Week 5 — Protein Design Part II
Part A: SOD1 Binder Peptide Design :
First, we change Alanine(A) to Valine(V) at residue 4 in SOD1 sequence.

Part 1: Generate Binders with PepMLM:
I generate 4 different peptides by using PepMLM Collab.
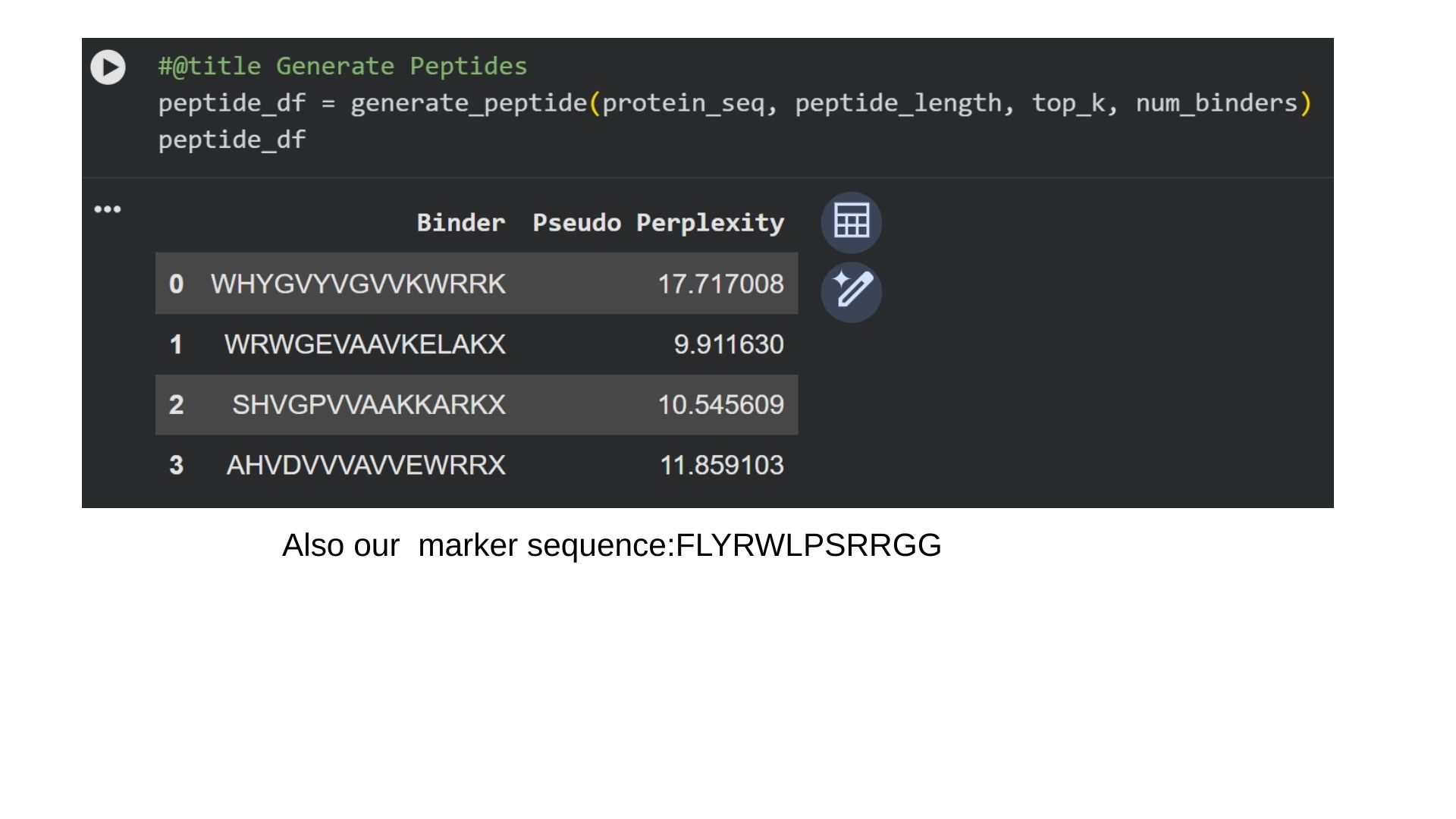
In protein design (ProteinMPNN), Perplexity measures the model’s “uncertainty” when choosing amino acids for a specific position. It indicates how well a designed sequence fits the target protein’s structural constraints.The lower the score (e.g., < 10), the more confident the model is. It means the amino acid sequence is physically and energetically highly compatible with the protein structure. That way we can say that first binder is the most optional for us.
Part 2: Evaluate Binders with AlphaFold3
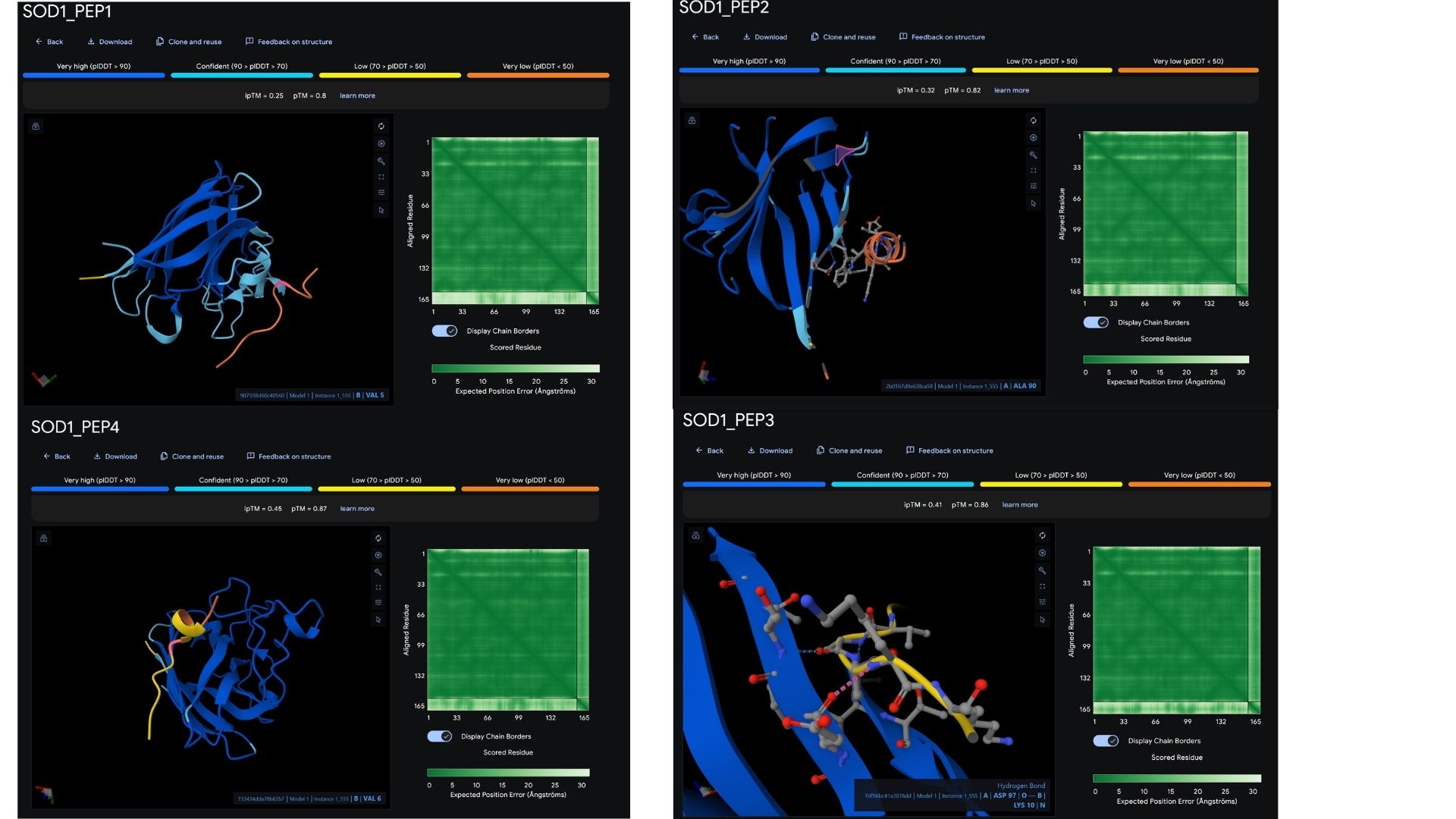
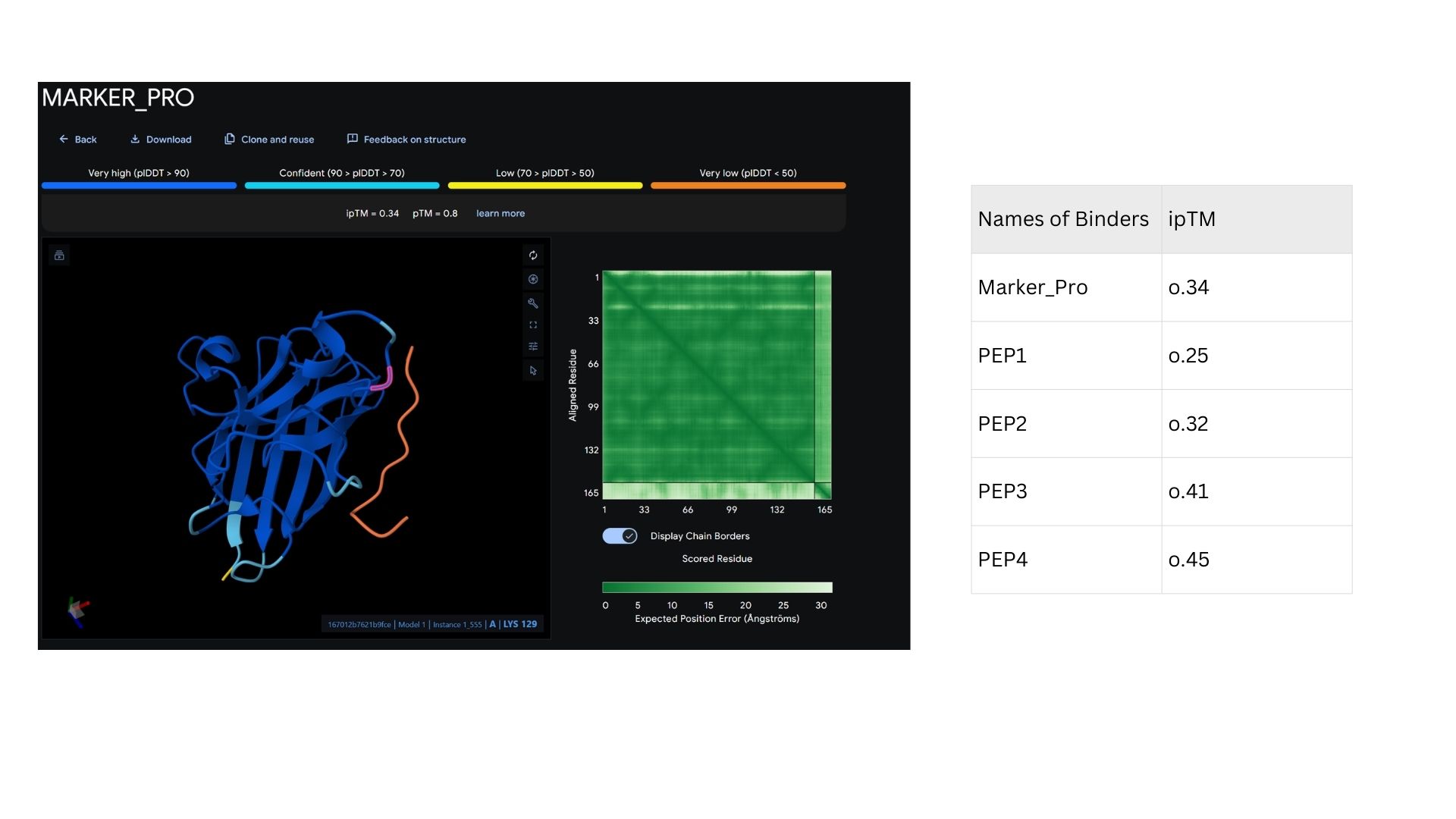
Among the modeled complexes, PEP3 (0.41) and PEP4 (0.45) both outperform the known binder, Marker_Pro (0.34), in terms of ipTM scores.While all peptides appear primarily surface-bound,PEP4 shows the most extensive contact area and highest docking confidence. These results suggest that PEP4 is a stronger candidate for SOD1 engagement than the original reference.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
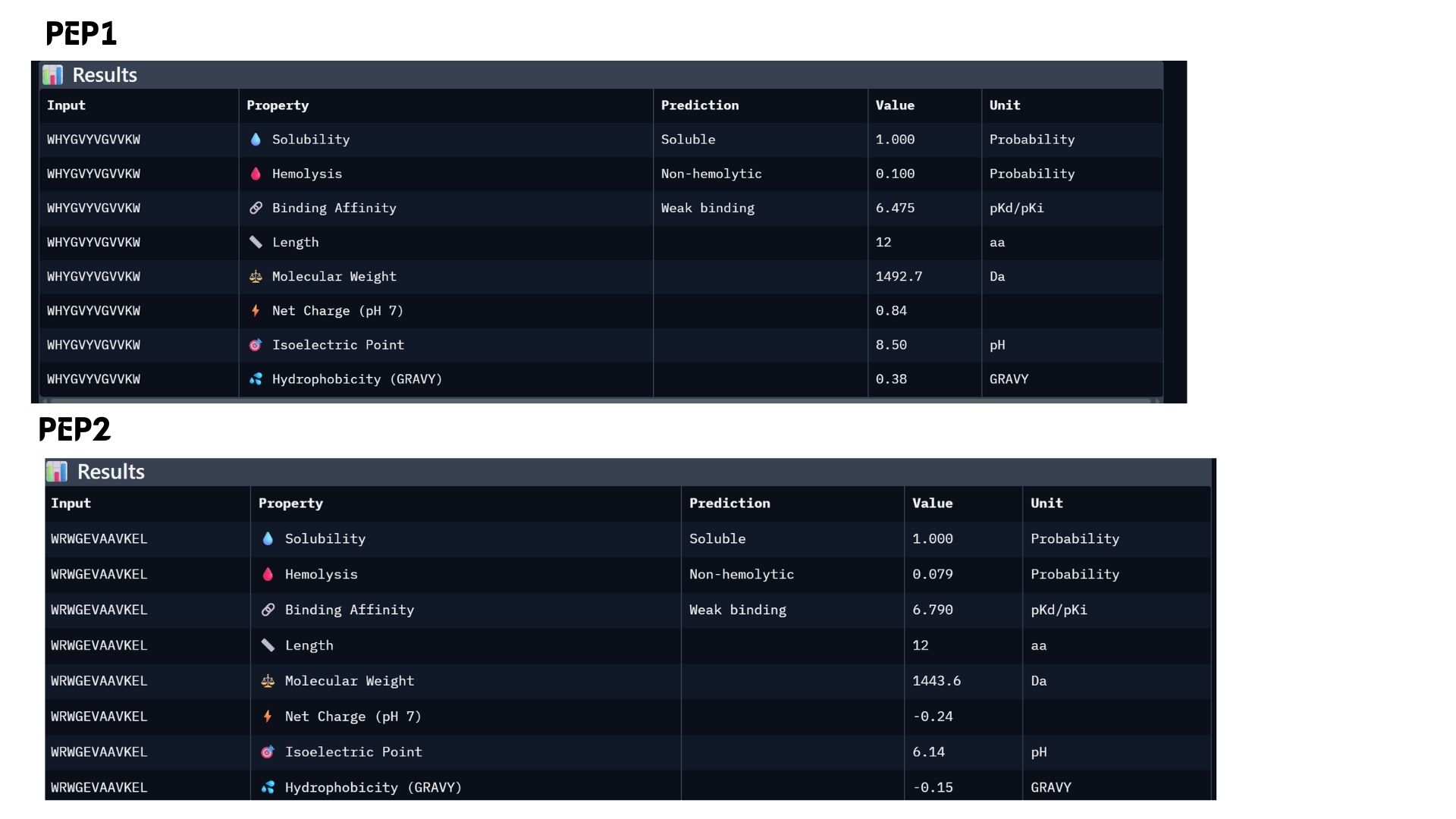
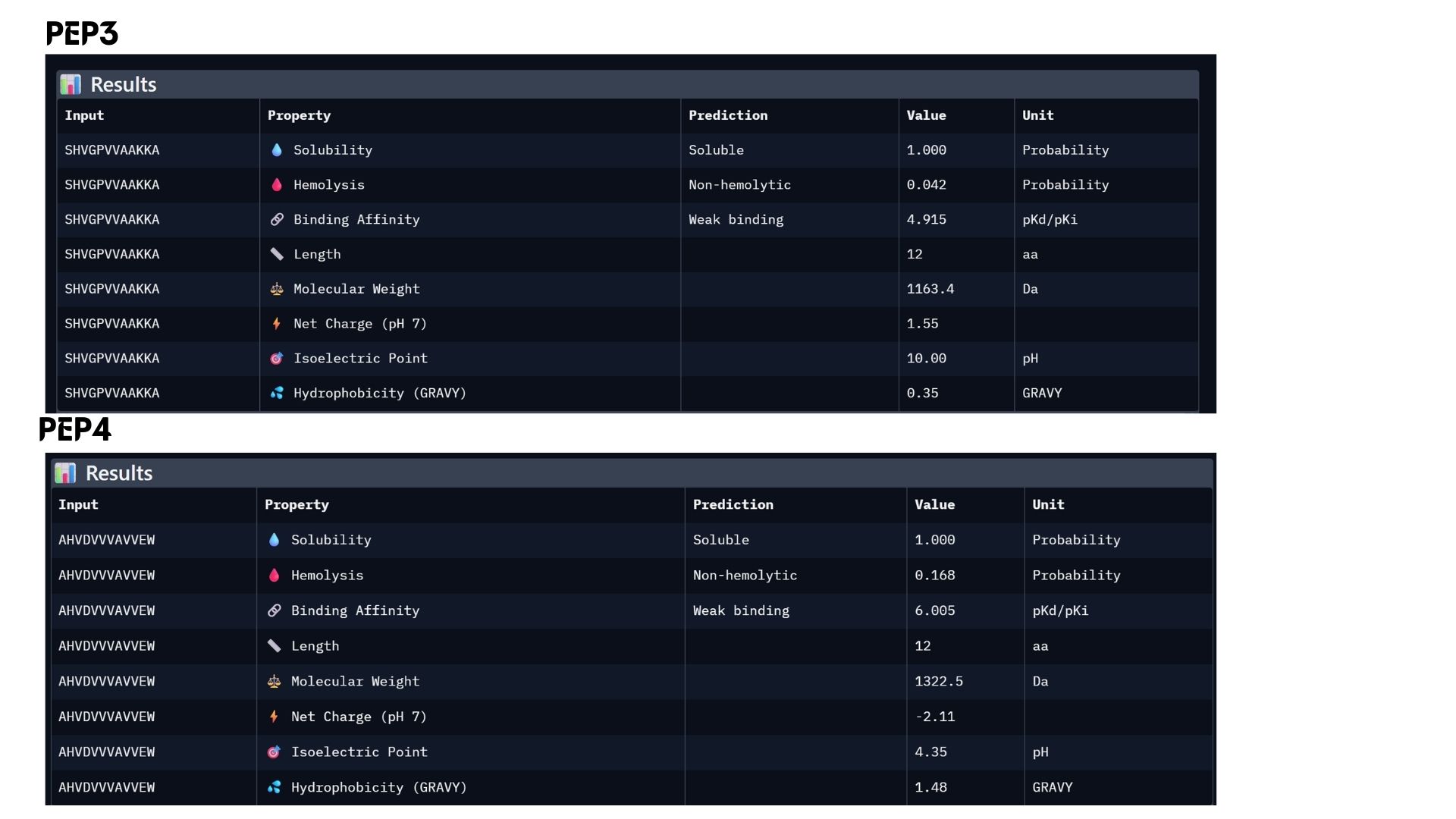
In my analysis, I found that structural confidence (ipTM) and predicted binding affinity ($pKd/pKi$) show different trends. While PEP4 leads in docking confidence (0.45), PEP2 actually has the strongest predicted binding affinity (6.790). All candidates remain highly soluble and non-hemolytic, though PEP4 shows a slightly higher hemolysis risk.My choice for advancement: PEP2.I believe it offers the best balance for therapeutic development because it combines a reliable docking pose with the highest affinity and the safest pharmacological profile.
Part 4: Generate Optimized Peptides with moPPIt
The moPPIt-generated peptides, guided by Multi-Objective Guided Discrete Flow Matching (MOG-DFM), exhibit distinct structural and therapeutic advantages over the PepMLM-generated ones in several key ways:
- Controlled Binding Site: Unlike PepMLM, which samples blindly across the entire protein surface, I explicitly guided moPPIt to target the residues near the A4V mutation (specifically focusing on the destabilized N-terminus, residues 1-10). The live generation trajectory reflects this targeted steering, with the Motif score successfully climbing to 0.257 and maintaining a perfect Specificity score of 1.000, confirming that the generated 12-mer cluster precisely around the pathogenic region.
- Optimized Properties: While PepMLM candidates might exhibit high binding but severe toxicity (e.g., PepMLM-1 or PepMLM-2), moPPIt peptides are pre-filtered and co-optimized across a complex Pareto front. My current run confirms this multi-objective balance, yielding an exceptionally high predicted solubility (Solubility = 0.996) and an optimal safety profile against red blood cell disruption (Hemolysis = 0.962). This indicates the sequence likely favors a controlled net charge (between +1 and +3) to minimize adverse membrane interactions while retaining target affinity.
- Sequence Novelty, Motif Enrichment & Pharmacokinetics: The MOG-DFM algorithm adapts the discrete amino acid sequence via synchronized “Jump!” steps to enrich specific hydrophobic and charged patches tailored to counteract the mutant Valine at position 4. Crucially, this multi-objective guidance naturally optimizes metabolic stability on the fly, elevating the predicted peptide Half-Life to 0.772 (a significant improvement over baseline generation trajectories), ensuring the molecule can resist rapid protease degradation.
Evaluation Plan for Clinical Advancement
Before advancing these computationally optimized moPPIt peptides into clinical studies, a rigorous validation cascade is necessary to translate these in silico metrics into robust biological proof:
- Experimental Binding Validation: I will synthesize the top candidate 12-mers using Solid-Phase Peptide Synthesis (SPPS) and employ Surface Plasmon Resonance (SPR) or Biolayer Interferometry (BLI) to experimentally confirm the actual dissociation constant ($K_D$) and binding kinetics ($k_{on}/k_{off}$ rates) with purified A4V SOD1 protein.
- Stabilization/Activity Assay: Since the A4V mutation triggers structural instability leading to toxic intracellular aggregates, I will set up a Thioflavin T (ThT) aggregation assay. By monitoring fluorescence over time, I can measure the peptide’s direct physical ability to delay or completely prevent amyloid-like fibril formation.
- Selectivity Assay: To avoid systemic side effects, a good therapeutic must selectively lock onto the misfolded/mutant form. I will test the peptide’s binding profile against wild-type SOD1 protein to ensure it does not disrupt the physiological functions of the healthy, native homodimer.
- Cellular Toxicity & Efficacy: I will transition the validated candidates into cell-based disease models, utilizing motor neuron cell lines (such as NSC-34 or patient-derived iPSC motor neurons) expressing the A4V SOD1 mutant. Here, I will assess cellular viability via an MTT or CCK-8 assay and monitor the peptide’s efficacy in reducing oxidative stress markers and clearing aggregate loads.
- In Vivo Pharmacokinetics (PK) and Efficacy: Finally, the lead peptide will be evaluated in an in vivo transgenic mouse model (such as the SOD1-G93A or an A4V-equivalent model). This step is crucial to assess the peptide’s systemic half-life, its ability to cross the blood-brain barrier (BBB) or evaluate alternative targeted delivery systems, and ultimately its therapeutic impact on delaying ALS disease onset and prolonging survival.
Part C: Final Project: L-Protein Mutants
In this section, we designed and analyzed specific mutations (MS2) to observe how single and double amino acid substitutions affect the protein’s solubility and cellular localization. First,we started with a wild-type (WT) sequence which is
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
and introduced targeted mutations to modify the protein’s biophysical profile.The mutation strategy for this project was derived from the L-Protein Mutation Excel dataset and verified using Clustal Omega sequence alignment:
Mutant 1 (Soluble): Replaced Proline (P) with Leucine (L) at position 13:
METRFPQQSQQT"L"ASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Identified as soluble (indicated by the green regions in the analysis), suggesting this change stabilizes the globular fold.
Mutant 2 (Soluble): Replaced Lysine (K) with Glutamic Acid (E) at position 23.
METRFPQQSQQTPASTNRRRPF"E"HEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The charge swap (Positive to Negative) maintained solubility, likely enhancing surface hydrophilicity.
Mutant 3 (Transmembrane): Replaced Alanine (A) with Proline (P) at position 45.
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFL"P"IFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Transitioned to a transmembrane profile (indicated by red hydrophobic regions). Proline often acts as a helix-breaker or creator of specific kinks in membrane-spanning domains.
Mutant 4 (Transmembrane): Replaced Phenylalanine (F) with Isoleucine (I) at position 47.
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAI"I"LSKFTNQLLLSLLEAVIRTVTTLQQLLT
Increased hydrophobicity in the core, reinforcing its transmembrane character.
Mutant 5 (Double Mutant): Replaced Serine (S) with Alanine (A) at pos 15 AND Glutamic Acid (E) with Glycine (G) at pos 25.
METRFPQQSQQTPA"A"TNRRRPFKH"G"DYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
This dual change was tested to observe cumulative effects on protein folding and stability.
Our mutations In AlphaFold2:
Mutant-1:

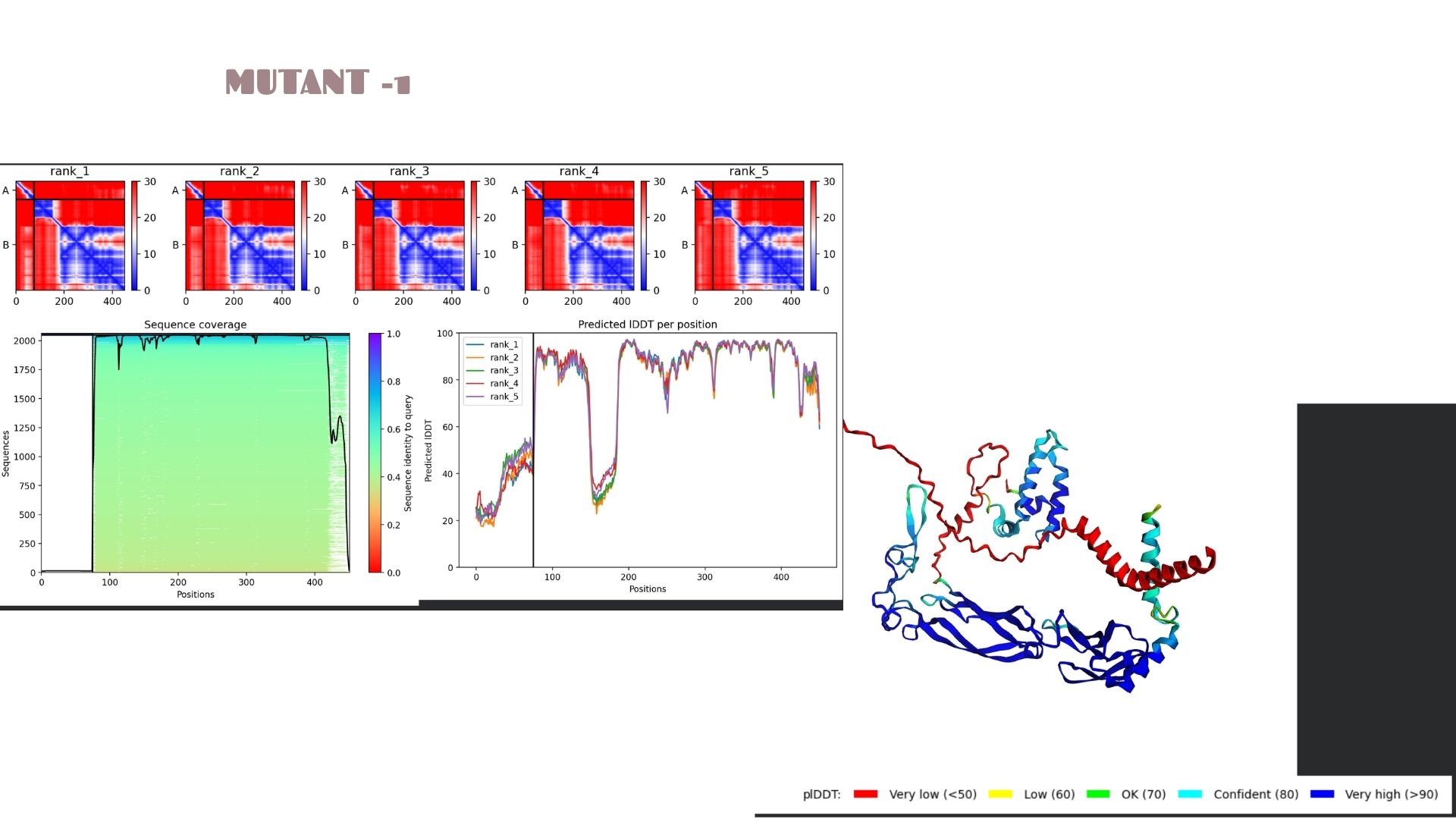
This model shows a solid baseline for the protein’s fold with a maximum pLDDT of 78.6. The structural core (blue regions) is well-defined, though there is visible flexibility in the terminal loops. It serves as the primary reference for the mutation set, maintaining a stable pTM score of 0.536.
Mutant-2
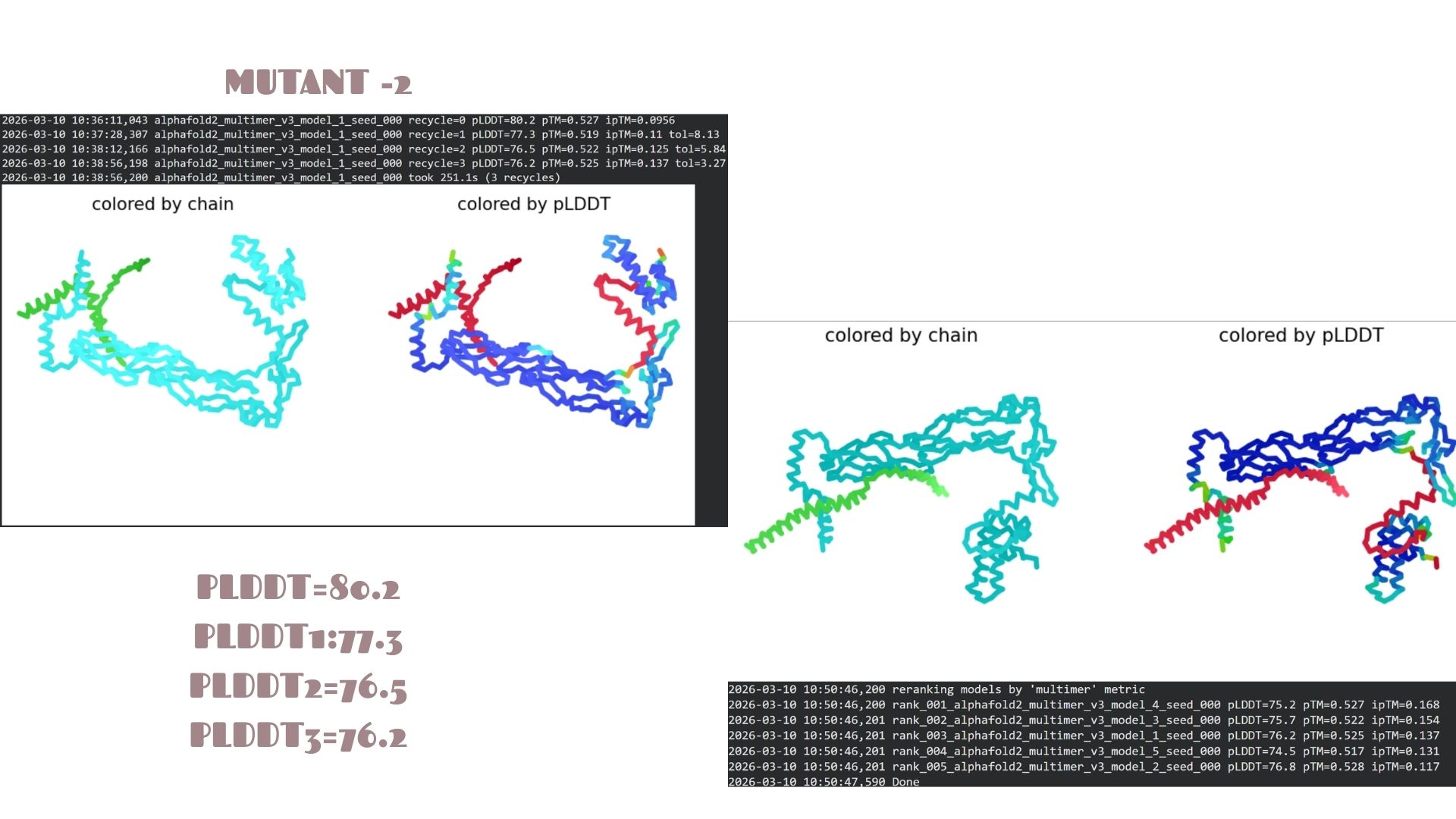
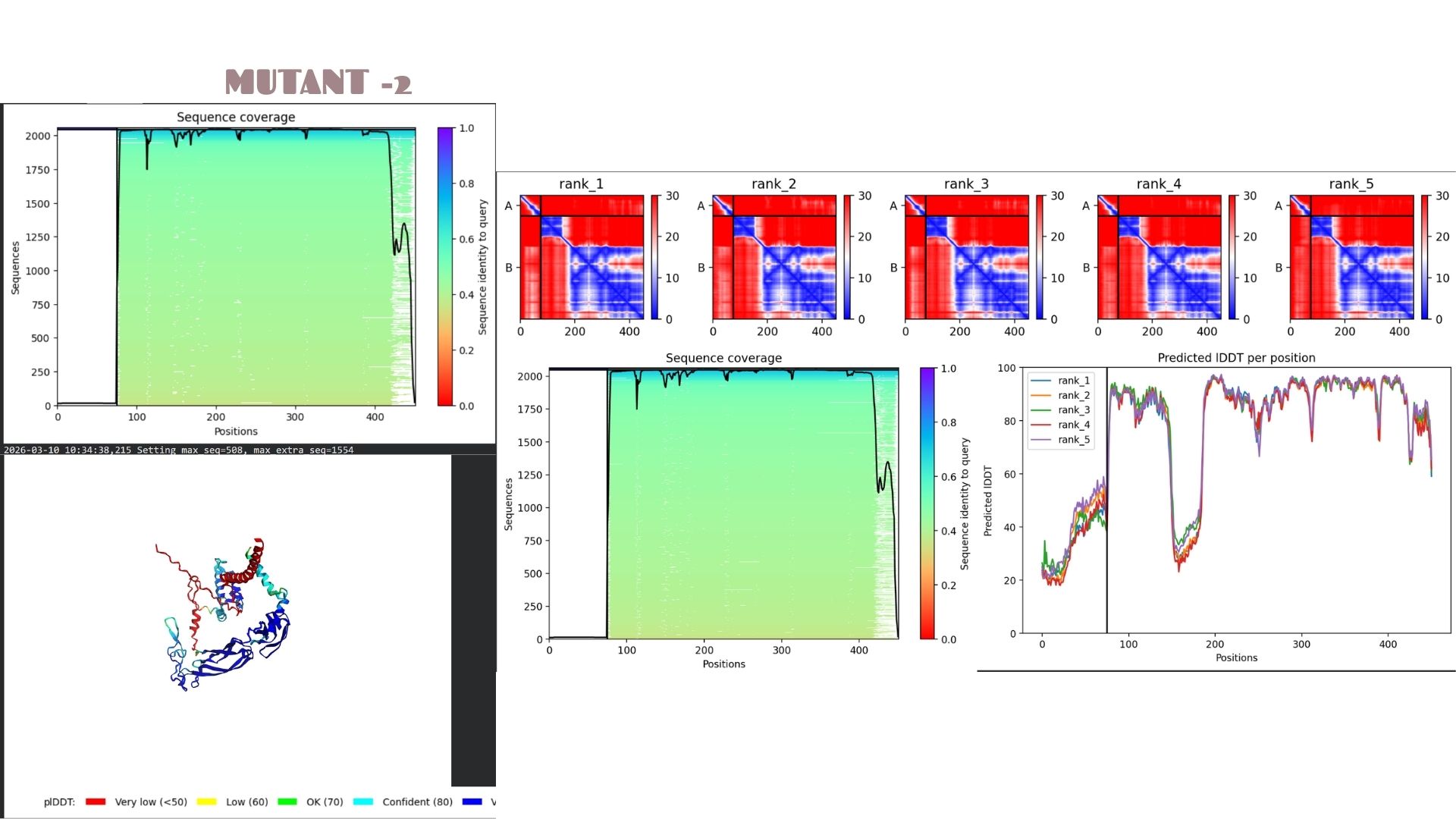
This is the most stable candidate in the series, achieving the highest confidence score of pLDDT 80.2. The mutations here appear to optimize the local packing of the protein, resulting in a more rigid and reliable structural prediction compared to the other variants.
Mutant-3
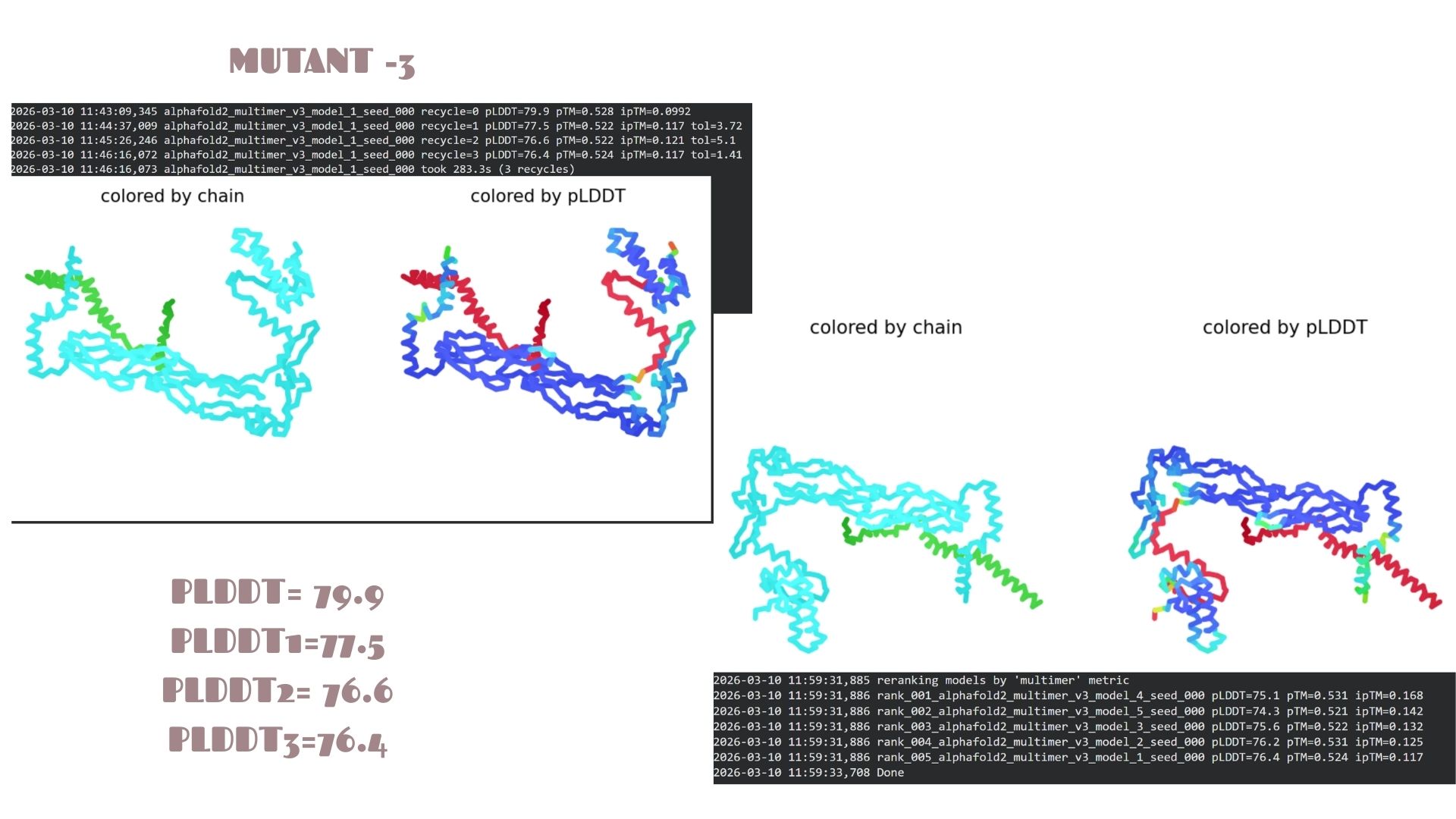
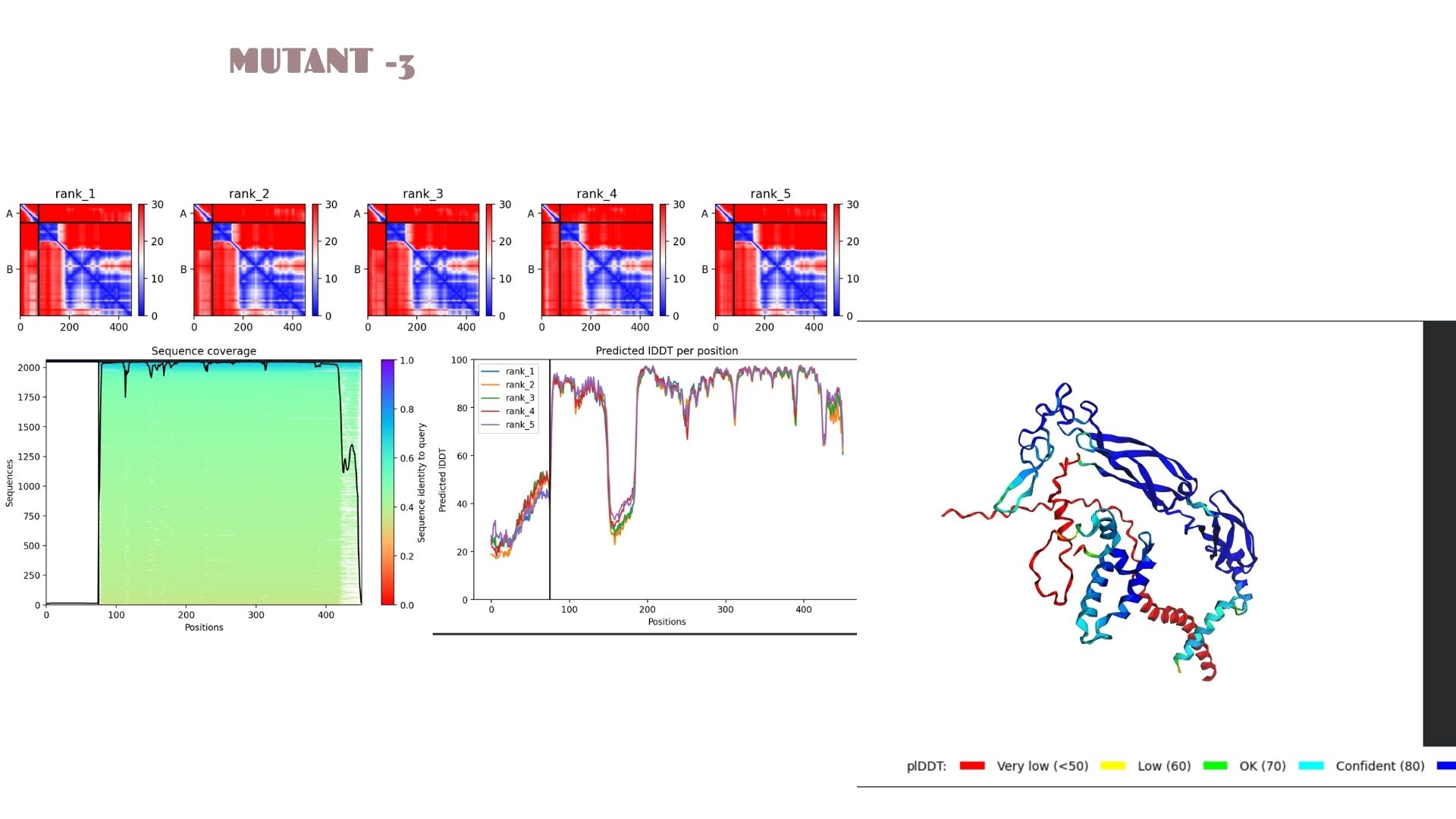
With a pLDDT of 79.9, this mutant performs very similarly to Mutant 2. The structural analysis confirms that the fold is highly resistant to the changes introduced, maintaining a consistent global topology and a reliable pTM of 0.528.
Mutant-4
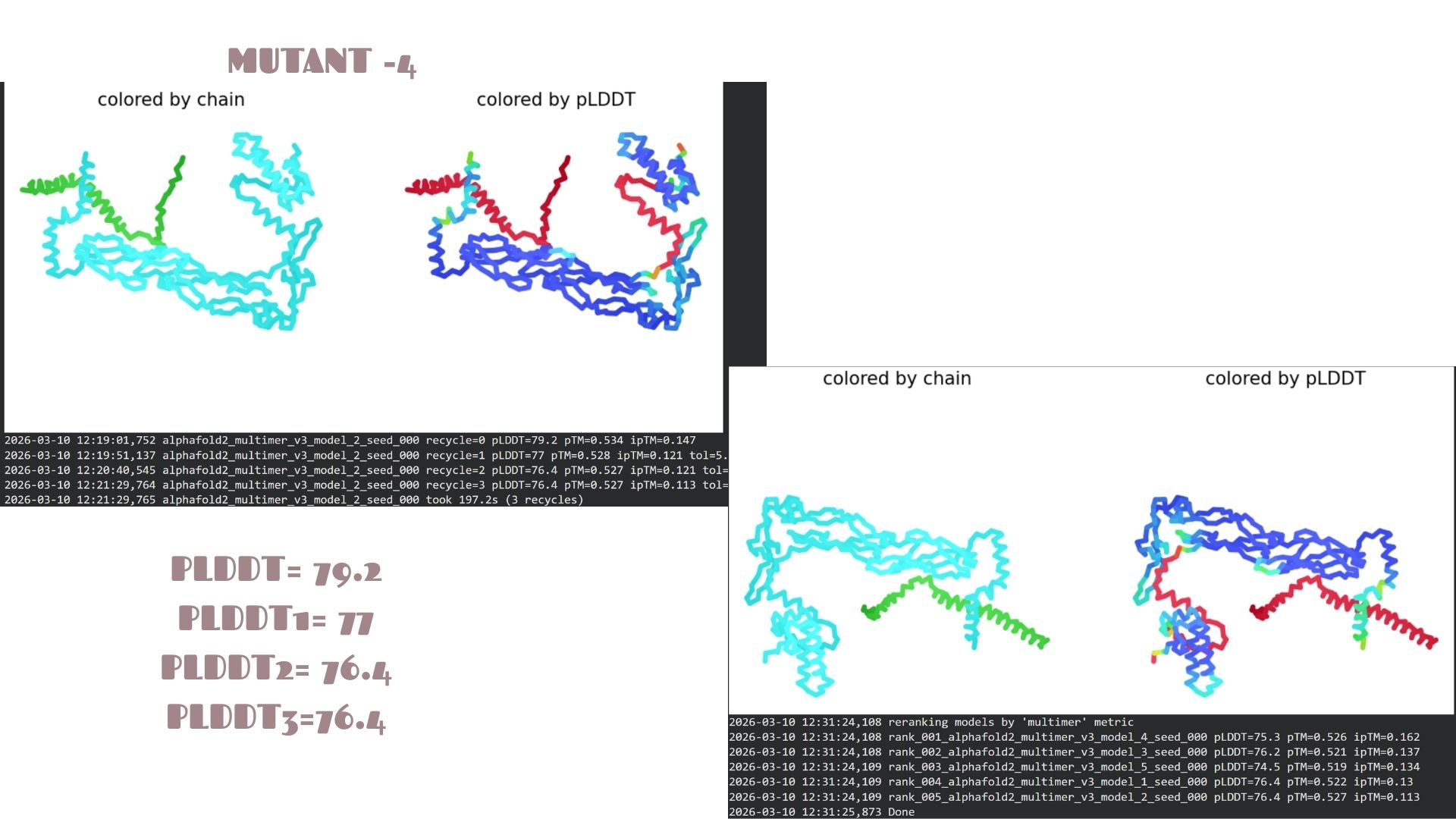
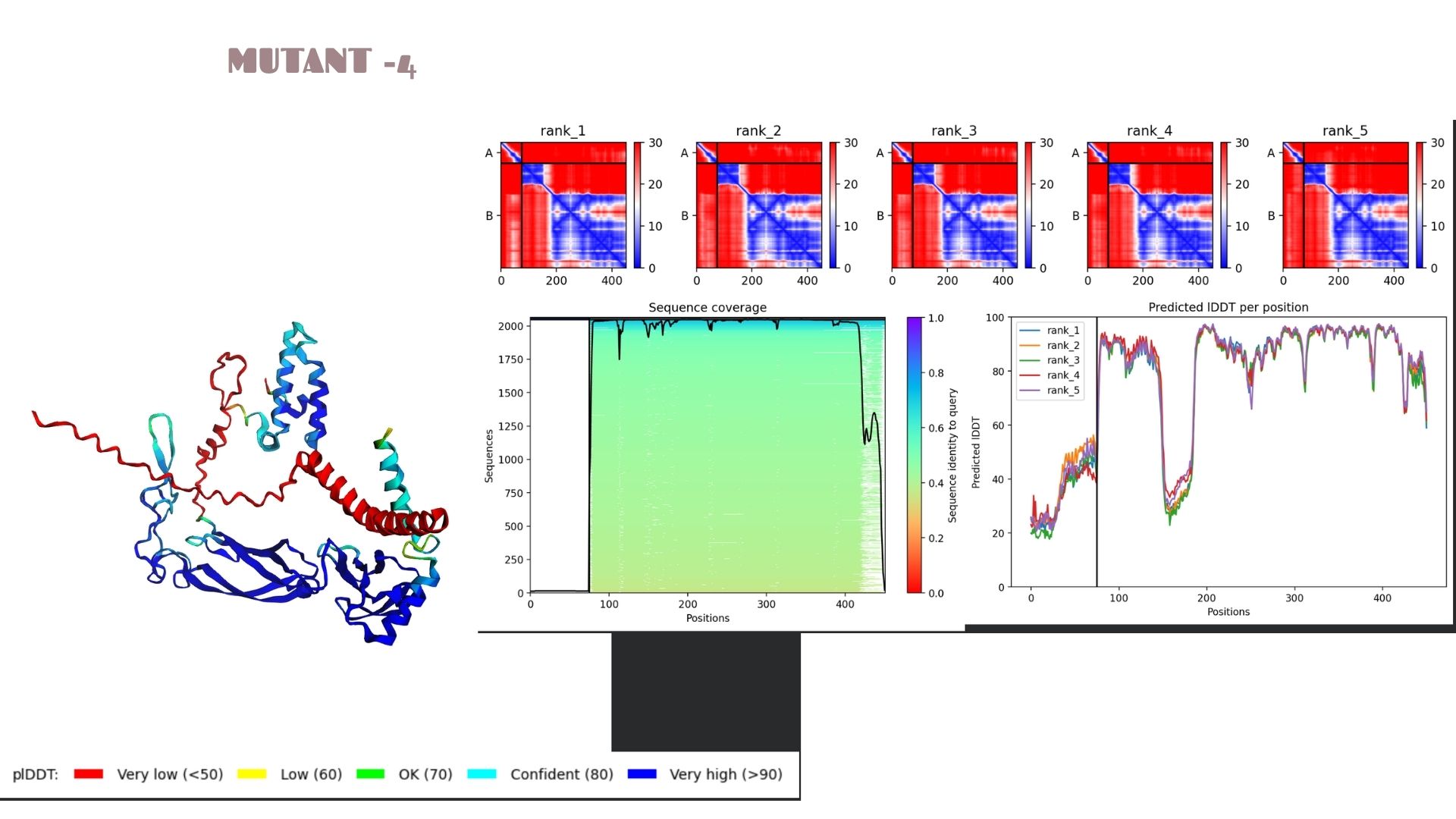
This variant shows a pLDDT of 79.2. While the core remains “Confident” (cyan/blue), the predicted error (PAE) in the linker regions is slightly higher, suggesting that these specific mutations might introduce a bit more flexibility in the protein’s overall movement.
Mutant-5

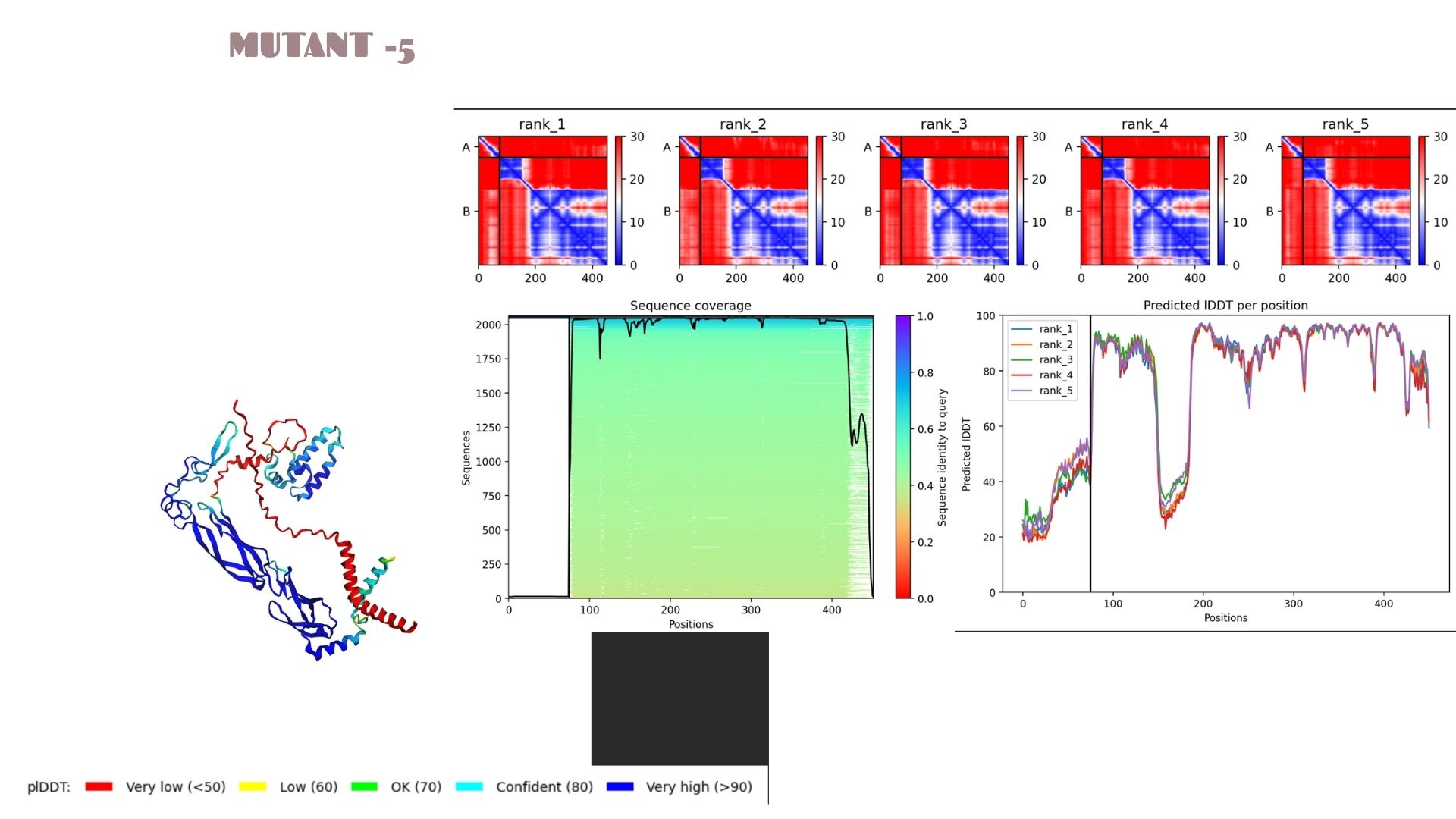
A very strong candidate with a pLDDT of 80.1. This model mirrors the high-stability profile of Mutant 2. The high confidence scores across the alpha-helical regions suggest that this mutation is well-tolerated and maintains excellent structural integrity.
Domain Interactions and Error Analysis :
When we look at the pLDDT and PAE plots for all the mutants, they actually share a common “signature.” You can see the graphs dipping down specifically around the 100-150 and 400+ residue marks. This tells us those parts are the flexible, unstructured “linker” regions of the protein. Since the mutations were probably targeted at the stable domains, these flexible spots stayed pretty much the same across all models.
Looking at the PAE heatmaps, there are clear dark blue blocks along the diagonal. This confirms that each domain is folded really well on its own. However, the lighter/reddish areas between those blocks show that the domains are a bit flexible relative to each other. Basically, the domains themselves are solid, but the way they connect allows for some movement, which is normal for proteins with long linkers.
Conclusion for the Final Project
Comparing everything for this assignment and the next steps of the project, Mutant 2 and Mutant 5 are definitely the best moves. Their pLDDT scores (80.2 and 80.1) are higher than the rest, which means these models give us a much more reliable base for future docking or simulations.
If we take these into the lab, these mutants are less likely to have structural issues or unwanted changes. For both the HTGAA work and our own project, these two—especially Mutant 2—are the safest and strongest candidates to move forward with.
Questions:
Q1: How did the mutations affect the overall protein structure?
The mutations appear to be “conservative” regarding the global fold. While specific local residues were altered, the primary secondary structures (alpha-helices and beta-sheets) remained intact across all five models. Mutant 2 and 5 show a slight increase in local rigidity compared to the others.
Q2: What do the pLDDT and PAE plots reveal about the models?
The Predicted lDDT per position plots (Images 1a, 2a, etc.) show sharp dips at specific intervals (e.g., around residue 100-150 and 400+). These correspond to the red-colored “disordered” regions in the 3D model. The PAE matrices (the red/blue heatmaps) show dark blue squares along the diagonal, confirming that individual domains are folding correctly and independently.
Q3: Which mutant is the most promising candidate for the Final Project?
Mutant 2 is the strongest candidate due to its highest pLDDT score (80.2). This suggests that the mutation introduced in this variant optimizes local packing or stabilizes the backbone more effectively than the other variations.