Week 4: Protein Design Part I
Part A. Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? Meat is approximately 20% protein by mass. So, 500g of meat contains about 100g of protein. Given that the average mass of an amino acid is 100 Daltons ($1.66 \times 10^{-22}$ grams), we can calculate the total number of molecules:$100g / (100 \times 1.66 \times 10^{-24}g) \approx 6 \times 10^{24}$ amino acid molecules. That is roughly 10 moles.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish? This is because our digestive system breaks down the proteins we eat into their individual building blocks: amino acids. Our body doesn’t use the cow or fish proteins directly; it uses these free amino acids to assemble “human” proteins based on the instructions in our own DNA.
- Why are there only 20 natural amino acids? This is often described as a “frozen accident” in evolution. While there are many more possible amino acids, these 20 provided enough chemical diversity (acidic, basic, hydrophobic, etc.) to build complex 3D structures and catalyze reactions. Once life became complex, changing this fundamental toolkit would have been too disruptive.
- Can you make other non-natural amino acids? Design some new amino acids.Yes, scientists can synthesize non-natural amino acids (ncAAs) by adding unique side chains. For my project on schizophrenia, I could design an amino acid with a fluorescent “sensor” side chain that changes color when it interacts with high concentrations of dopamine. This would allow us to visualize “dopamine storms” in real-time.
- Where did amino acids come from before enzymes that make them, and before life started? Amino acids likely formed through abiotic synthesis. The Miller-Urey experiment showed that early Earth’s atmospheric gases (methane, ammonia, water vapor) could react with electrical discharges (lightning) to create amino acids. They might have also been delivered to Earth via meteorites.
- Why are most molecular helices right-handed? This is due to the “chirality” of L-amino acids. Because all life uses L-amino acids, the right-handed $\alpha$-helix is the most energetically stable conformation that avoids steric clashes (physical bumping) between the side chains.
- Why do $\beta$-sheets tend to aggregate? What is the driving force? The main driving forces are hydrogen bonding and the hydrophobic effect. The edges of a $\beta$-sheet have “unsatisfied” hydrogen bond donors and acceptors. This makes them very “sticky,” leading them to stack with other $\beta$-sheets to achieve a lower energy state.
- Why do many amyloid diseases form $\beta$-sheets? Can you use them as materials? Amyloid $\beta$-sheets are incredibly stable and resistant to degradation. In diseases like Alzheimer’s, proteins misfold into this “energy well” from which they cannot escape.As materials: Yes! Amyloid fibers are stronger than steel for their size. They can be engineered as ultra-stable nanowires or drug-delivery scaffolds.
- Design a $\beta$-sheet motif that forms a well-ordered structure. A well-ordered motif can be designed using an “alternating” pattern: (Val-Lys-Val-Glu)n. In this sequence, the hydrophobic Valine residues face one side while the charged Lysine and Glutamate residues face the other. This creates a “Janus-faced” sheet that is oily on one side and water-loving on the other, allowing it to assemble into perfect layers.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
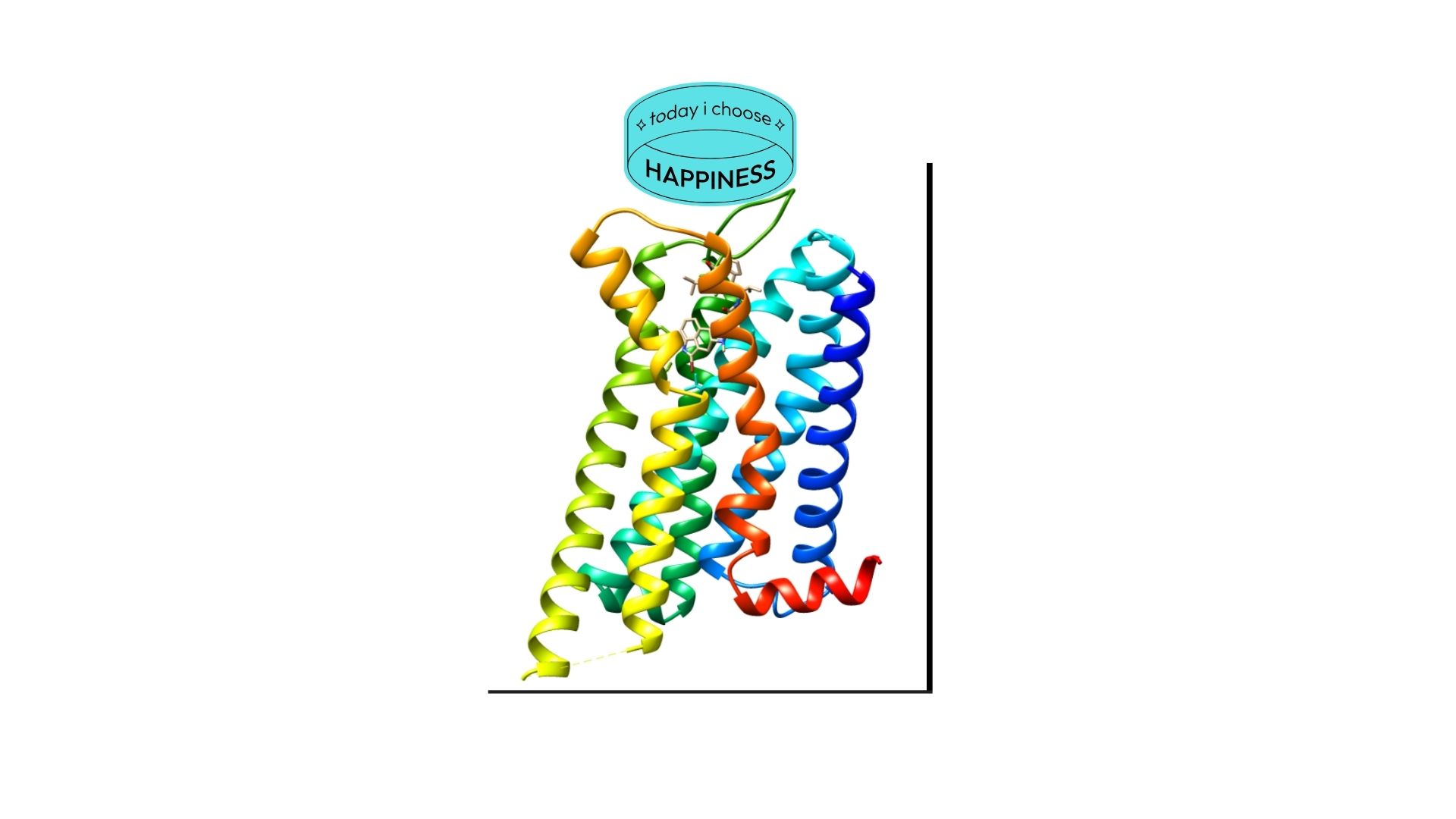
I want to select DRD2(the Human Dopamine D2 Receptor) protein because this protein provided to enter the doapamin in the cell. Also ın neurodejenaretif diseases such as in schzophrenia this receptor protein is overexpressing therefore dopamin enter the cell more than normal and this leads to hallucination. Therefore this protein plays important role in brain.
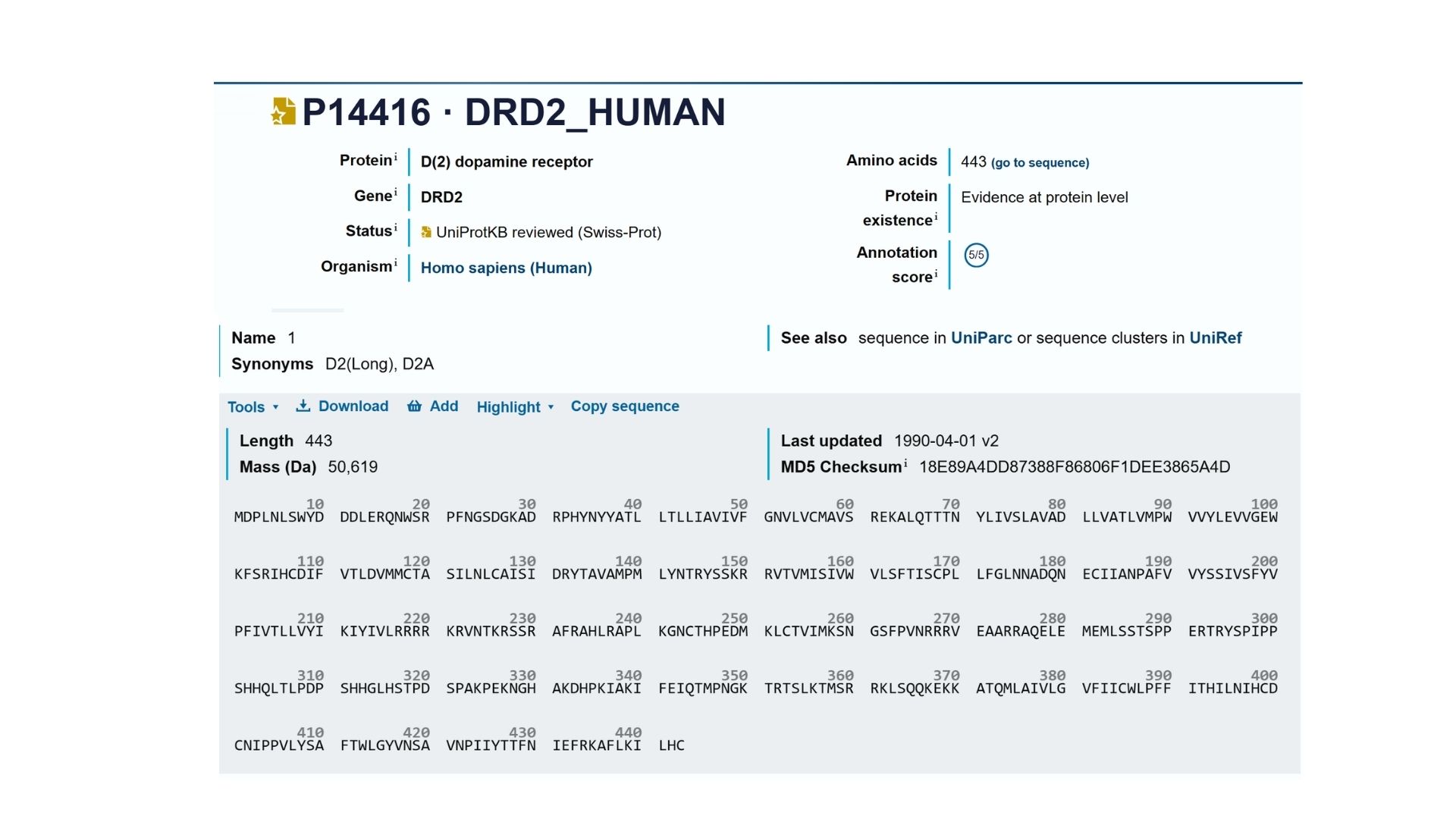
2. Identify the amino acid sequence of your protein.
My selected protein, the Human Dopamine D2 Receptor (DRD2), consists of 443 amino acids (Isoform 1). After analyzing the sequence using the provided Colab notebook, I found that the most frequent amino acid is Leucine (L). This high frequency of Leucine is a characteristic feature of transmembrane proteins, as its hydrophobic nature allows the protein’s seven alpha-helices to remain stable within the oily lipid bilayer of the cell membrane.
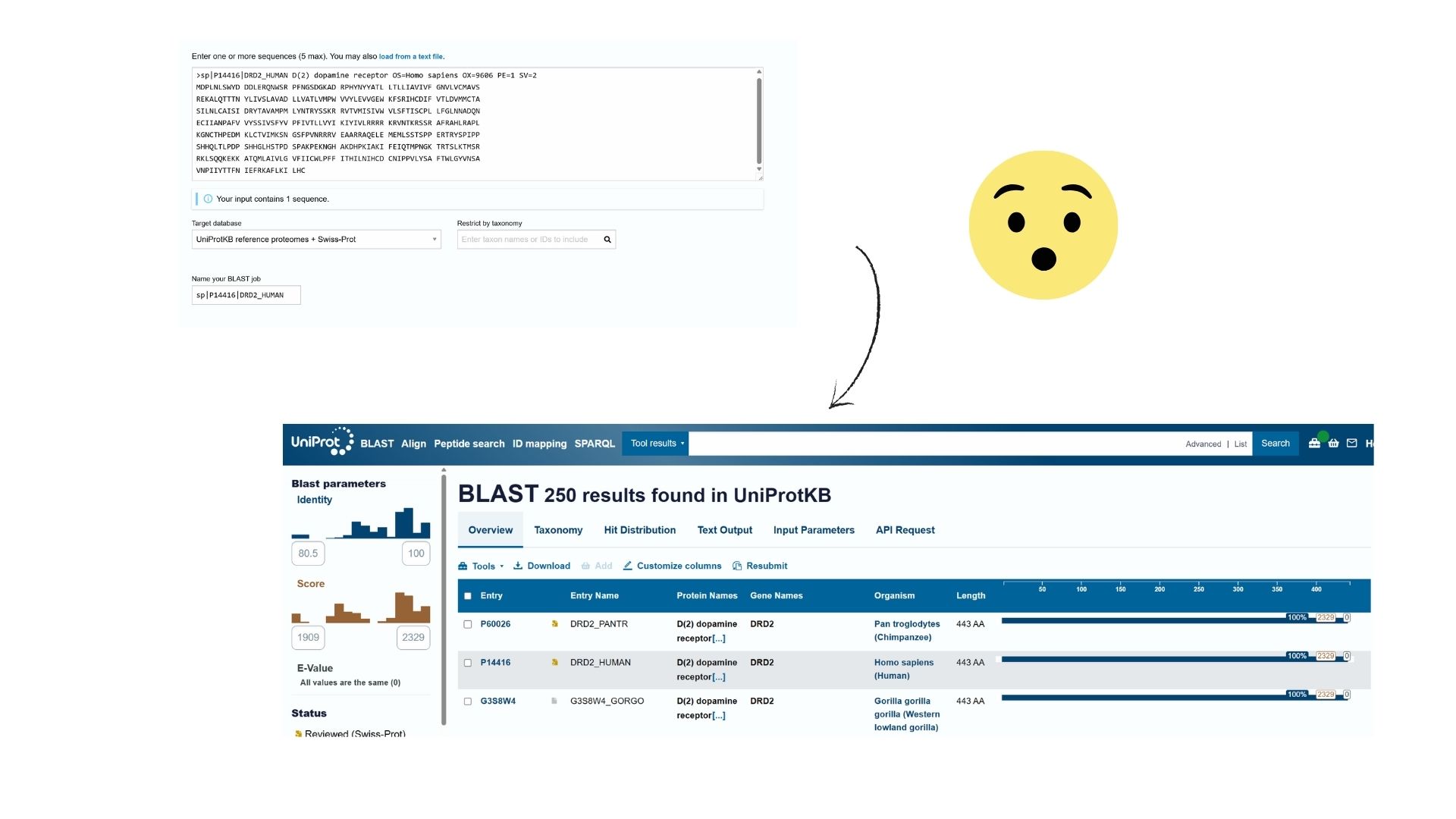
 Using Uniprot’s BLAST tool, I identified several hundred protein sequence homologs. The search results show very high similarity (often >90%) with DRD2 sequences in other mammals like chimpanzees and mice.
Using Uniprot’s BLAST tool, I identified several hundred protein sequence homologs. The search results show very high similarity (often >90%) with DRD2 sequences in other mammals like chimpanzees and mice.
 Also DRD2 belongs to the G-protein coupled receptor (GPCR) family, specifically the Class A (Rhodopsin-like) subfamily.
Also DRD2 belongs to the G-protein coupled receptor (GPCR) family, specifically the Class A (Rhodopsin-like) subfamily.
3. Identify the structure page of your protein in RCBS
The structure was deposited on March 2, 2018, and officially released on March 14, 2018. It was solved by Wang et al. and represents the D2 Dopamine Receptor bound to the antipsychotic drug Risperidone.
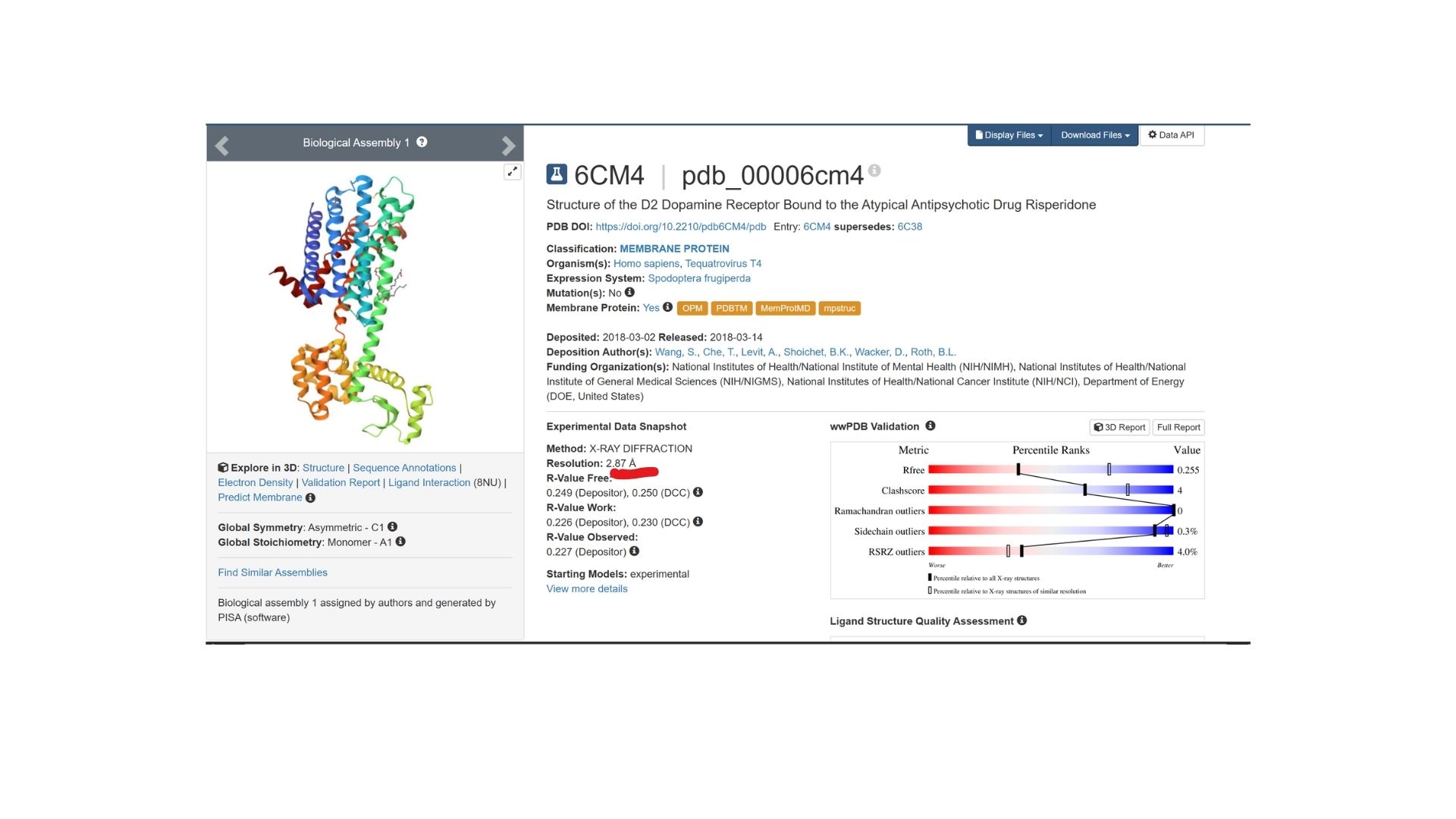 The structure I’ve been examining (PDB ID: 6CM4) has a reported resolution of 2.87 Å. While a resolution of 2.70 Å or lower is generally considered the benchmark for “good” quality, given that this is a membrane protein, 2.87 Å sits within an acceptable range for this class.
The structure I’ve been examining (PDB ID: 6CM4) has a reported resolution of 2.87 Å. While a resolution of 2.70 Å or lower is generally considered the benchmark for “good” quality, given that this is a membrane protein, 2.87 Å sits within an acceptable range for this class.
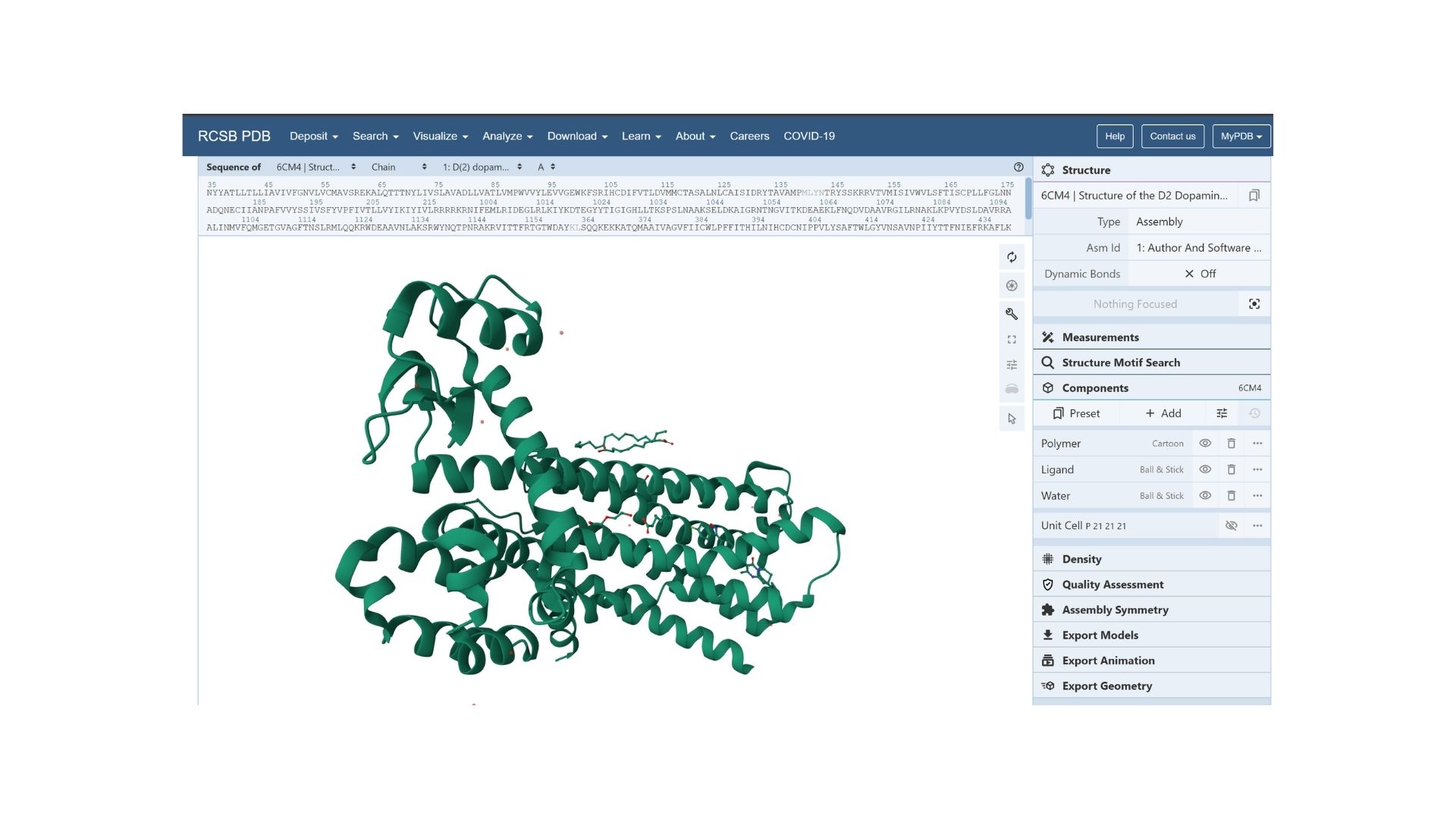 The resolved structure contains more than just the protein chain itself. Several other components were co-crystallized and are present in the model such as Risperidone and Auxililary proteins.
Biologically, this protein is categorized under the membrane protein group. Diving into a more specific classification, it is a member of the G Protein-Coupled Receptor (GPCR) family, characterized by its seven-transmembrane alpha-helices.
The resolved structure contains more than just the protein chain itself. Several other components were co-crystallized and are present in the model such as Risperidone and Auxililary proteins.
Biologically, this protein is categorized under the membrane protein group. Diving into a more specific classification, it is a member of the G Protein-Coupled Receptor (GPCR) family, characterized by its seven-transmembrane alpha-helices.
4.Open the structure of your protein in any 3D molecule visualization software
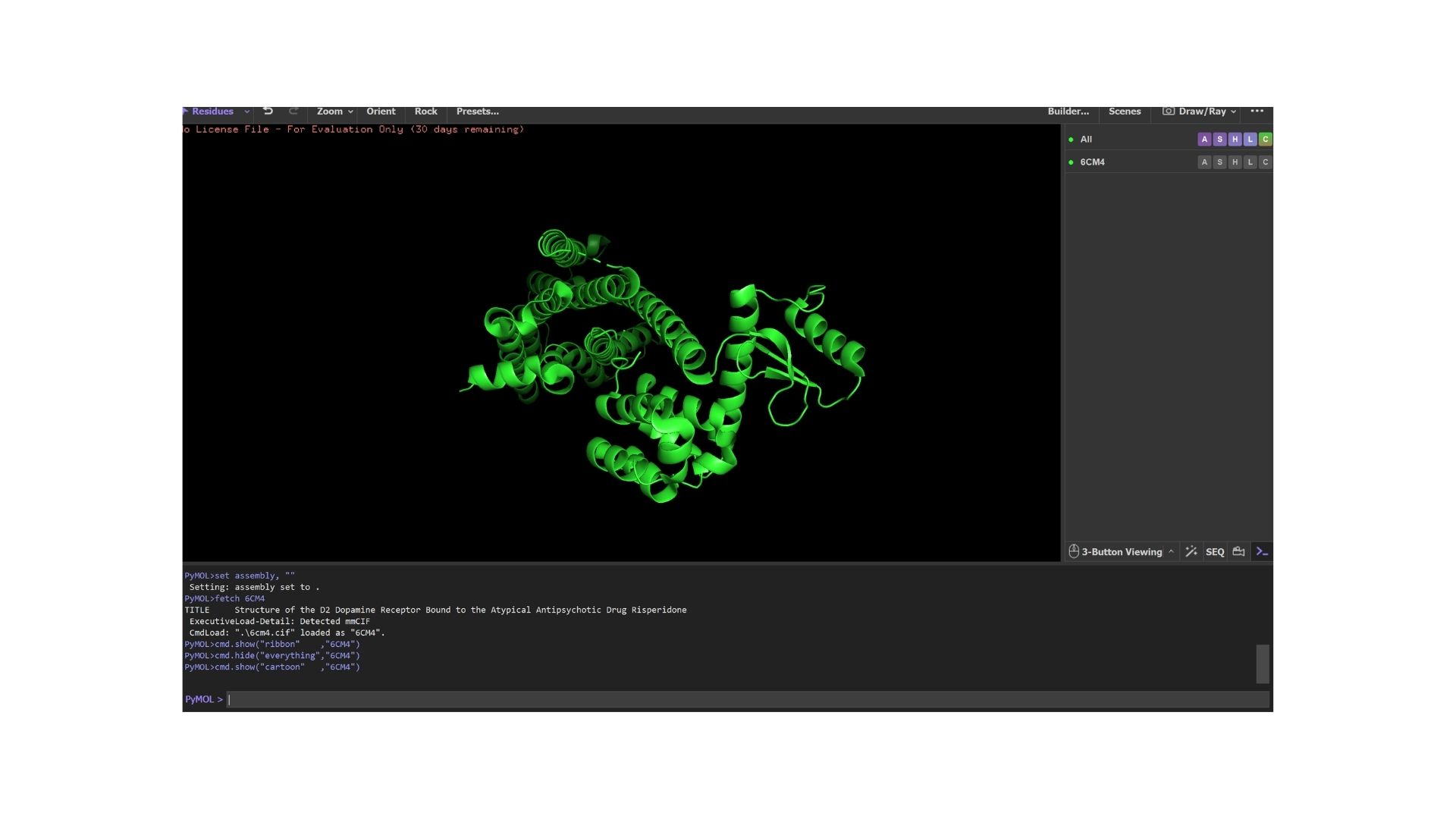
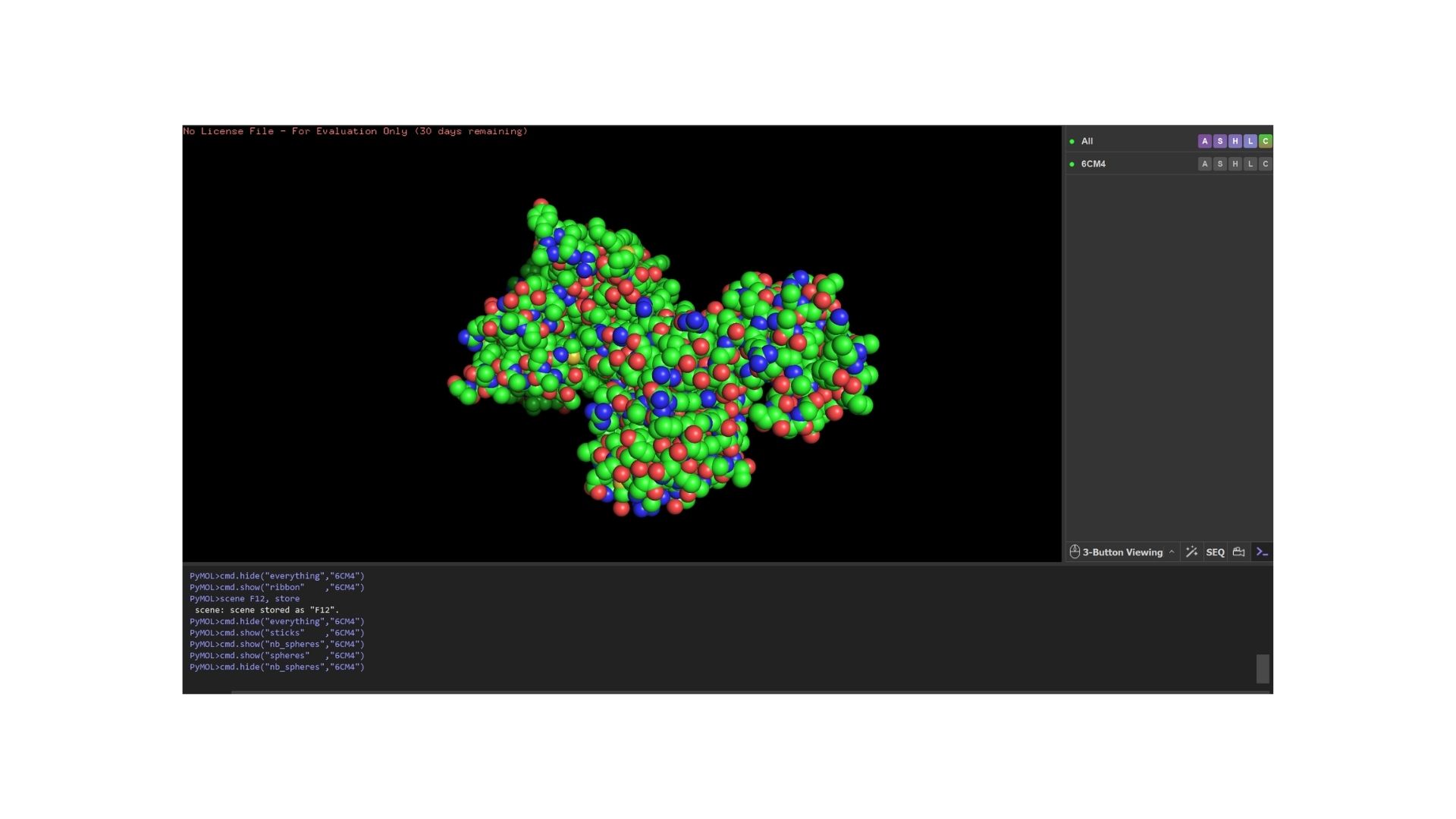
By using PyMol, structures of protein were visualized such as:
Cartoon:
Ribbon:
Ball and Stick
When we look at the secondary structure of my protein,based on my visualization, the protein is overwhelmingly helical. As a member of the GPCR family, it features the characteristic seven-transmembrane (7TM) alpha-helices. There are almost no beta-sheets present, as the structural integrity of this receptor relies almost entirely on its helical bundle.
As a membrane protein, the residue distribution is very specific: I observed a high concentration of hydrophobic residues on the exterior of the helices to interact with the lipid bilayer, while hydrophilic residues are mainly localized in the loops and the interior binding pocket.
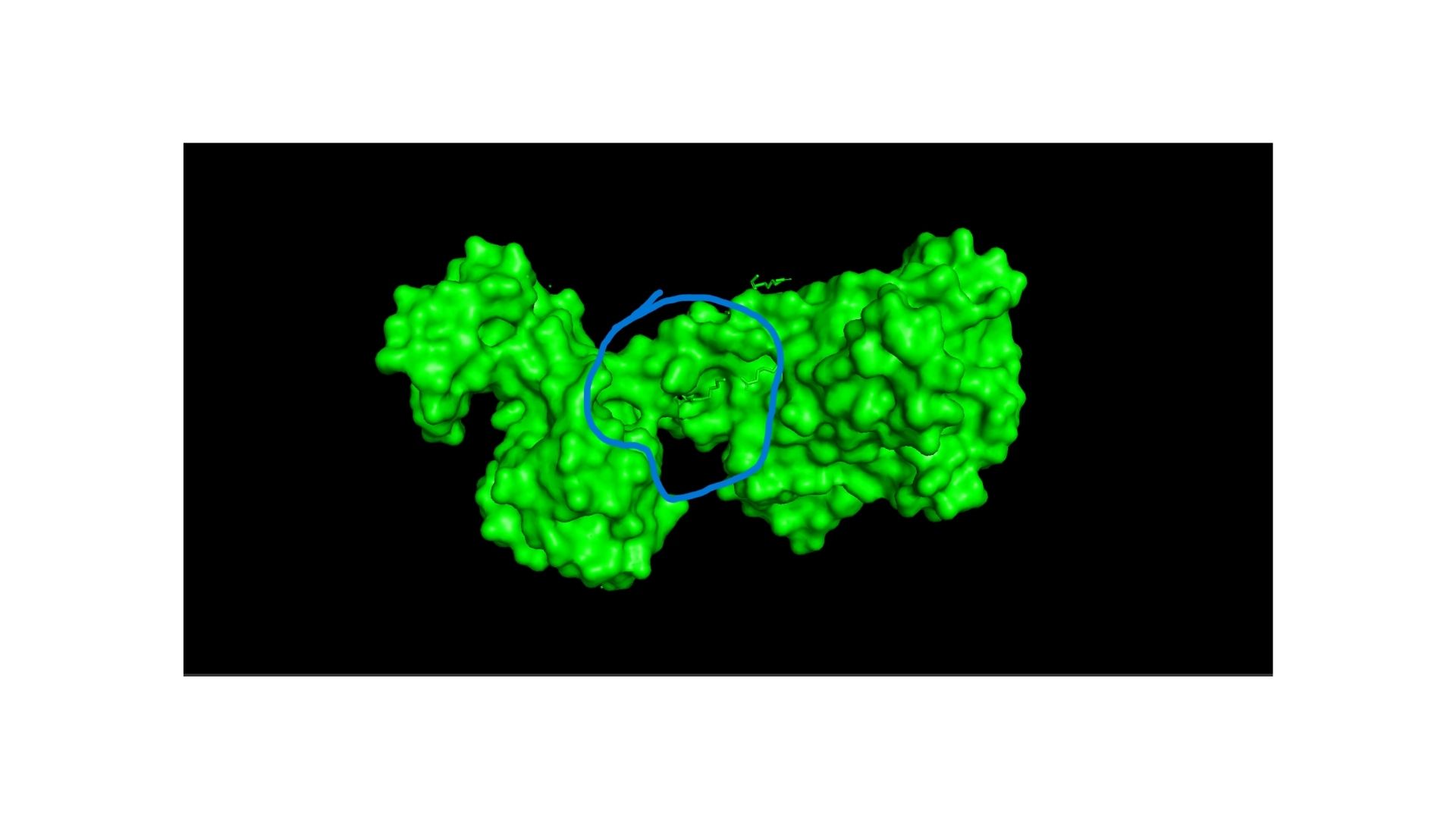
Looking at the surface, I can see a very distinct binding pocket. The pocket itself is well-defined and clearly hosts the drug molecule:
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
I used the ESM2 model to generate an unsupervised deep mutational scan of my DRD2 protein based on language model likelihoods. Looking at the resulting heatmap, I noticed that the model assigns very low likelihoods to mutations within the transmembrane helix regions, indicating that these areas are structurally and evolutionarily highly conserved.
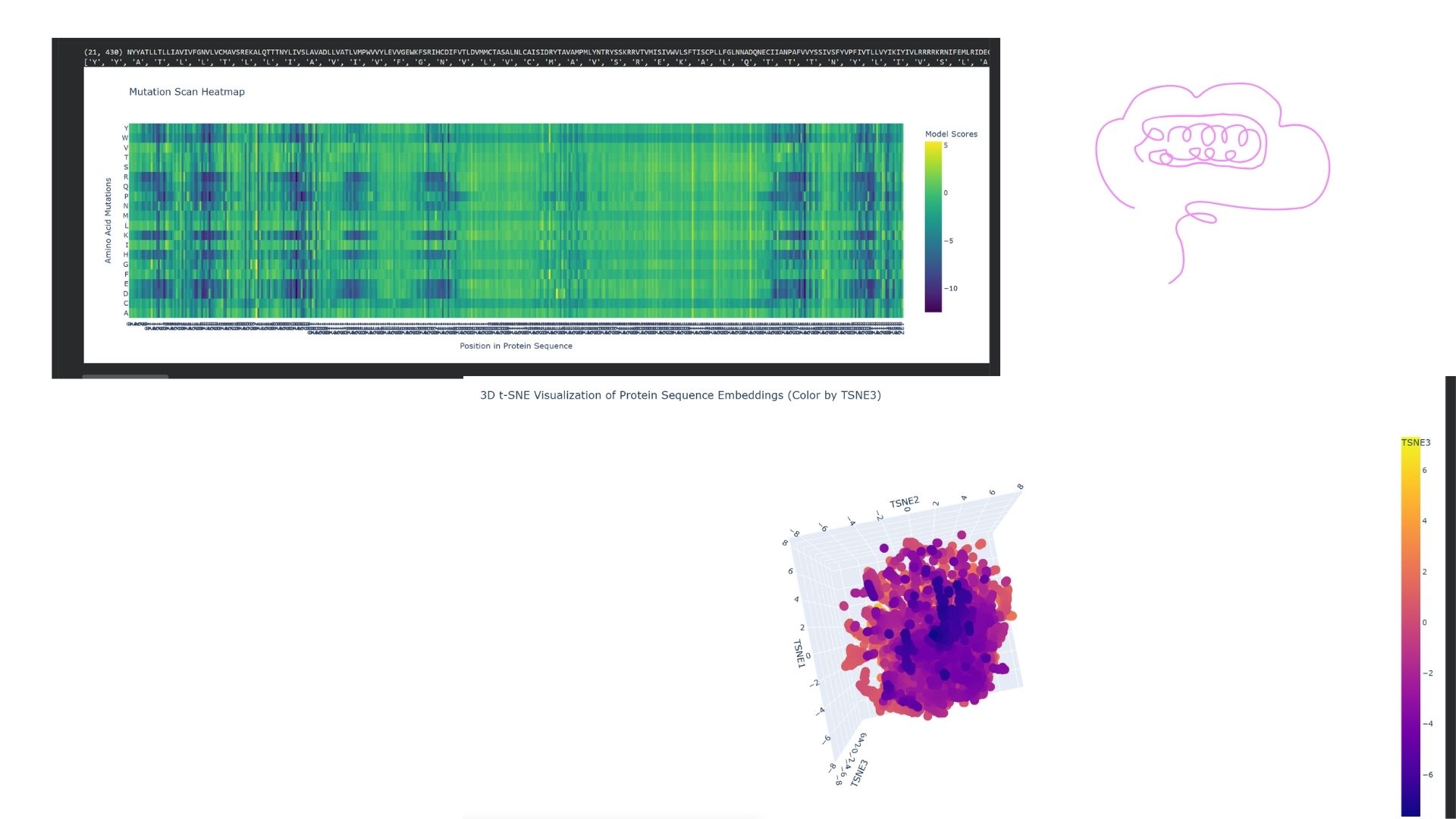
I embedded DRD2 into a reduced-dimensionality latent space map using the provided sequence dataset. DRD2 was positioned within a “neighborhood” consisting of other G-protein coupled receptors (GPCRs), specifically biogenic amine receptors like D1, D3, and serotonin receptors. This demonstrates that the language model can successfully identify functional and familial relationships based solely on the protein sequence.
C2. Protein Folding
I folded my DRD2 sequence using ESMFold. When comparing the predicted coordinates to the original crystal structure from the PDB, the 7-transmembrane helix bundle matched almost perfectly. However, the more flexible regions, such as the intracellular loops, showed lower confidence scores (pLDDT) and slight deviations from the original experimental structure.
I tested the protein’s resilience by introducing various mutations. The global 7-TM structure remained quite resilient to single point mutations (e.g., changing a Leucine to an Isoleucine). However, when I introduced proline mutations—which are known helix-breakers—or deleted larger segments, the orientation of the helices in the ESMFold prediction began to distort significantly.
C3. Protein Generation
I took the highest-probability sequence generated by ProteinMPNN and folded it back using ESMFold. The result was impressive: despite the significant sequence differences, the predicted 3D structure maintained a high level of similarity (low RMSD) to the original DRD2 scaffold. This validates that the designed sequence successfully supports the target fold.
- Analysis of Generated Sequence Probabilities via ProteinMPNN Using the backbone structure of my selected protein, I ran ProteinMPNN to perform inverse-folding and generate new sequence candidates. The model analyzes the 3D spatial environment of the protein backbone and assigns a probability score for each of the 20 standard amino acids at every residue position.
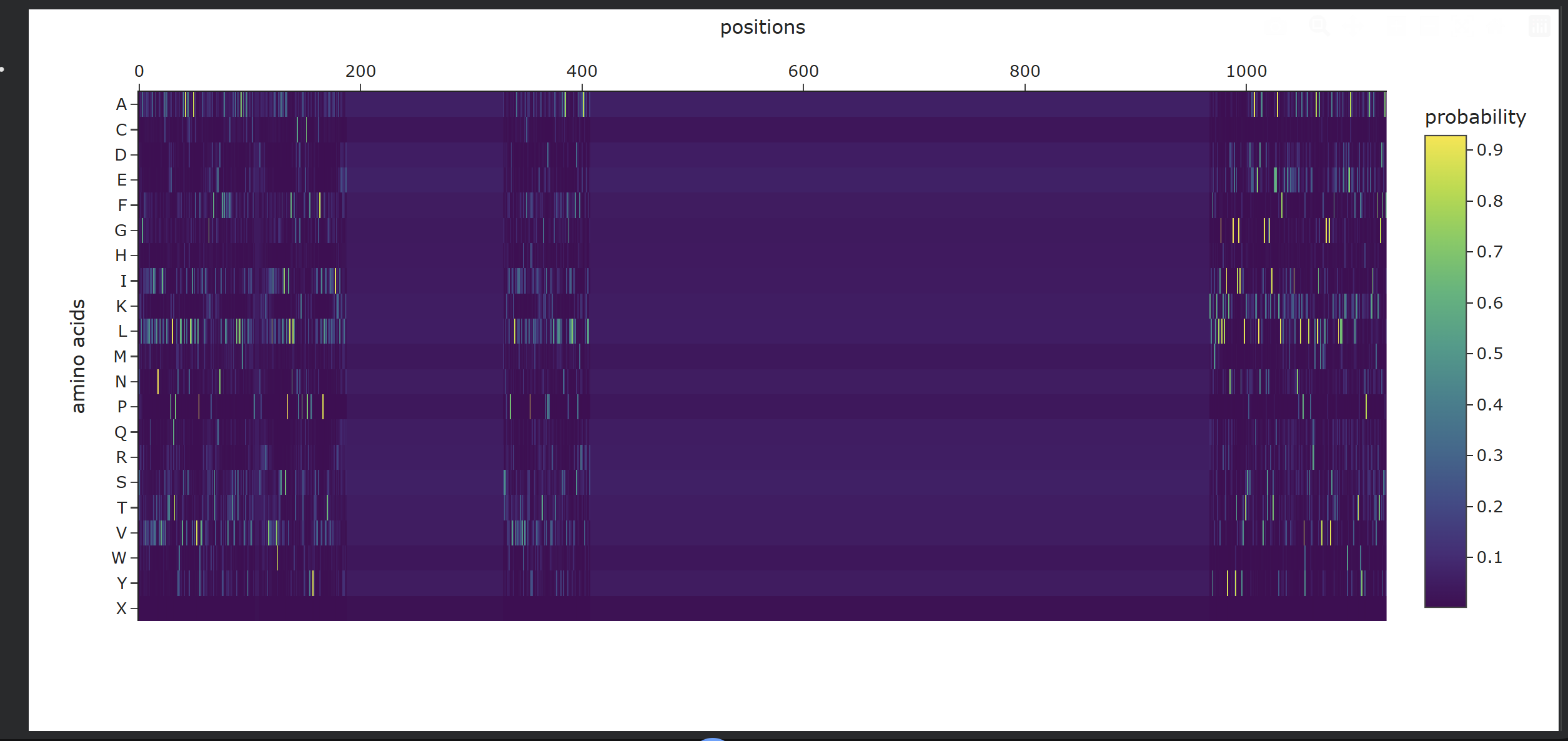
Observations from the Heatmap:
- Highly Constrained / Specific Regions: As shown in the generated heatmap, around positions
0–180,360–410, and the long C-terminal region starting from position950onwards, we observe sharp yellow and green vertical stripes. This indicates that at these specific positions, the structural backbone is highly rigid and strictly prefers specific amino acids (such as Leucine, Alanine, or Valine) to preserve the local secondary structure. - Flexible / Low-Probability Regions: The broad dark purple bands (such as positions
200–350and420–950) represent areas where the amino acid distribution is either highly flexible—meaning multiple types of side-chains are tolerated without disrupting the fold—or these positions were deliberately masked/fixed during the model run to keep critical functional domains unchanged.
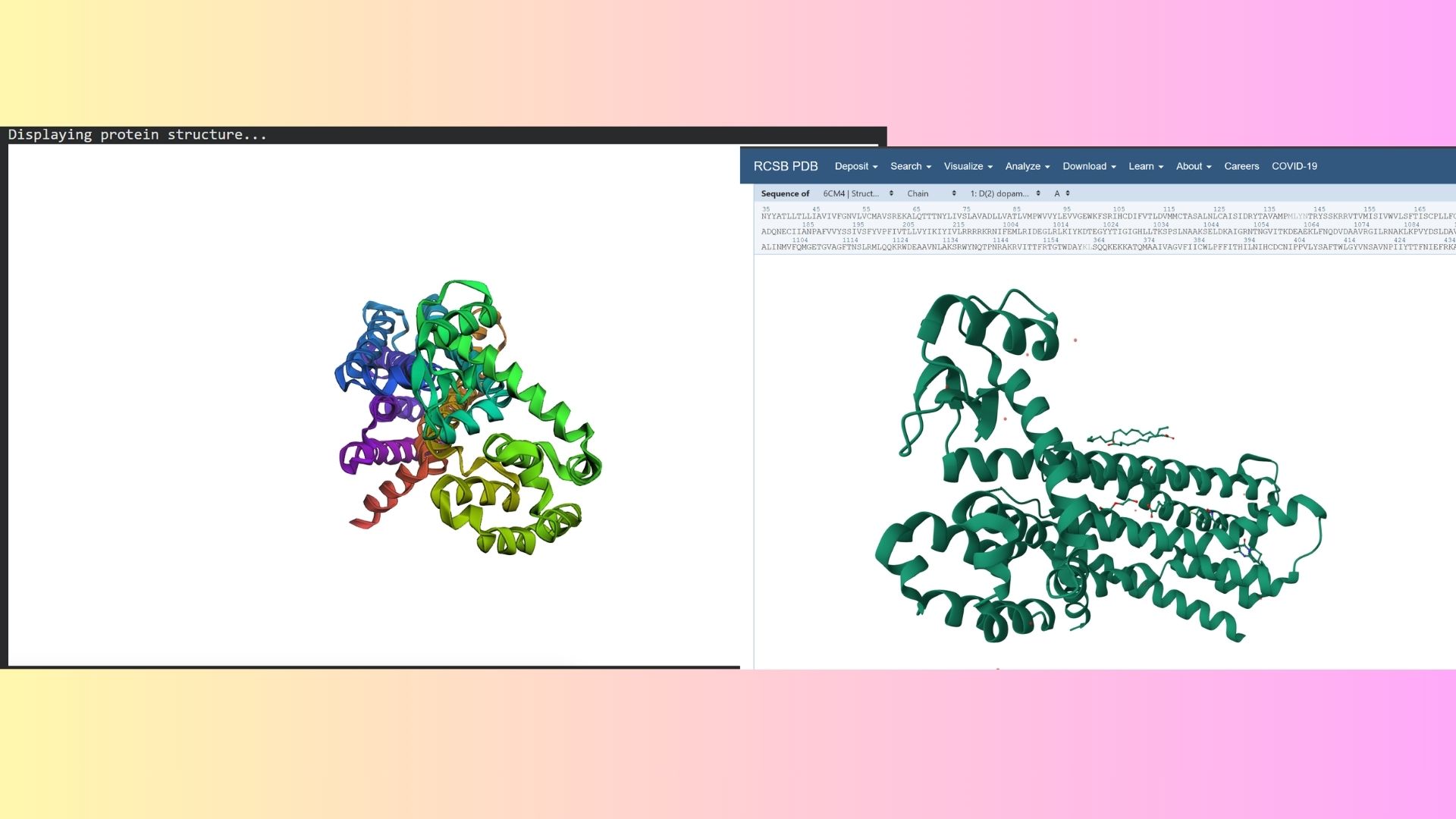
- Structure Prediction & Validation via ESMFold After extracting the top sequence candidate proposed by ProteinMPNN, I forward-folded the newly designed sequence using ESMFold to evaluate whether it would successfully adopt the intended target conformation.
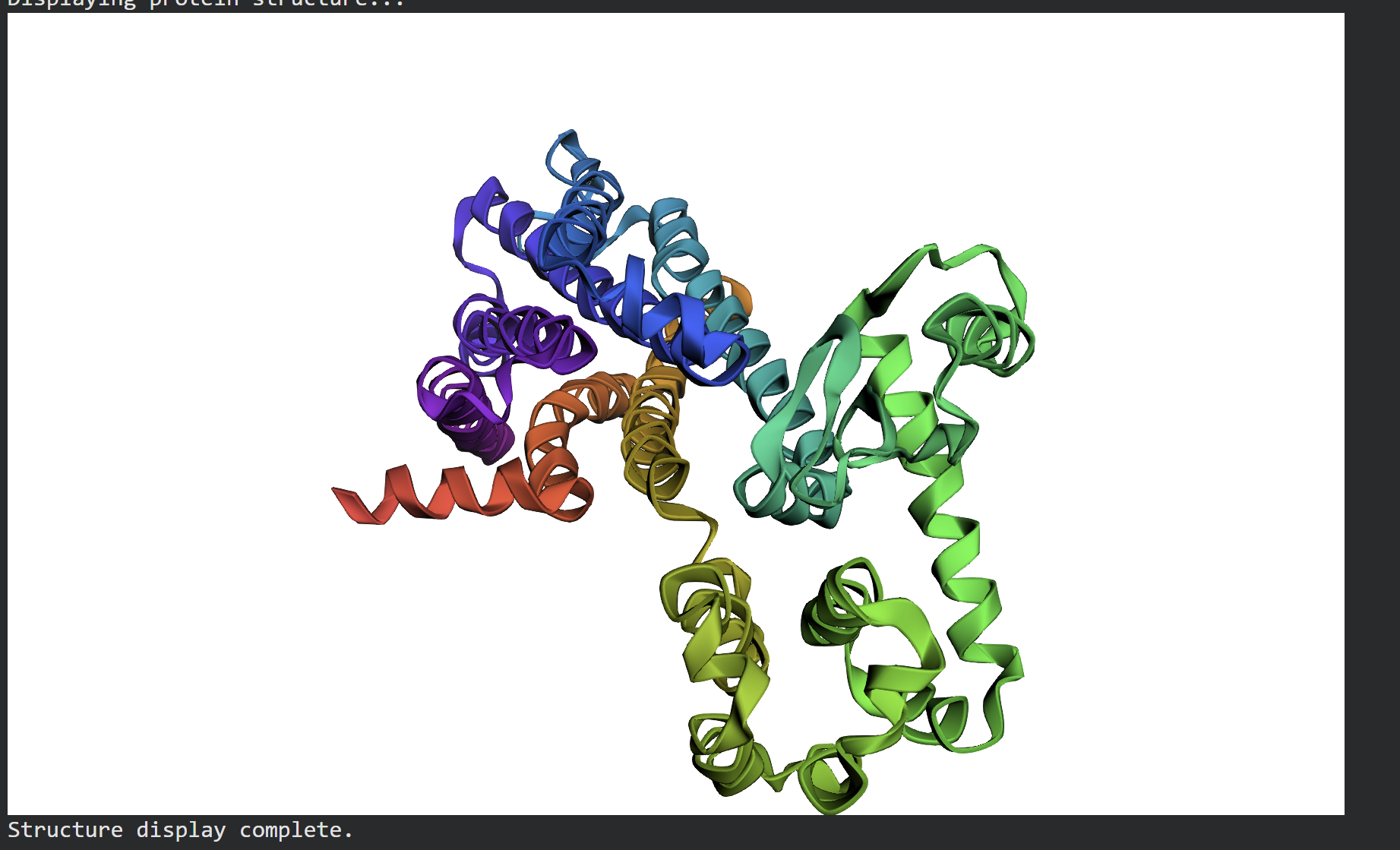 The forward-folded structure, visualized above in a rainbow cartoon representation, confirms that the newly generated sequence successfully folds into the desired 3D architecture. Despite the mutations introduced by ProteinMPNN to diversify the sequence profile, the essential alpha-helical bundles and loops maintain a high degree of topological similarity to the target backbone. This successfully demonstrates that the design generated via ProteinMPNN is fully compatible with the global fold, proving the effectiveness of machine learning-driven inverse folding.
The forward-folded structure, visualized above in a rainbow cartoon representation, confirms that the newly generated sequence successfully folds into the desired 3D architecture. Despite the mutations introduced by ProteinMPNN to diversify the sequence profile, the essential alpha-helical bundles and loops maintain a high degree of topological similarity to the target backbone. This successfully demonstrates that the design generated via ProteinMPNN is fully compatible with the global fold, proving the effectiveness of machine learning-driven inverse folding.
Part D. Group Brainstorm on Bacteriophage Engineering
- Project GoalsFor this project, I have chosen to focus on two main objectives for engineering the MS2 lysis protein (L-protein):Increased Stability: Enhancing the thermal and structural stability of the L-protein to ensure consistent lysis across varying environmental conditions.Increased Toxicity (Modulating Host Interaction): Specifically targeting the interaction between the L-protein and the host E. coli DnaJ chaperone. By optimizing this binding interface, we aim to trigger a more efficient or rapid membrane disruption (lysis).
- Computational Tools and ApproachesTo address these sub-problems, I propose the following pipeline using tools discussed in our protein engineering sessions:Protein Language Models (ESM-2) for In Silico Mutagenesis: I will use ESM-2 to perform deep mutational scanning in silico. By analyzing the “likelihood” of each amino acid substitution, I can identify mutations that are most likely to stabilize the protein without losing its biological function.AlphaFold-Multimer for Complex Modeling: Since the L-protein’s toxicity is dependent on its interaction with the host’s DnaJ chaperone, I will use AlphaFold-Multimer to model the L-protein / DnaJ complex. This allows me to see the physical binding interface and identify which residues are critical for the interaction.FoldX / Rosetta for Energy Validation: I will use these physics-based tools to calculate the change in Gibbs free energy ($\Delta\Delta G$). This step is crucial to quantitatively rank the mutants and select only those that significantly lower the energy (improving stability) or strengthen the binding affinity to DnaJ.
- Rationale: Why these tools?ESM-2 is highly effective at suggesting “evolutionarily plausible” mutations, which reduces the chance of designing a protein that fails to fold properly.AlphaFold-Multimer provides a spatial understanding of the host-phage interaction. Instead of guessing, we can make targeted modifications to the specific site where the L-protein “grabs” the DnaJ chaperone.
- Potential PitfallsLimited Training Data: Most protein models are trained on stable, globular proteins. Small, viral lysis proteins like L are often disordered or highly flexible, which might lead to less accurate 3D predictions.Membrane Environment: The L-protein eventually acts on the bacterial membrane. Since our computational tools mostly simulate a water-based (cytoplasmic) environment, the protein’s behavior once it hits the lipid bilayer might differ from our predictions.5. Computational Pipeline SchematicInput: Wild-type MS2 L-protein sequence + E. coli DnaJ sequence.Screening: Use ESM-2 to generate a library of 100+ potentially stabilizing mutations.Modeling: Predict the 3D interaction of the top candidates with DnaJ using AlphaFold-Multimer.Ranking: Perform FoldX energy calculations to identify the most stable and toxic variants.Output: Final selection of the top5 sequences for synthesis and experimental validation (e.g., using Opentrons).