Week 2: DNA Read, Write and Edit
Part 1: Benchling & In-silico Gel Art
First I start to stimulate Restriction Enzyme in Lambda DNA:
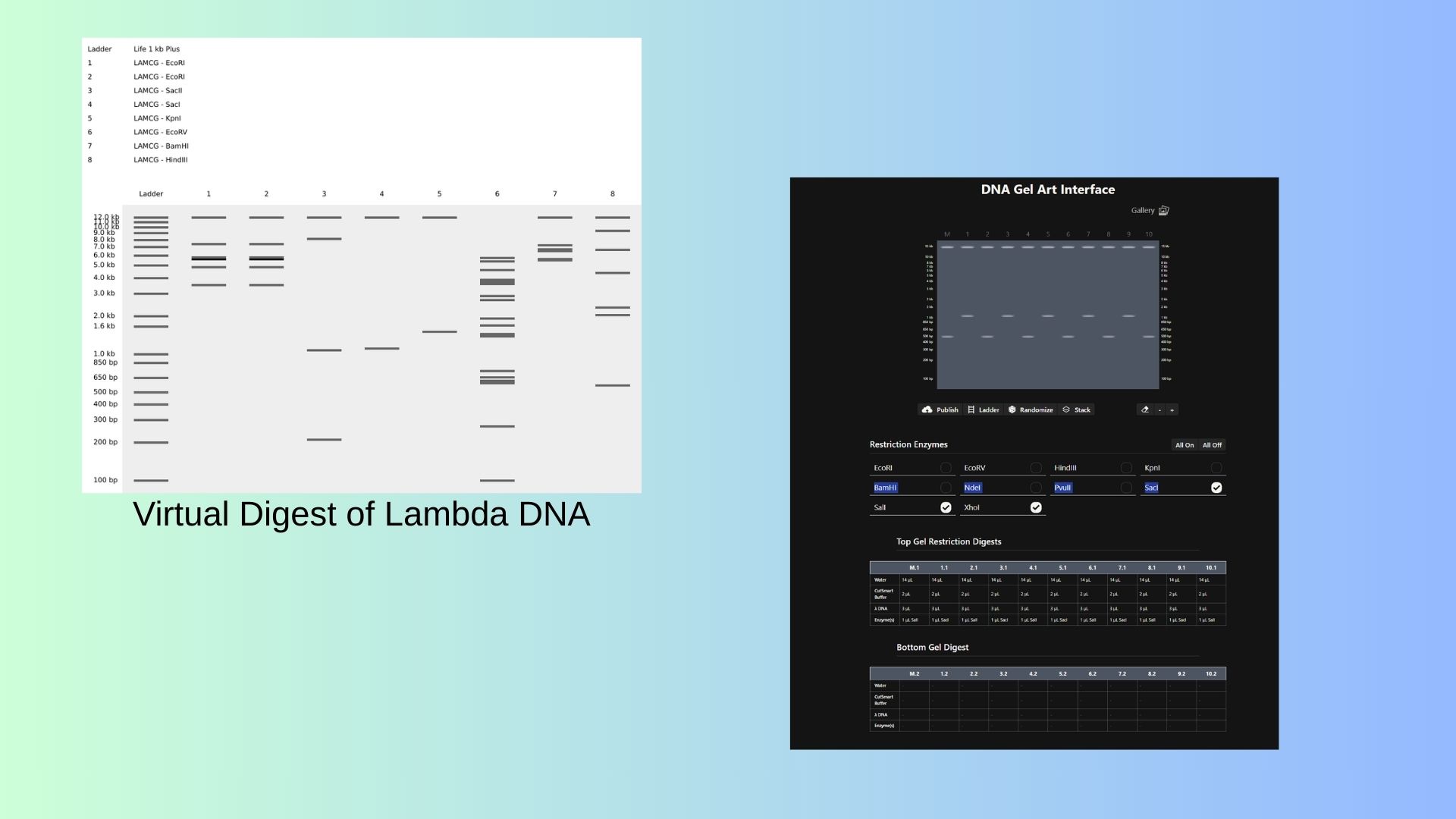
Also shown above,with Ronan’s website, I try to pattern with enzymes.
Part 3: DNA Design Challenge
3.1. Choose your protein:
I decided to choose GFP protein in water jellyfish(Also called “Aqua Victoria”). Because with GFP protein I can see that organization of cell. I can track protein and find to where to go in the cell. On the shown below, you can see GFP Protein Sequence:

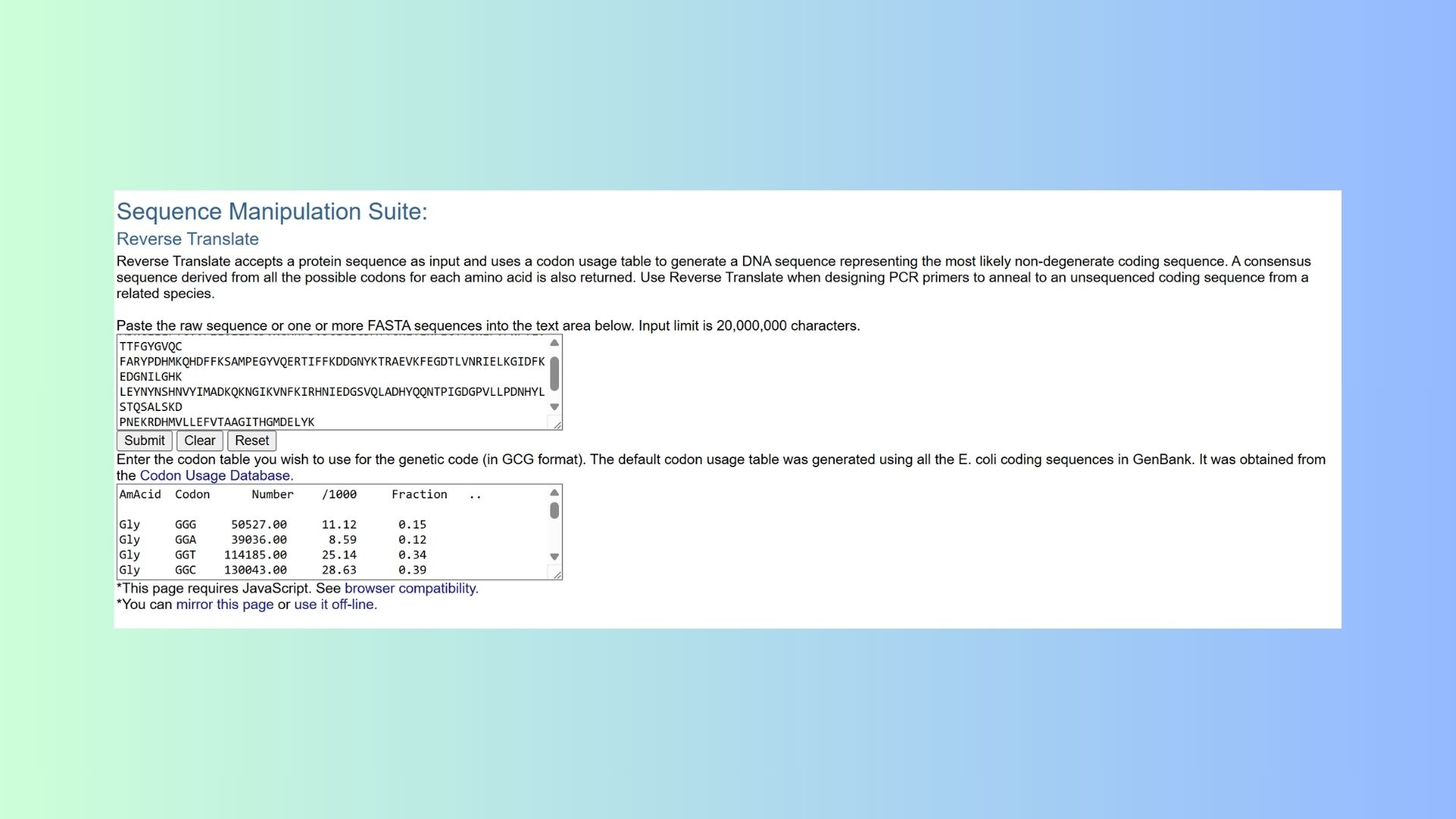
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence:
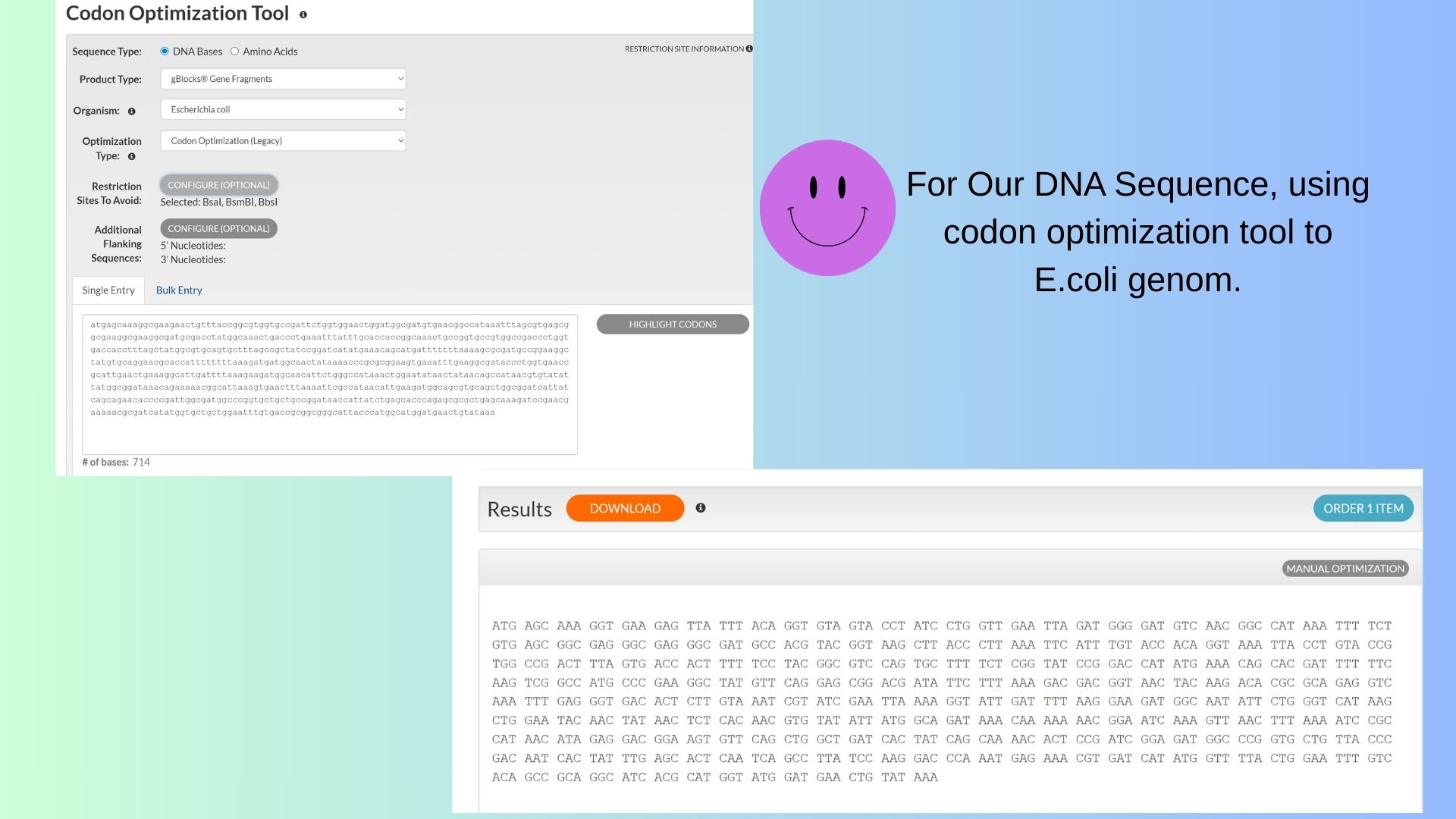
3.3. Codon optimization:
I want to use codon optimization tool for E.coli genom.
3.4 You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
To make GFP protein from DNA, the DNA first needs to be transcribedinto mRNA and then translated into protein. The DNA has a promoter, the GFP coding sequence, and other regulatory parts. RNA polymerase reads the DNA to make mRNA, and ribosomes read the mRNA to assemble the amino acids into the GFP protein, which then folds and becomes fluorescent.
Ways to produce GFP:
In cells (cell-dependent):
In bacteria like E. coli, you put the GFP DNA in a plasmid and the bacteria make the protein for you—fast and cheap.
For more complex proteins, yeast or mammalian cells can be used to get proper folding and modifications.
Cell-free (in vitro):
You can use a test-tube system with the cell’s transcription and translation machinery. Add GFP DNA, and the protein is made directly. This is fast and easy to control.
Both methods let you get functional GFP for experiments or imaging.
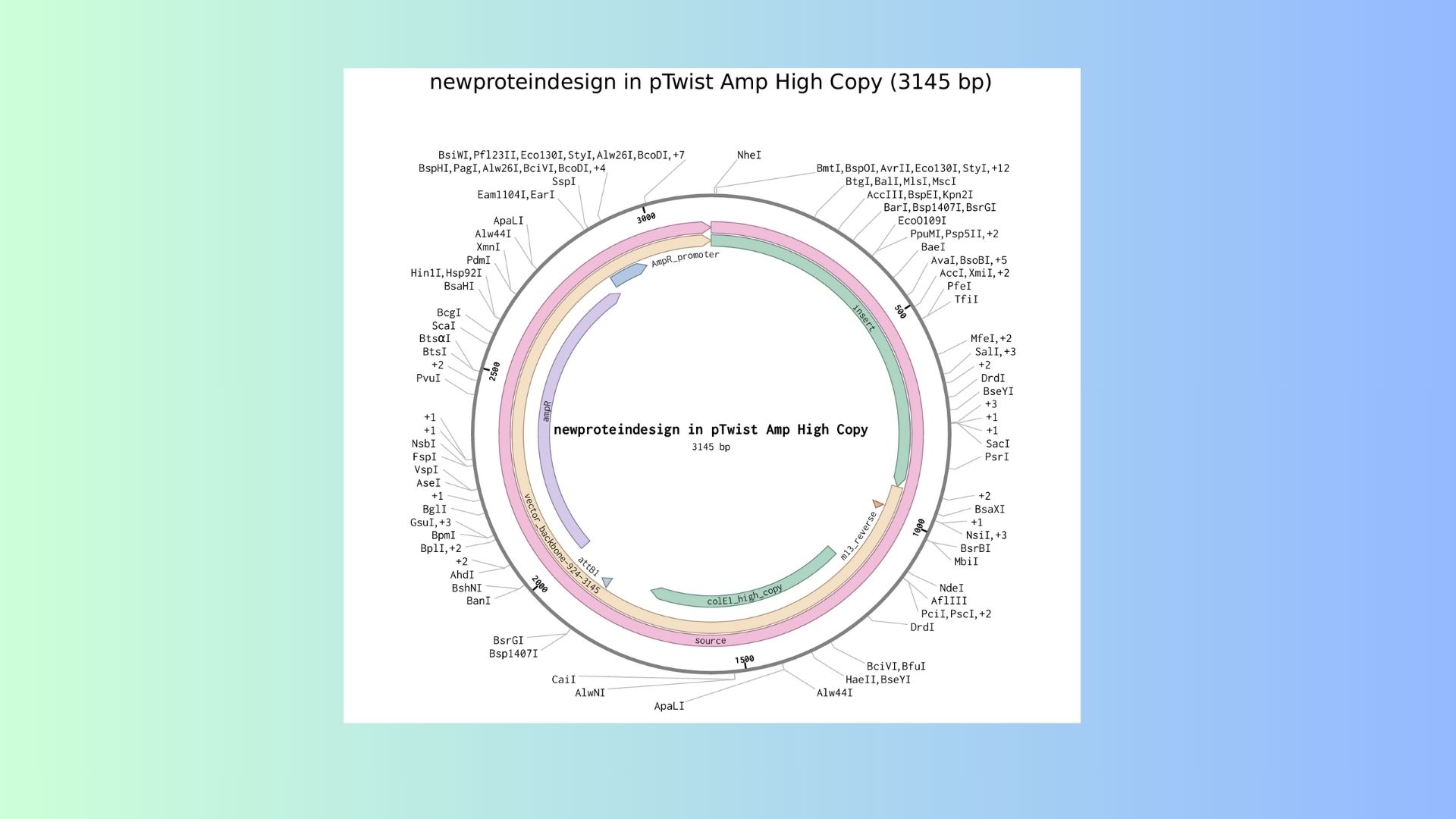
Part 4: Prepare a Twist DNA Synthesis Order:
Here are expression cassed and its vector DNA:

Part 5: DNA Read/Write/Edit:
5.1 DNA Read:
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank). I want to sequence DNA related to Alzheimer’s disease which is neurodegenerative diseases that affects memorh, cognition and behavior. The main risk genes are APP, PSEN1 and PSEN2 and these genes are linked to early-onset familial Alzheimer’s disease. And sequencing these genes would help identify pathogenic mutations, assess genetic risk etc.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I use Whole Exome Sequencing (WES) on an Illumina platform.This method focuses on protein-coding regions (exons), where most disease-causing mutations occur, and is cost-effective compared to whole genome sequencing. Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
This is a second-generation sequencing method because it relies on massively parallel short-read sequencing and requires PCR amplification of DNA fragments.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is genomic DNA extracted from patient samples, such as blood or saliva.
Essential preparation steps include:
Fragmentation of DNA into short fragments (~150–200 bp).
Adapter ligation to allow binding to the sequencing flow cell.
PCR amplification to increase the quantity of DNA library.
Exome capture using hybridization probes to enrich for coding regions.
Library quality control to check fragment size and concentration.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
DNA fragments are attached to a flow cell and amplified into clusters (bridge amplification).
Sequencing by synthesis: fluorescently labeled nucleotides are added one at a time.
Each incorporated nucleotide emits a specific color that is captured by a camera.
The software converts these fluorescence signals into nucleotide sequences (A, T, G, C) — this process is called base calling.
What is the output of your chosen sequencing technology?
The sequencing produces FASTQ files, containing millions of short DNA reads with associated quality scores. After bioinformatic analysis, variant calling identifies single nucleotide polymorphisms (SNPs), insertions, or deletions, producing variant call files (VCFs) for interpretation of disease-associated mutations.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? Selected DNA to Synthesize: A Synthetic Genetic Circuit for Detecting Inflammation
I would design and synthesize a synthetic genetic circuit capable of sensing inflammatory signals and generating a controlled therapeutic response. Specifically, this construct would be compatible with human cells and engineered to detect activation of the NF-κB signaling pathway, which is a central regulator and biomarker of inflammation. Upon activation, the circuit would trigger the expression of either an anti-inflammatory cytokine, such as IL-10, or a fluorescent reporter protein for diagnostic purposes.
Rationale for Synthesizing This DNA
This synthetic DNA construct has both therapeutic and diagnostic potential. From a therapeutic perspective, engineered immune cells or stem cells containing this circuit could respond locally to inflammatory signals and produce anti-inflammatory molecules in conditions such as autoimmune diseases or inflammatory bowel disease (IBD). From a diagnostic standpoint, the system could function as a living biosensor, providing real-time detection of inflammatory activity through measurable reporter expression. Additionally, this project exemplifies key principles of synthetic biology, including modular genetic design, where distinct elements—such as promoters, sensing modules, logic components, and output genes—are assembled into a programmable biological system.
Simplified Genetic Components of the Circuit
The proposed construct would include:
An NF-κB–responsive promoter to sense inflammatory signaling
A minimal promoter combined with enhancer elements to regulate transcription
Coding sequence (e.g. IL10 or GFP) • PolyA signal • Insulator sequences
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Chosen Technology: Array-based Chemical DNA Synthesis (Phosphoramidite Method)
This is the dominant commercial method used by companies like Twist.
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
In silico DNA design Everything starts on the computer. First, I would digitally design the full DNA sequence of the genetic circuit. During this step, I would optimize the codons to ensure efficient expression in human cells. I would also remove problematic elements such as repetitive sequences, unwanted restriction sites, and regions that might form strong secondary structures. The goal here is to make sure the DNA is not only correct in theory but also stable and easy to synthesize and express.
Chemical oligonucleotide synthesis Once the design is finalized, the DNA is synthesized in small fragments called oligonucleotides. Using phosphoramidite chemistry, nucleotides are added one at a time in a controlled, step-by-step process. In simple terms, the DNA strand is chemically built base by base.
Oligo amplification and error correction Because chemical synthesis is not perfectly error-free, the short DNA fragments are amplified using PCR to increase their quantity. At this stage, error-correction methods can be applied to remove sequences that contain mismatches or synthesis mistakes, improving the overall accuracy of the final product.
DNA assembly After obtaining the correct short fragments, they are assembled into the full-length gene or genetic circuit. Techniques such as Gibson Assembly or Golden Gate Assembly are used to seamlessly join overlapping DNA pieces. This step is where the smaller parts come together to form the complete functional construct.
Cloning and validation Finally, the assembled DNA is inserted into a plasmid vector and introduced into host cells for replication. To confirm that the sequence matches the original design, sequencing is performed. This verification step ensures that the synthesized DNA is accurate and ready for downstream applications.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Array-based chemical DNA synthesis using phosphoramidite chemistry is widely used, but it has several limitations in terms of speed, accuracy, and scalability.
In terms of speed, the process is relatively slow because nucleotides are added one at a time through sequential chemical reactions. Although many oligonucleotides can be synthesized in parallel on an array, additional steps such as amplification, assembly, and validation increase the overall time required.
Regarding accuracy, synthesis errors accumulate as the DNA strand gets longer. Since each base addition is not 100% efficient, longer sequences are more likely to contain deletions or substitutions. For this reason, DNA is usually synthesized in short fragments and later assembled, followed by sequence verification.
In terms of scalability, the method is highly efficient for producing large numbers of short oligonucleotides. However, synthesizing very long genes or complex genetic circuits becomes more expensive and technically challenging due to assembly and error-correction requirements.
Overall, while the method is reliable and scalable for short sequences, it becomes less efficient and more error-prone as sequence length increases.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit disease-causing mutations in the human genome, specifically monogenic disorders such as sickle cell disease caused by mutations in the HBB gene. Since this condition results from a single nucleotide substitution, it is an ideal candidate for precise genome editing. The goal would be therapeutic—to correct the mutation in a patient’s hematopoietic stem cells and restore normal hemoglobin production. Similar strategies could be applied to other inherited diseases. The purpose of the edit would be to treat or cure genetic disorders rather than enhance human traits.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas9 genome editing because it allows precise, targeted modification of specific DNA sequences.
Also answer the following questions: How does your technology of choice edit DNA? What are the essential steps?
CRISPR works by using a guide RNA that directs the Cas9 enzyme to a specific location in the genome. Cas9 creates a cut in the DNA, and the cell repairs it. If a correct DNA template is provided, the mutation can be replaced through homology-directed repair.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
The main inputs include Cas9, a guide RNA, a repair template, and patient-derived cells. After delivery into the cells (e.g., via electroporation), edited cells are screened and verified by sequencing.
What are the limitations of your editing methods (if any) in terms of efficiency or precision? Limitations include possible off-target effects, limited editing efficiency, and challenges in achieving precise repair. Despite these limitations, CRISPR remains one of the most promising tools for treating genetic diseases.