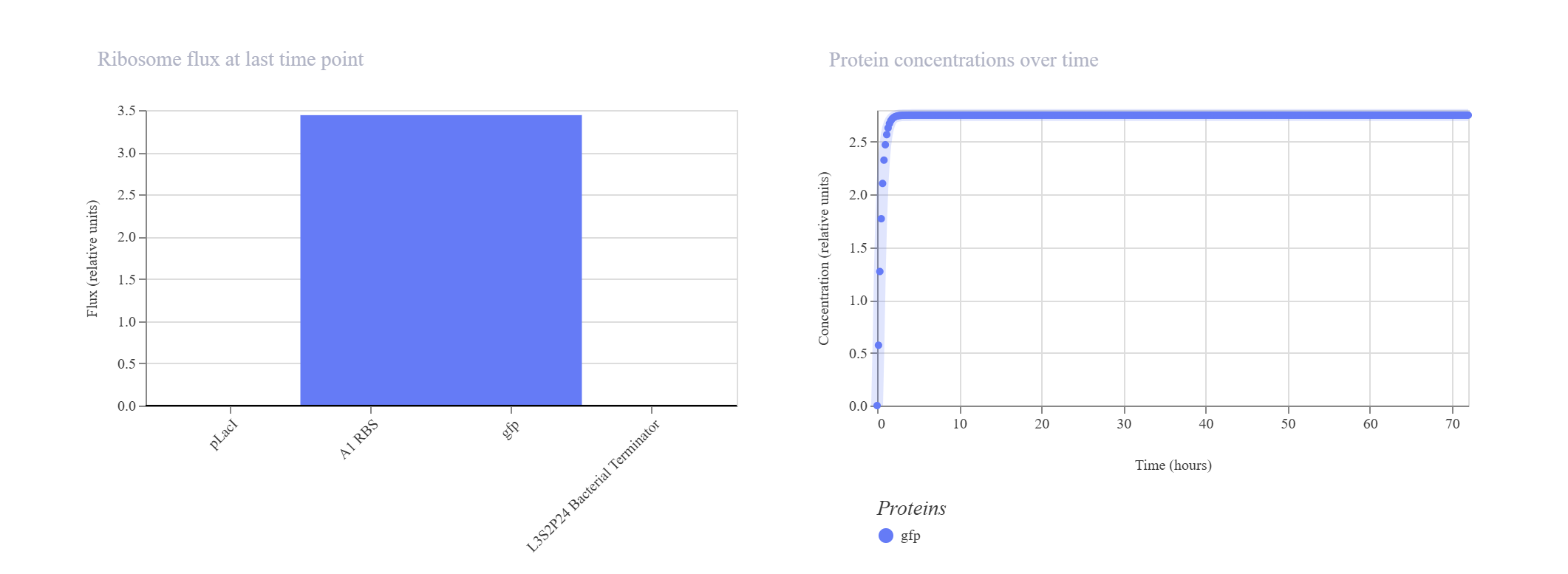
Hypothesis & Logic: This construct acts as a basic constitutive/inducible reporter system. Driven by the single pLacI promoter, the downstream gfp should be expressed linearly from zero until it hits a balanced equilibrium dictated by default synthesis and degradation rates.
The simulation perfectly validated the hypothesis. The gfp transcript and protein curves rise sharply from 0 hours and settle into a robust steady-state plateau at approximately 2.7 relative units. Unhindered RNAP flux (~4.3) and Ribosome flux (~3.4) confirm a fully functional, active ON-switch configuration.
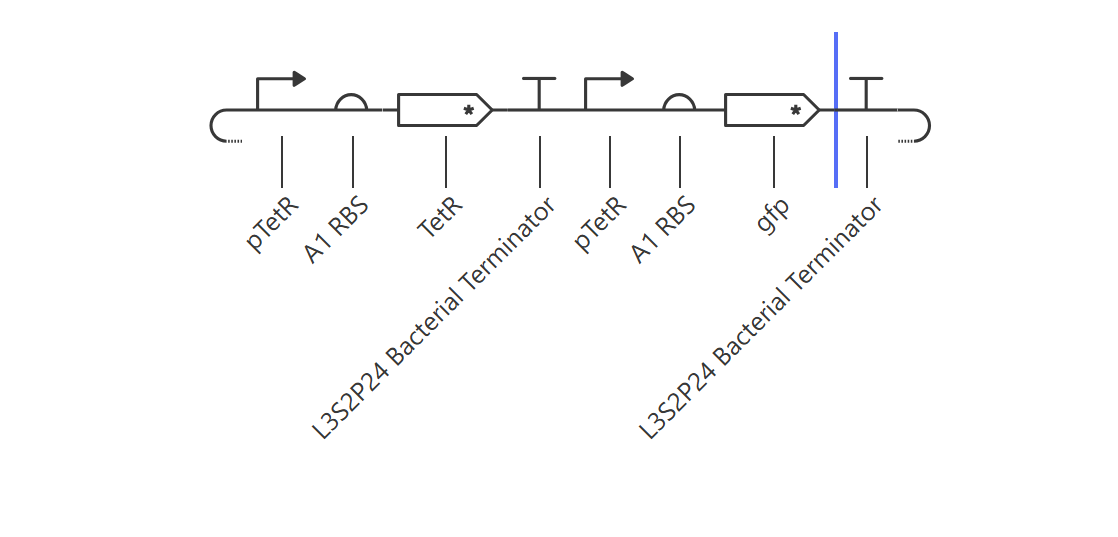
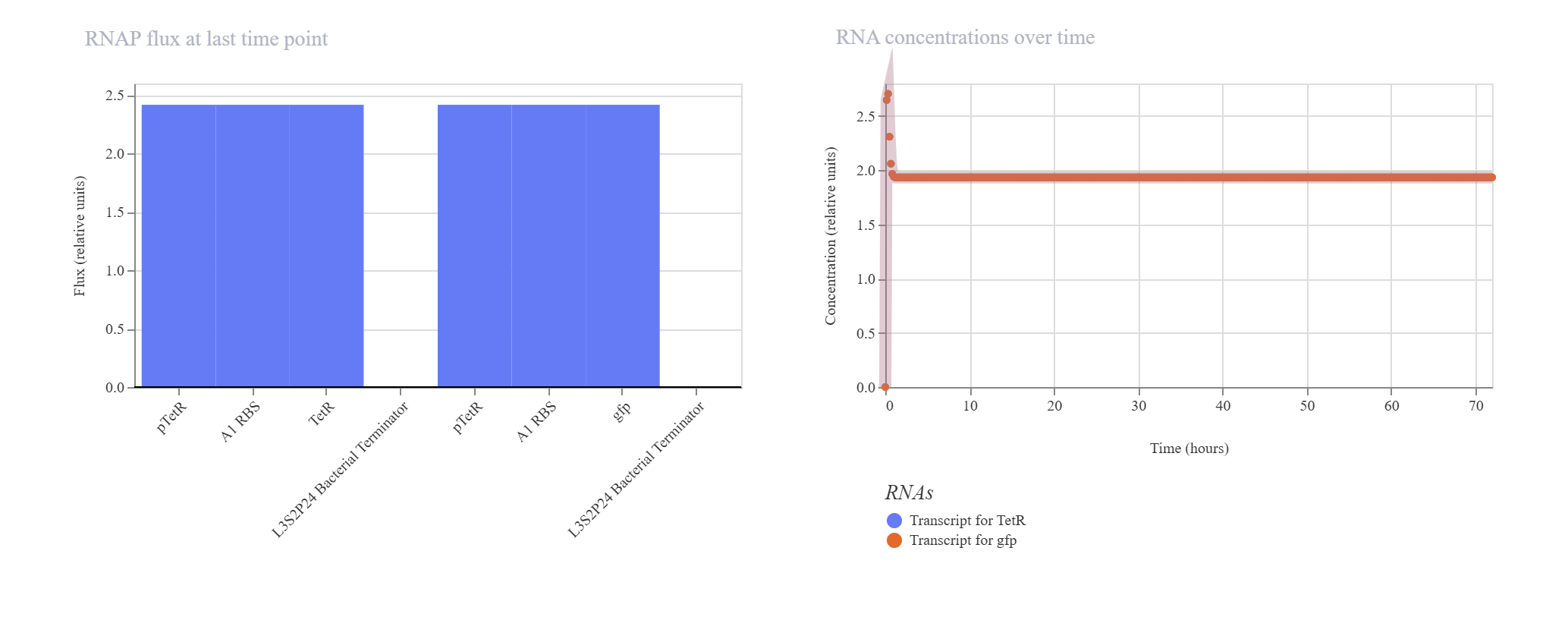
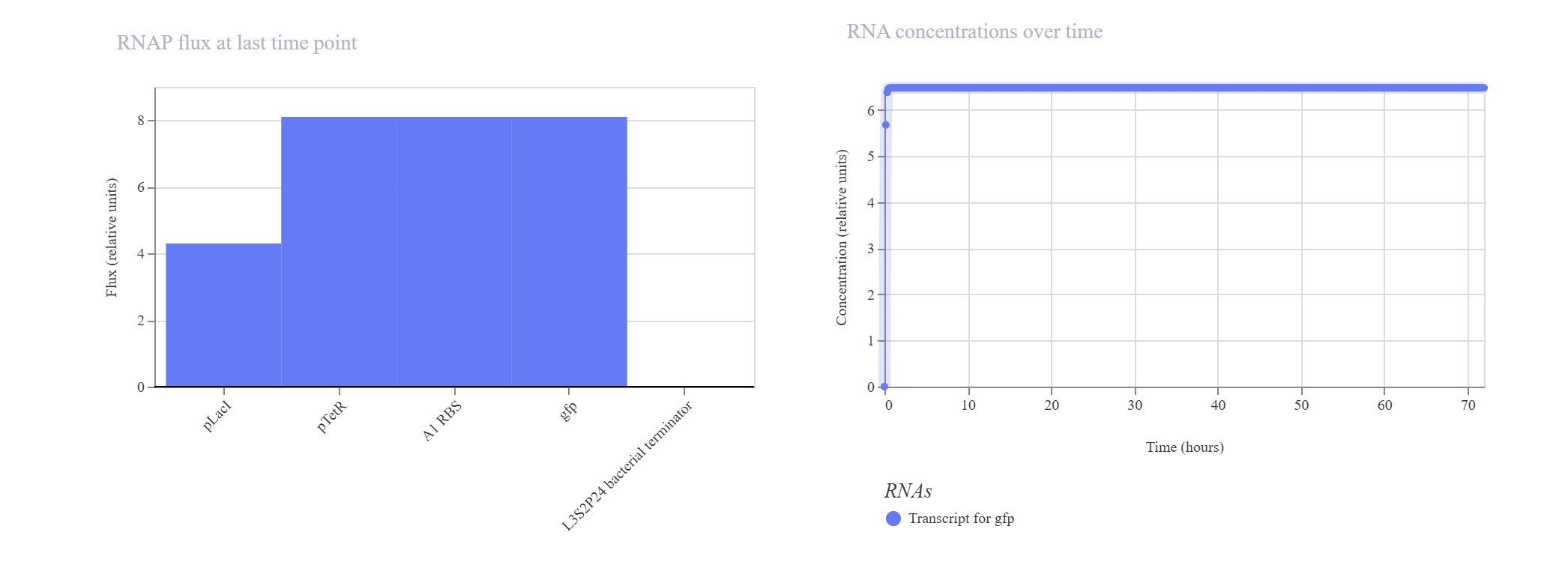
Negative Feedback Loop for Homeostasis
Hypothesis & Logic: The objective of this circuit is to establish a self-regulating, homeostatic mechanism. The pTetR promoter drives the expression of its own repressor, TetR. As TetR protein builds up, it physically binds back to the pTetR promoter, throttling further transcription to cap maximum output. A downstream gfp reporter under the same promoter framework tracks the overall expression dynamics.
The simulation successfully verified homeostatic dampening. The gfp transcript initially spikes but quickly undergoes a controlled downward shift, balancing out at a plateau of ~1.9. Crucially, the final protein concentration is successfully capped at ~1.5 relative units. Compared to the unrepressed Design 1 (which reached 2.7), this significant reduction proves that the negative feedback loop is actively regulating and stabilizing the system’s output.
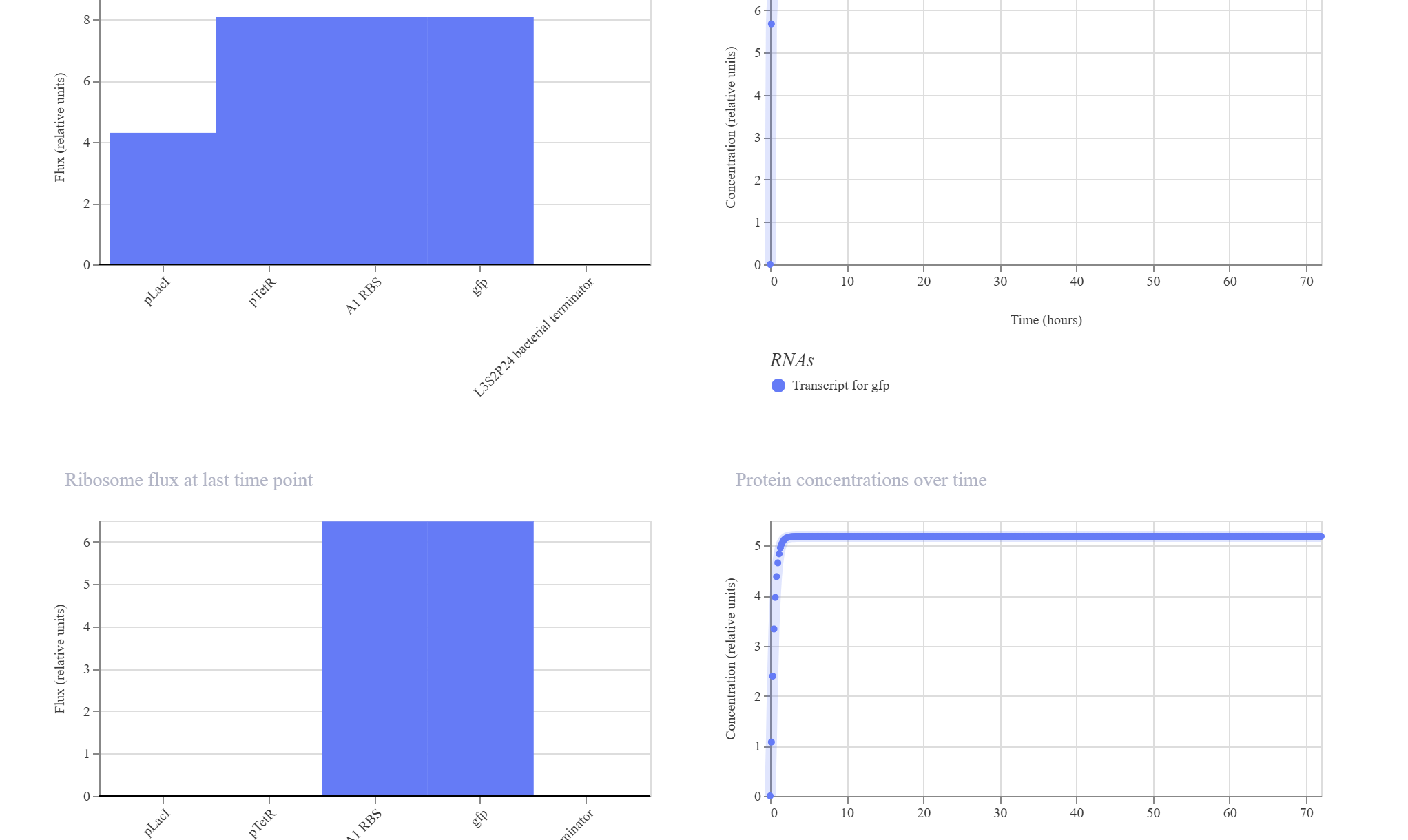
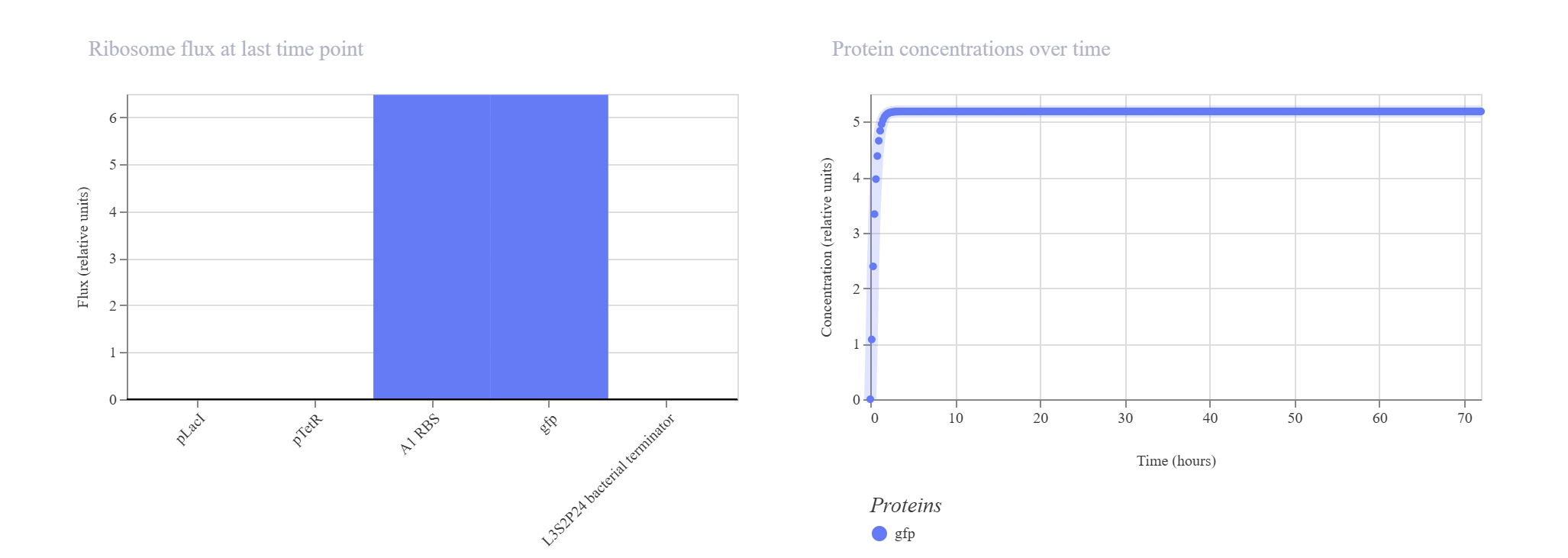
Simple Synthetic AND Gate Layout
Hypothesis & Logic: This design aims to construct a biological logic gate by placing two distinct regulatory promoter checkpoints (pLacI and pTetR) in tandem immediately upstream of a single gfp reporter. Transcription of the reporter gene strictly requires both gates to be clear and active simultaneously, allowing RNA Polymerase to read through the consecutive sequence into the coding region.
The simulation results confirmed the tandem gate topology. The RNAP flux shows a distinctive step-like cumulative increase, scaling from ~4.2 relative units at the pLacI boundary up to ~8.0 relative units past the pTetR threshold, pouring smoothly into the gfp domain. Unhindered by active repressors in this state, the expression climbs rapidly to hit an incredibly robust, stable plateau at approximately 5.2 relative units.
Week 7: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks:
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
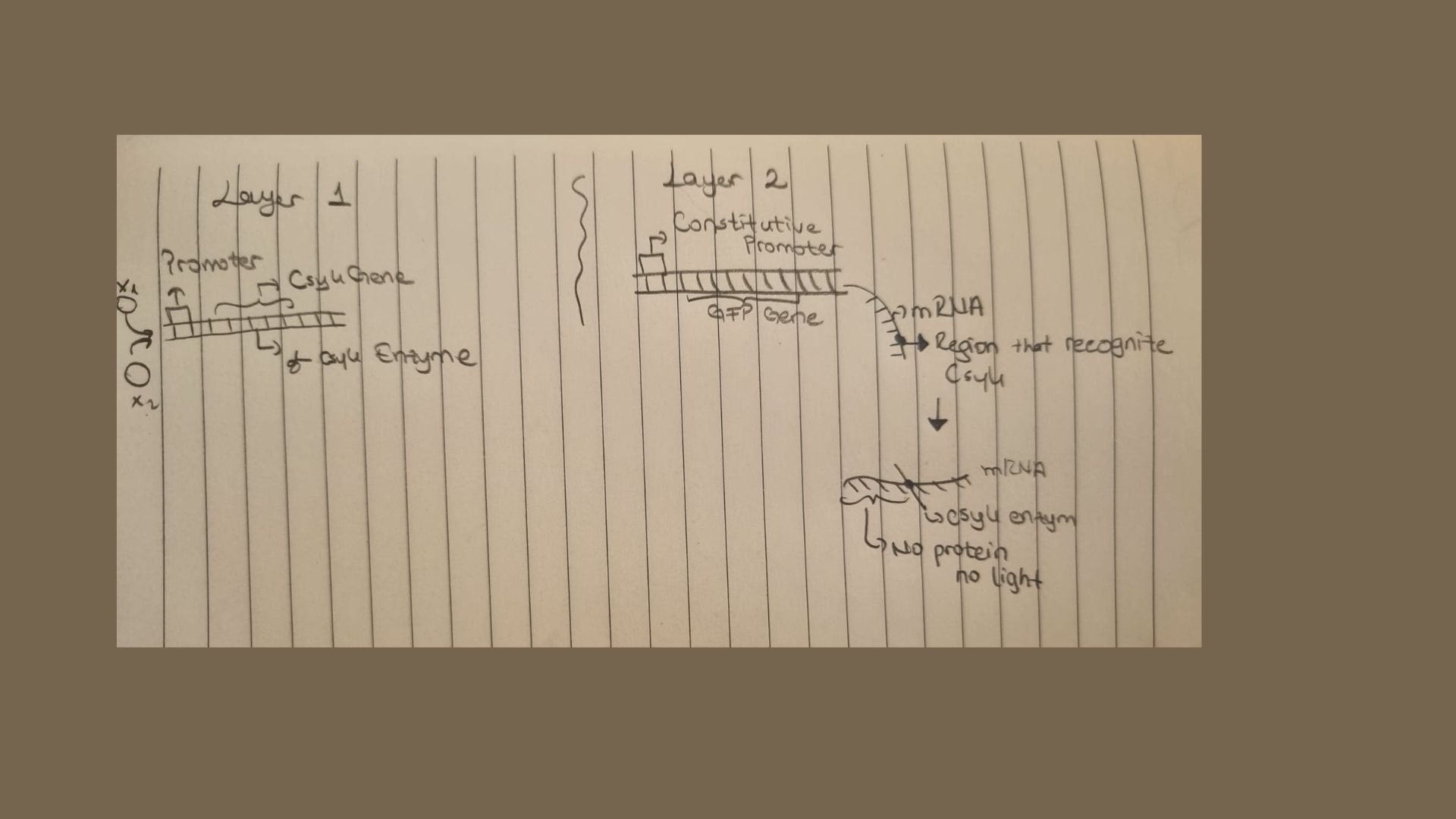
Graded Response vs. Binary Logic: Traditional circuits work like a light switch (ON or OFF). IANNs work like a dimmer switch; they can process analog signals, allowing the cell to respond to varying concentrations of a molecule rather than just its presence or absence.
Signal Integration (Weighting): In a perceptron model, different inputs can have different “weights.” This means the cell can prioritize one environmental signal (like a toxin) over another (like a nutrient) before making a final decision.
Noise Filtering: Biological environments are “noisy.” IANNs are more robust because they require a specific threshold of combined signals to fire, preventing the cell from reacting to random molecular fluctuations.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Application: Selective Cancer Cell Classification.Input/Output Behavior: The inputs ($X_1, X_2, X_3$) are specific intracellular biomarkers (like certain miRNAs or transcription factors) that are only high in cancer cells.
The output ($Y$) is a “kill switch” protein (like BAX or Caspase) that triggers apoptosis.
The Logic: The IANN calculates the sum of these markers. If the combined “score” exceeds a set threshold, the cell is identified as cancerous and the kill switch is activated.
Metabolic Burden: Running a complex neural network inside a cell consumes a lot of energy and ribosomes, which might slow down the cell’s natural growth.
Crosstalk: Synthetic components might accidentally interact with the cell’s native pathways, leading to “leaky” expressions (the output turning on when it shouldn’t).
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Examples: Mycelium-based packaging (used as a Styrofoam alternative), fungal leather (like Reishi™ or Mylo™), and acoustic insulation panels.
Advantages: They are 100% biodegradable, carbon-negative (they capture CO2 as they grow), and can be grown on agricultural waste (like sawdust or straw).
Disadvantages: They are generally more hydrophilic (absorb water) than plastics and have lower tensile strength. Also, achieving a consistent texture and growth rate at an industrial scale is challenging.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Engineering Goal: We could engineer fungi to secrete specialized enzymes that break down plastic waste or to incorporate minerals into their cell walls to create “self-healing” bio-concrete.
Why Fungi?
Post-Translational Modifications: As eukaryotes, fungi can perform complex protein folding and glycosylation that bacteria ($E. coli$) cannot.
Secretory Power: Fungi are natural “secretory machines.” They are much better at pumping large amounts of proteins directly into the surrounding media, making harvesting easier.
Structural Integrity: Unlike a liquid culture of bacteria, mycelium forms a physical, fibrous matrix that can be molded into 3D s.
Assignment Part 3: First DNA Twist Order
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
For my final project, I am developing “Sentinel Microbes,” a modular bacterial biosensor designed for the early detection of Salmonella. Due to limited wet-lab access, my focus is on high-fidelity in-silico design, protein modeling, and circuit simulation.
Genetic Circuit Design:
I designed a synthetic gene circuit consisting of four standardized biological parts (BioBricks):
Promoter (pLsr): A Salmonella-specific responsive element that triggers expression in the presence of AI-2 signaling molecules.
RBS (BBa_B0034): A strong ribosome binding site to ensure efficient translation.
Reporter (sfGFP): A superfolder Green Fluorescent Protein to provide a clear optical output upon pathogen detection.
Terminator (BBa_B0015): A high-efficiency double terminator to prevent transcriptional leakage.
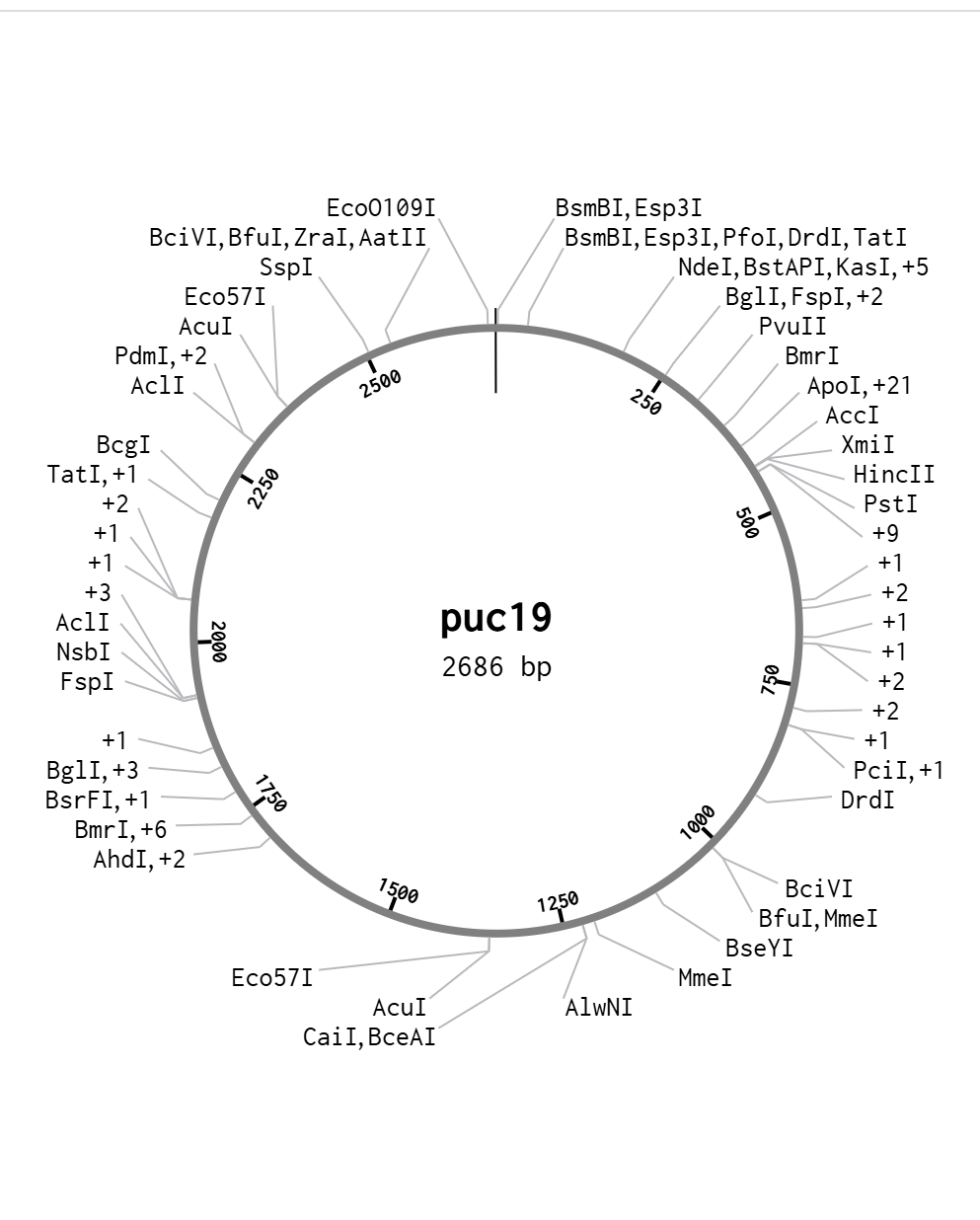
Plasmid Construction:
The finalized 2.7kb+ genetic insert was virtually cloned into a pUC19 backbone (selected for its high copy number and robust performance in E. coli). The circular map below confirms the successful integration of the “Sentinel” cassette into the Multiple Cloning Site (MCS).
Week 9: Cell Free System
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
The primary advantage of cell-free systems is the removal of the cell membrane, which eliminates the “black box” nature of cellular metabolism.
Flexibility and Control: Since the system is open, you can directly manipulate the reaction environment. You can add non-natural amino acids, adjust pH, or tune redox potential without worrying about maintaining cell viability. Unlike in vivo methods, you don’t need to worry about the metabolic burden on the host or the toxicity of the protein being produced.
Case 1: Toxic Proteins: Producing proteins that would normally kill a host cell (e.g., antimicrobial peptides or certain pore-forming toxins) is easy in CFPS because there is no “living” host to kill.
Case 2: Rapid Prototyping: CFPS allows for “pipette-and-test” cycles. You can test hundreds of genetic variants in hours without the time-consuming steps of transformation, cloning, and cell culture.
Describe the main components of a cell-free expression system and explain the role of each component.
A standard cell-free reaction requires three main categories of components:
Component Role
Crude Extract (Lysate): Provides the “machinery”: ribosomes, aminoacyl-tRNA synthetases, translation factors, and often RNA polymerase.
Energy Solution: Contains an energy source (like phosphoenolpyruvate or glucose) and nucleotide triphosphates (ATP, GTP, UTP, CTP) to fuel the reaction.
Reaction Buffer: Contains essential ions ($Mg^{2+}$ and $K^{+}$), amino acids, and chemical additives to maintain pH and osmotic balance.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Why it’s critical: Protein synthesis is energetically expensive. Every peptide bond requires the hydrolysis of multiple high-energy phosphate bonds. Without a regeneration system, the accumulated inorganic phosphate ($P_i$) inhibits the reaction, and ATP levels drop rapidly, leading to very low yields.
Method for Continuous Supply: One common method is the use of secondary energy substrates combined with specific enzymes. For example, using Creatine Phosphate and Creatine Kinase. The kinase transfers a phosphate group from creatine phosphate back to ADP, constantly replenishing the ATP pool.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
The choice depends entirely on the complexity of the protein.
Prokaryotic (e.g., E. coli): High yield, fast, and inexpensive. However, it lacks post-translational modification (PTM) machinery.
Protein Choice: GFP (Green Fluorescent Protein). It is a simple, robust protein that doesn’t require complex folding or glycosylation, making it ideal for the high-speed E. coli system.
Eukaryotic (e.g., CHO or Wheat Germ): Lower yields but capable of complex folding and PTMs (like glycosylation or disulfide bond formation).
Protein Choice: Human Erythropoietin (EPO). As a therapeutic glycoprotein, EPO requires specific glycosylation patterns to be biologically active, which only a eukaryotic system (like CHO cell extract) can provide.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Challenges: Membrane proteins are hydrophobic. In a standard aqueous cell-free mix, they often misfold or aggregate because there is no lipid environment to stabilize their transmembrane domains.
Design Strategy:
Supplement with Surfactants/Lipids: Add nanodiscs, liposomes, or mild detergents directly to the reaction. This provides a “home” for the protein as it is translated.
Addressing the Challenge: To ensure proper orientation, use pre-formed liposomes. As the ribosome translates the mRNA, the hydrophobic regions of the protein can spontaneously insert into the lipid bilayer of the liposome.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Reason for Low Yield Troubleshooting Strategy
mRNA Degradation: Add RNase inhibitors or ensure all equipment is RNase-free. You can also check mRNA stability by running a gel.
Magnesium Imbalance: Perform a Magnesium Titration. CFPS is extremely sensitive to Mg{2+} concentration; test a range (e.g., 8mM to 20mM) to find the “sweet spot” for your specific template.
Codon Bias: If using a human gene in an E. coli extract, the tRNA pools may not match. Use codon-optimized DNA or supplement the reaction with specialized tRNA mixtures (e.g., Rosetta-style extracts).
Homework question from Kate Adamala:
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
a. What would my synthetic cell do? What is the input and what is the output?
My synthetic cell will function as a Smart Antioxidant Biosensor. It is designed to detect environmental oxidative stress and respond by producing and releasing protective enzymes.
Input: High concentrations of Reactive Oxygen Species (ROS), specifically Hydrogen Peroxide ($H_2O_2$).
Output: The production and secretion of the enzyme Catalase.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
While the chemical reactions for transcription and translation (Tx/Tl) would occur in an open solution, the system would lack stability. Without encapsulation in a lipid bilayer, the DNA, enzymes, and ribosomes would be immediately diluted and degraded in a complex environment (like a biological tissue). Encapsulation is essential to maintain a “protected reaction space.”
c. Could this function be realized by a genetically modified natural cell?
Yes, it could. However, a synthetic cell is a “minimal” and safer alternative. Natural cells have complex metabolic backgrounds that might interfere with the specific task, and they pose a risk of uncontrolled replication. A synthetic cell is a programmable, non-living machine that performs only the desired function.
d. Describe the desired outcome of your synthetic cell operation.
The goal is for these cells to act as localized therapeutic agents. When they encounter an area of inflammation (marked by high ROS), they “turn on” their internal factory, produce Catalase, and neutralize the H2O2, thereby preventing oxidative damage to surrounding healthy cells.
Design all components that would need to be part of your synthetic cell
a. What would be the membrane made of?
The membrane will be a Lipid Bilayer composed of POPC (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine) for basic structure and Cholesterol to enhance membrane stability and mimic human cell fluidity.
b. What would you encapsulate inside?
Transcriptional/Translational Machinery: Ribosomes, T7 RNA Polymerase, amino acids, and tRNAs.
Energy Regeneration System: Creatine Phosphate and Creatine Kinase to ensure a steady supply of ATP.
Genetic Material: A custom-designed plasmid DNA.
c. Which organism will your Tx/Tl system come from?
I will use a Bacterial (E. coli) system.
Reasoning: Bacterial extracts (like PURE system or S30 lysate) are much more robust and offer significantly higher protein yields compared to mammalian systems. Speed is critical when the goal is to neutralize oxidative stress quickly.
d. How will your synthetic cell communicate with the environment?
Communication will occur via Membrane Channels. Since the Catalase enzyme is too large to diffuse through the lipid bilayer, I will incorporate a pore-forming protein into the membrane to allow the output to exit the synthetic cell.
Experimental details
a. List all lipids and genes.
Lipids: POPC and Cholesterol.
Gene 1 (The Sensor): The oxyS promoter. This is an E. coli promoter that is specifically activated in the presence of oxidative stress.
Gene 2 (The Effector): The katG gene, which encodes for the enzyme Catalase to break down H2O2.
Gene 3 (The Channel): The alpha-Hemolysin (hlyA) gene. This produces a small pore (1.4 nm) in the membrane, allowing the produced enzymes to be released into the environment.
b. How will you measure the function of your system?
I will use a two-step verification process:
Fluorescence Reporting: I will include a GFP (Green Fluorescent Protein) gene under the same oxyS promoter. If the synthetic cells glow green when H2O2 is added, it proves the sensing and translation mechanisms are working.
Activity Assay: I will use an Amplex Red Assay. This reagent changes color in the presence of H2O2. If the color intensity decreases after adding my synthetic cells to the sample, it confirms that Catalase is being successfully produced and released to neutralize the oxidant.
Homework question from Peter Nguyen:
I chose Textiles/Fashion because it allows us to turn everyday clothing into a life-saving diagnostic tool.
Pitch Sentence: I propose the “Bio-Active Rescue Suit,” a garment embedded with freeze-dried cell-free sensors designed to detect toxic environmental chemicals and change color to warn the wearer of invisible dangers.
How it works: The cell-free reaction components (ribosomes, DNA templates, and energy buffer) are freeze-dried onto the textile fibers. When the fabric is exposed to a specific liquid or moisture containing a target contaminant (like heavy metals or nerve agents), the water rehydrates the system. This triggers the transcription and translation of a “reporter protein” like mCherry or GFP, causing a visible color change on the clothing surface within minutes.
Societal Challenge: This addresses the need for real-time, wearable safety monitoring for industrial workers, first responders, and soldiers who often lack bulky diagnostic equipment in high-risk zones.
Addressing Limitations: To handle one-time use and stability, the sensors are designed as modular, replaceable “bio-patches” that can be attached to the suit. To prevent accidental activation by rain, the patches are covered by a semi-permeable membrane that only allows specific target molecules to pass through while keeping pure water out.
Homework question from Ally Huang:
This proposal uses the BioBits® system to solve a critical healthcare challenge for long-term space travel.
A major challenge for deep-space missions is “Radiation-Induced Oxidative Stress.” High-energy cosmic rays damage human DNA, leading to a build-up of reactive oxygen species (ROS) that can cause cancer or organ failure. Currently, we cannot monitor an astronaut’s cellular response to radiation in real-time. Developing a portable, low-resource method to detect cellular stress markers is vital for protecting astronaut health during a journey to Mars. This research is significant because it paves the way for “on-demand” diagnostics in environments where traditional labs are impossible.
The genetic target is the human HMOX1 (Heme Oxygenase-1) promoter fused to a green fluorescent protein (GFP) reporter gene. HMOX1 is a primary biomarker for oxidative stress.
In space, radiation causes systemic inflammation. The HMOX1 gene is naturally up-regulated by the body to fight this stress. By using the HMOX1 promoter in a BioBits® system, we can create a “living” sensor that mimics the human body’s response. If an astronaut’s blood or saliva sample contains stress-induced signaling molecules, the BioBits® system will activate the promoter, indicating that the radiation levels have reached a dangerous threshold for human biology.
Hypothesis / Research GoalHypothesis: I hypothesize that a freeze-dried BioBits® cell-free system, utilizing a stress-responsive promoter, can accurately detect biomolecular markers of radiation damage in a microgravity environment as effectively as it does on Earth.
Reasoning: Microgravity affects molecular diffusion and protein folding. By testing this system on the ISS, we can determine if cell-free sensors remain reliable diagnostic tools for astronauts.
My goal is to validate that the “Bio-Bits + P51 Viewer” setup can provide a “Yes/No” visual signal for cellular damage without needing complex liquid handling.
I will test freeze-dried BioBits® pellets containing the HMOX1-GFP reporter DNA.
Samples: One set of pellets will be activated with a “stressed” sample (containing H2O2 or radiation-mimetic chemicals), and the Control set will be activated with pure water.
Measurements: After incubation, I will use the P51 Molecular Fluorescence Viewer to measure the intensity of green light produced.
Data: I will record the time it takes for a visible signal to appear and compare the brightness of the ISS samples against Earth-based controls to account for microgravity effects.
Week 10:Advanced Imaging & Measurement Technology
Final Project
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
For my “Sentinel Microbes” project, which aims to detect foreign pathogens in the gut, the most critical aspect to measure is the quantitative expression of the reporter protein. When the engineered microbes encounter a specific pathogen (like Salmonella), they trigger a genetic circuit to produce a visual signal. I need to measure the concentration and identity of this protein to confirm that the “Detected!” signal is accurate and strong enough to be noticed.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
I plan to measure three main elements:
- Reporter Protein Fidelity: To ensure the microbe is producing the exact reporter protein (e.g., eGFP) without mutations, I will measure its molecular weight.
- Detection Sensitivity: I will measure the threshold concentration of the pathogen needed to activate the reporter. This is done by exposing the microbes to varying amounts of the "foreign substance" and recording the output intensity.
- Kill-Switch Efficiency: I will measure the degradation of the microbe's DNA and the presence of "toxin" peptides once the detection task is complete to verify that the biocontainment system worked.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Mass Spectrometry (Waters Xevo G3 QTof MS): I will use LC-MS to perform Intact Mass Analysis and Peptide Mapping. This provides the “molecular fingerprint” of the reporter proteins and ensures they are correctly synthesized in the harsh gut environment.
Fluorometry (Microplate Readers): This technology will be used to quantify the fluorescence intensity of the reporter signals, allowing me to calibrate the sensitivity of the biosensor.
Opentrons Automation: I will utilize the Opentrons OT-2 robot to automate the sampling and serial dilutions of pathogens. This ensures that my measurements are highly reproducible and that the simulated gut environment is precisely controlled.
DNA Sequencing (NGS): I will use sequencing to monitor the stability of the synthetic genetic circuits within the microbes over multiple generations to prevent “genetic drift.”
Waters Part I — Molecular Weight
Waters Part II — Secondary/Tertiary structure
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
The primary difference between native and denatured protein conformations is the accessibility of ionizable residues.
Native proteins are tightly folded, meaning most of the basic amino acid residues are buried inside the protein core and cannot be easily protonated.
When a protein unfolds (denatures), it loses its 3D structure and becomes an open, linear chain. This exposes many more residues to the solvent, allowing them to pick up more positive charges.
In a mass spectrometer, this is determined by observing the charge state distribution. In Figure 2, the denatured protein (top) shows peaks at much lower m/z values compared to the native protein (bottom). This shift occurs because m/z is an inverse relationship; as the charge (z) increases due to unfolding, the m/z ratio decreases. Additionally, the denatured state shows a much broader distribution of peaks because the unfolded chain can exist in many different charge states.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
Yes, I can discern the charge state of the peak at ~2800 m/z by analyzing the isotopic spacing shown in the zoomed-in inset of Figure 3.To determine the charge state (z), I look at the distance between the individual isotope peaks. In the inset, the peaks are approximately 0.1 m/z units apart (for example, moving from 2545.03 to 2545.13).
Since isotopes of a protein differ by approximately 1 Dalton (1 Da) in mass, we use the formula:
- Delta m/z ={1}/{z}
Given that the spacing (Delta m/z) is 0.1, the calculation is 0.1 = 1/z, which results in z = 10. Therefore, the charge state of this protein peak is +10.
Waters Part III — Peptide Mapping - primary structure Assignees for this section
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Looking at Figure 5a, between the retention times of 0.5 and 6 minutes, I can identify approximately 15 to 17 chromatographic peaks that are above 10% relative abundance.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Usually, the number of observed peaks is fewer than the total number of predicted peptides from the PeptideMass tool. This is because some peptides might be too small to be retained, some may co-elute (come out at the same time), and some may not ionize well in the mass spectrometer.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]) based on its m/z and z.
m/z of the most abundant peak: According to Figure 5b, the most abundant peak is at 525.76712.
Charge state (z): Looking at the inset, the isotopic spacing is 0.5 m/z units (525.76 to 526.25). Using the formula Delta m/z = 1/z, we find that z = 2.
Calculation of singly charged mass ([M+H]⁺):To find the [M+H] (where z=1), we first find the neutral mass (M) and add one proton.
Neutral Mass (M) = (Observed\ m/z \times z) - (z \times 1.008)$$M = (525.76712 \times 2) - (2 \times 1.008) = 1051.53424 - 2.016 = 1049.51824 Da.
So, $[M+H]^+ = 1049.51824 + 1.008 = 1050.52624\ Da.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. (Recall that Accuracy Formula)
By comparing the calculated mass (1050.52 Da) to the PeptideMass results, this matches the eGFP peptide sequence: “LPDNHYLSTQSALSK”.
And ppm calculation is:7.6ppm.
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Based on Figure 6, the amino acid coverage map shows that 88% of the eGFP sequence is confirmed by the peptide mapping data.
Waters Part IV — Oligomers
To identify the oligomeric species, we multiply the subunit mass by the number of units in the complex and match them with the peaks on the CDMS spectrum (Figure 7).
Waters Part V — Did I make GFP?
Theoritical
Observed/ Measured on the Intact LC-MS
ppm Mass Error
Molecular Weight
27.935 kDa
27.981 kDa
1600 ppm
Week 11:Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
E.Coli Lysate:
It is acted as the catalytic core of the reaction; it provides the ribosomes for translation and T7 RNA Polymerase for high-level transcription.
Salts and Buffers:
Potassium Glutamate: It provides necessary ionic strength and potassium ions for protein folding without inhibiting DNA-protein interactions like chloride salts might.
HEPES-KOH pH 7.5: It is pH indicator.Also it prevents reaction from becoming too acidic due to metabolic byproducts.
Magnesium Glutamate: It supplies magnesium ions required for stabilizing ribosome assembly and acting as a cofactor for RNA polymerase.
Potassium Phosphate (Monobasic- Dibasic): It provides secondary buffering and source of inorganic phosphate needed to regenerate ATP.
Energy and Nucleotide System:
Ribosome
Guanine: It serves as specific region for nitrogenous base that can be salvaged by lysate enzymes to create GTP for transcription when direct GMP levels are low.
Translation Mix (Amino Acids):
they are provide fundamental building blocks for synthesizing the polypeptide chain.
Additives:
Nicotinamide: Stabilizes metabolis cofactors like NAD+, preventing their degradation and ensuring energy generating pathways reman active.
Backfill
Nuclease Free Water: Acts as solvent for reaction, ensuring no DNA/RNA-degrading enzymes interfere with genetic templates.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above.
The 1-hour optimized PEP-NTP master mix utilizes a high-energy phosphate donor (PEP) for rapid, burst-like ATP production, which is ideal for short, high-speed reactions but leads to quick energy depletion and pH drops. In contrast, the 20-hour NMP-Ribose-Glucose master mix relies on a slower, more sustainable metabolic recycling of nucleotides and sugars, allowing the reaction to maintain productivity and pH stability for nearly a day, resulting in a much higher total protein yield.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur through nucleotide salvage pathways present in the E. coli lysate. Enzymes such as phosphoribosyltransferases can take the Guanine base and chemically link it to a ribose-phosphate molecule to form GMP, which is then further phosphorylated by the energy system into GTP, the required substrate for RNA polymerase.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP (Superfolder GFP): Known for its extremely fast folding and high stability; it is the “gold standard” for cell-free systems because it folds correctly even when fusion tags are attached.
mRFP1 (monomeric Red Fluorescent Protein): While a classic red marker, it is known for having a relatively slow maturation time and lower brightness compared to newer red variants like mScarlet.
mKO2 (monomeric Kusabira Orange 2): A bright orange protein that is highly pH-stable but can be sensitive to oxygen levels for proper chromophore maturation.
mTurquoise2: An exceptionally bright cyan protein with a high quantum yield and high photostability, though it requires specific filter sets for accurate readout.
mScarlet-I: One of the brightest red fluorescent proteins available; its main advantage in cell-free systems is its high brightness and faster maturation compared to mRFP1.
Electra2: A newer generation protein designed for high-level expression; it is often optimized for minimal metabolic burden on the cell-free machinery.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Protein: mScarlet-I
Reagent Adjustment: Increase the concentration of the 17 amino acid mix and magnesium glutamate.
Expected Effect: Since mScarlet-I is a highly bright and “demanding” protein, increasing the amino acid supply will ensure that translation doesn’t stall during the long 36-hour incubation. Boosting Magnesium will further stabilize the ribosomes for this extended run, ideally resulting in a steeper fluorescence curve and a higher final signal-to-noise ratio.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
For my custom 2 $\mu$L supplement in the 20 $\mu$L reaction, I plan to focus on molecular crowding agents (like PEG) or additional energy boosters (like extra Glucose) to see if we can sustain the reaction beyond the typical 20-hour window.