Week 4 HW: Protein Design I
Part A
How many molecules of amino acids do you take with a piece of 500 grams of meat?
Let’s assume that meat is on average 20% protein. so 500g of meat would mean 100g of protein.
After calculations we can assume that 1Da/molecule = 1g/mol, so by having around 100 Da on a amino acid we can say that we have approximatelly 1 mol of amino acid residue in 500g of meat.
Therefore in total we have 6.022*10^23 amino acid molecules.
Why are there only 20 natural animo acids?
DNA uses triplet codons so there are 64 possible codons. Out of these, 3 are stop codons so there remain only 61 available code for amino acids. These 61 codons map redundantly to 20 core amino acids. Early life likely used fewer than 20. As new amino acids were gradually incorporated, the code expanded—then “froze.” Once the translation machinery (ribosomes, tRNAs, enzymes) became complex, changing the code would have been catastrophic. So evolution locked in the set. It’s just like a code that wants to become more and more complex but instead of adding new data types, they are forced to use already existing data types to form something else which would most likely be useless and would just become more difficult to use.
Also each amino acid uses a specific tRNA and a matching animoacyl-tRNA synthetase enzyme, so adding new ones require an even more complex and risky path to not accidentally cause cross-reactions between them.
The already existing 20 amino acids can achieve hydrophobic cores, charged interactions, structural rigidity, a stable 3D structure after folding.
Can you make other non-natural amino acids? Design some new amino acids.
Yes you can design non-natural amino acids and scientist have already done so. We can design new ones by modifying the side chain while keeping the core amino acid backbone: NH₂–CH(R)–COOH. Instead of R I can place something else.
For example, by replacing one single methyl group with a trifluoromethyl group in Leucine (R = –CH₂–C(CH₃)(CF₃)) we will obtain a superhydrophonic core builder that is more resitant to oxidation.
Another example would be an nnAA that changes shape when exposed to light allows light-controlled protein folding. We can obtain this by replacing the side chain with an anzobenzene group (R = –CH₂–C₆H₄–N=N–C₆H₄).
Where did amino acids come from before enzymes that make them, and before life started?
The first thing we need to understand is that amino acids can form abiotically, they can be synthesized through chemical reactions under certain conditions, whithout needing enzymes or life. Some of the key ways scientists think amino acids could have formed before life include Miller-Urey type synthesis (simulated early Earth’s atmosphere and electric sparks and managed to obtain several amino acids), hydrothermal vents (Dee-sea hydrothermal vents release hot, mineral-rich water that can catalyze chemical reactions to produce amino acids from simpler molecules like CO₂, H₂, and NH₃) and extraterrestrial delivery (amino acids found in meteorites).
Enzymes are proteins that can catalyze reactions in life, but this doesn’t mean that reactions don’t work without them. They still do, slower and less specific, but under the right conditions they can still happen.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A D-amino acid is the mirrored version of an L-amino acid, so the α-helix headedness of a D-amino acid will be the opposite of one of a L-amino acid.
Since almost all amino acids in natural protein are L-amino acids we can understand easier how they form.
The α-helix is defined by specific torsion angles (ϕ and ψ) along the peptide backbone:ϕ (phi) = rotation around N–Cα bond and ψ (psi) = rotation around Cα–C bond. For a helix to form, the backbone must adopt a set of angles that allows regular hydrogen bonding between the carbonyl oxygen of residue i and the amide hydrogen of residue i+4. The specific geometry of L-amino acids makes this hydrogen-bonding network more favorable in a right-handed twist.
L-amino acids have the Cα in the S configuration (except glycine). When you try to twist the backbone into a helix a right-handed twist positions the side chains away from the helix axis without clashing with the backbone. On the other hand, a left-handed twist would force side chains into steric clashes with the backbone or the other side chains. A left-handed helix is possible but it will require a lot more energy to form.
So L-amino acids form right-handed helices, so if we mirror it we can say that R-amino acids form left-handed helices
Can you discover additional helices in proteins?
Yes you can discover additional helices. An α-helix is a secondary structure element characterized by: Backbone hyrogen bonds (between carbonyl oxygen of residue i and the amide hydrogen of residue i+4), Specific φ and ψ angles, and right-handed twist. If a stretch of amino acids satisfies these geometric and hydrogen-bonding criteria, it can be classified as an α-helix.
There are several ways to identify α-helices in proteins:
X-ray crystallography & cryo-EM
- These methods give atomic-resolution structures
- You can look at the backbone position and see repeating helical patterns.
Software and algorithms
- Like DSSP, STRIDE and PyMol
Sequence prediction
- Certain amino acids have high helix-forming propensity
- Predictive algorithms like PSIPRED or JPred can guess where helices are based on sequence alone.
There also exist other types of helices like 3₁₀ helices (tighter, 3 residues per turn) and π-helices (looser, ~4.4 residues per turn)
Why are most molecular helices right-handed?
Almost all natural amino acids are L-amino acids. I’ve talked a little bit earlier about how do L-amino acids make right-handed helices. Shortly, when the peptide chain coils to form an α-helix, the side chains naturally point outward, and the backbone hydrogen-bonding pattern is energetically most favorable when the helix is right-handed.
Most molecular helices are right-handed because the chirality of their building blocks makes right-handed twisting energetically favorable, avoiding steric clashes and stabilizing hydrogen bonds.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
First of all let’s see what a β-sheet. A β-sheet is a common secondary structure in proteins, formed by β-strands linked by hydrogen bonds. Some characteristics of β-sheet are Backbone hydrogen bonding (Between the C=O of one strand and the NH of another strand), side chain orientation (Side chains alternate above and below the sheet) and its arrangement (parallel β-sheet strands run in the same N→C direction, antiparallel β-sheet strands run in opposite directions).
β-sheets have an inherent tendency to stack into extended, layered structures, and this is driven by several forces:
- Hydrogen bonding:
- Exposed backbone NH and C=O groups at the edges of β-sheets can form additional hydrogen bonds with other β-sheets. This allows β-sheets to “zipper” together, forming extended sheets.
- Hydrophobic interactions:
- Side chains alternate above and below the sheet.
- Hydrophobic side chains on one face can pack against hydrophobic side chains of another sheet.
- This minimizes exposure to water, driving aggregation.
- Van der Waals forces:
- Close packing of side chains in stacked β-sheets leads to favorable van der Waals contacts, further stabilizing aggregates.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloid disease involve proteins that misfold and aggregate into β-sheet–rich fibrils.
The backbone always wants to form hydrogen bond. Every protein backbone has repeating carbonyl and amide groups. If these groups are not satisfied by internal folding, they seek hydrogen bonds elsewhere.
When a protein partially unfolds, its backbone becomes exposed and neighbouring molecules can form hydrogen bonds with it and it forms a cross-β structure (since the easiest repeating pattern for intermolecular H-bonding is a β-sheet alignment), the hallmark of amyloid fibrils.
Another factor could be the fact that β-sheets allow infinite extension. Unlike α-helices, that are stabilized within 1 molecule, β-sheets cam form between different molecules, in an extended reapeting way, without needing complex folding. This means they can stack and grow into long fibrils.
Also, amyloid β-sheets have high thermodynamic stability and are often more stable than the original folded protein. So under stress, mutation or aging, proteins can fall into deep energy minimum.
Amyloid β-sheets can be used as materials due to their mechanical strength and self-assembly mechanisms. There are some applications being explored right now, such as tissue engineering scaffolds, drug delivery systems and sustainable biomaterials.
Part B
chosen protein: Elastin
Elastin is a protein encoded by the ELN gene in humans and several other animals. Elastin is a key component in the extracellular matrix of jawed vertebrates. It is highly elastic and present in connective tissue of the body to resume its shape after stretching or contracting. Elastin helps skin return to its original position whence poked or pinched. Elastin is also in important load-bearing tissue of vertebrates and used in places where storage of mechanical energy is required.
I was done with fibroin and silk, but I wanted to keep a sort of continuity between my proteins so I chose Elastin due to its stretching functionality.
Here is the elastin sequence of the human body found on Uniprot:
sp|P15502|ELN_HUMAN Elastin OS=Homo sapiens OX=9606 GN=ELN PE=1 SV=4 MAGLTAAAPRPGVLLLLLSILHPSRPGGVPGAIPGGVPGGVFYPGAGLGALGGGALGPGGKPLKPVPGGLAGAGLGAGLGAFPAVTFPGALVPGGVADAAAAYKAAKAGAGLGGVPGVGGLGVSAGAVVPQPGAGVKPGKVPGVGLPGVYPGGVLPGARFPGVGVLPGVPTGAGVKPKAPGVGGAFAGIPGVGPFGGPQPGVPLGYPIKAPKLPGGYGLPYTTGKLPYGYGPGGVAGAAGKAGYPTGTGVGPQAAAAAAAKAAAKFGAGAAGVLPGVGGAGVPGVPGAIPGIGGIAGVGTPAAAAAAAAAAKAAKYGAAAGLVPGGPGFGPGVVGVPGAGVPGVGVPGAGIPVVPGAGIPGAAVPGVVSPEAAAKAAAKAAKYGARPGVGVGGIPTYGVGAGGFPGFGVGVGGIPGVAGVPGVGGVPGVGGVPGVGISPEAQAAAAAKAAKYGAAGAGVLGGLVPGAPGAVPGVPGTGGVPGVGTPAAAAAKAAAKAAQFGLVPGVGVAPGVGVAPGVGVAPGVGLAPGVGVAPGVGVAPGVGVAPGIGPGGVAAAAKSAAKVAAKAQLRAAAGLGAGIPGLGVGVGVPGLGVGAGVPGLGVGAGVPGFGAGADEGVRRSLSPELREGDPSSSQHLPSTPSSPRVPGALAAAKAAKYGAAVPGVLGGLGALGGVGIPGGVVGAGPAAAAAAAKAAAKAAQFGLVGAAGLGGLGVGGLGVPGVGGLGGIPPAAAAKAAKYGAAGLGGVLGGAGQFPLGGVAARPGFGLSPIFPGGACLGKACGRKRK
It has 786 amino acids, the most frequent one being G, which appears 225 times.
There are 250 homologs for my protein.
Elastin belongs to the elastic fiber protein family, alongside elaunin and oxytalan.
Here you can see the 3D representation I found on RCSB of Elastin found in human body:
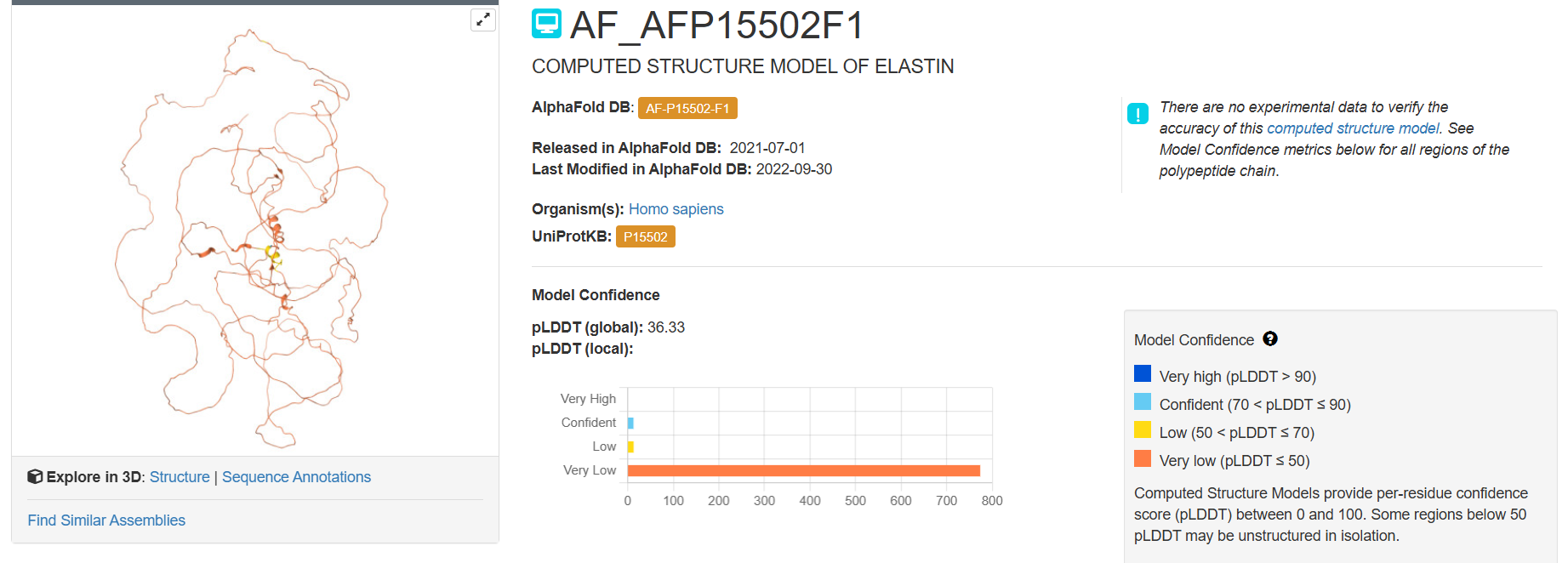
It was release firstly in 2021.07.01 and lastly modified in 2022.09.30. as you can see on the bottom right, the confidence score is very low. There aren’t any other molecules in the solved structure apart from my protein.
Elastin belongs to the fibrous protein family and is structurally classified as a structural extracellular matrix (ECM) protein.
For the next part I couldn’t find from RCSB the PDB file needed so I searched for it and found the exact protein on Alphafolds protein structure database.
Ribbon
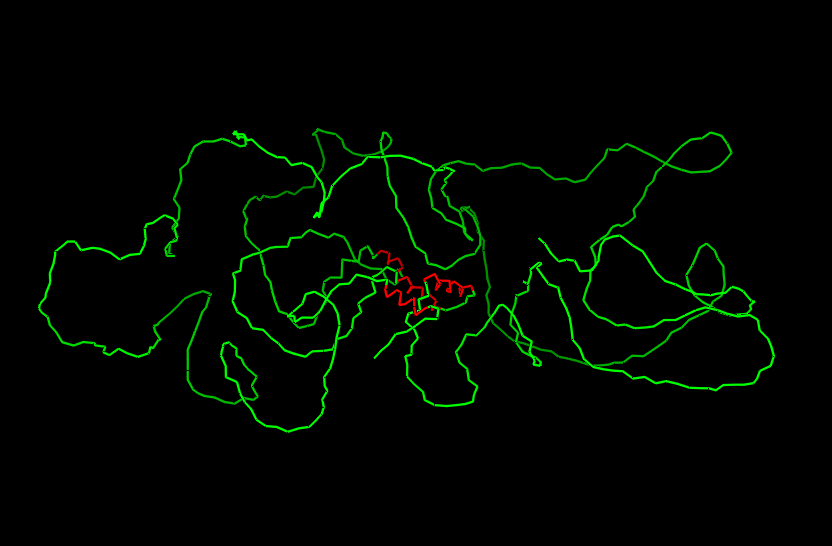
Cartoon
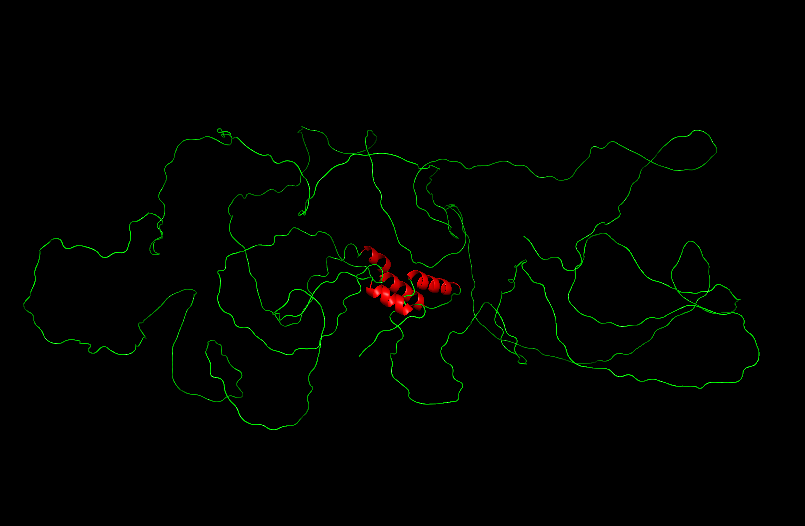
Ball and Sticks

After coloring the secondary structure (colored in cyan and purple) we can clearly see that it has more helices than sheets.
Here is the Protein after highlighting the residues:
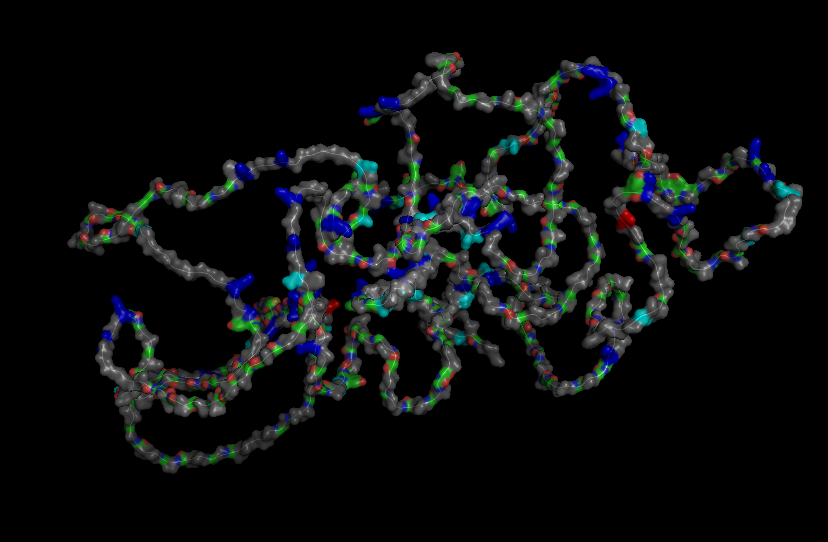
With grey I have the Hydrophobic residue and with the everything else is the Hydrophilic residue. The hydrophilic residue is divided into Polar uncharged (green), positively charged (blue) and negatively charged (red) residues. We can see that Elastin is highly hydrophobic due to how many grey regions are spread across the whole protein. There are very few hydrophilic parts as we can see and they are mostly positively charged around the edges. Inside the helixes of the secondary structures we can see yet again how most of it are hydrophobic which is understandable, due to Elastins ability to coacervate and form elastic fibers.
The Elastin has little to no binding pockets which is perfect due to its nature. Elastin is an intrinsically disordered protein (IDP). It doesn’t fold into a compact globular shape like enzymes or receptors, instead, it forms long, flexible, coiled chains that assemble into elastic fibers. This is also a reason for the Hydrophobic core.
Part C
C1. Protein Language modelling
At first I ran two DMS (deep mutational scans) with the given options of relative and absolute to also analyze the difference between them.
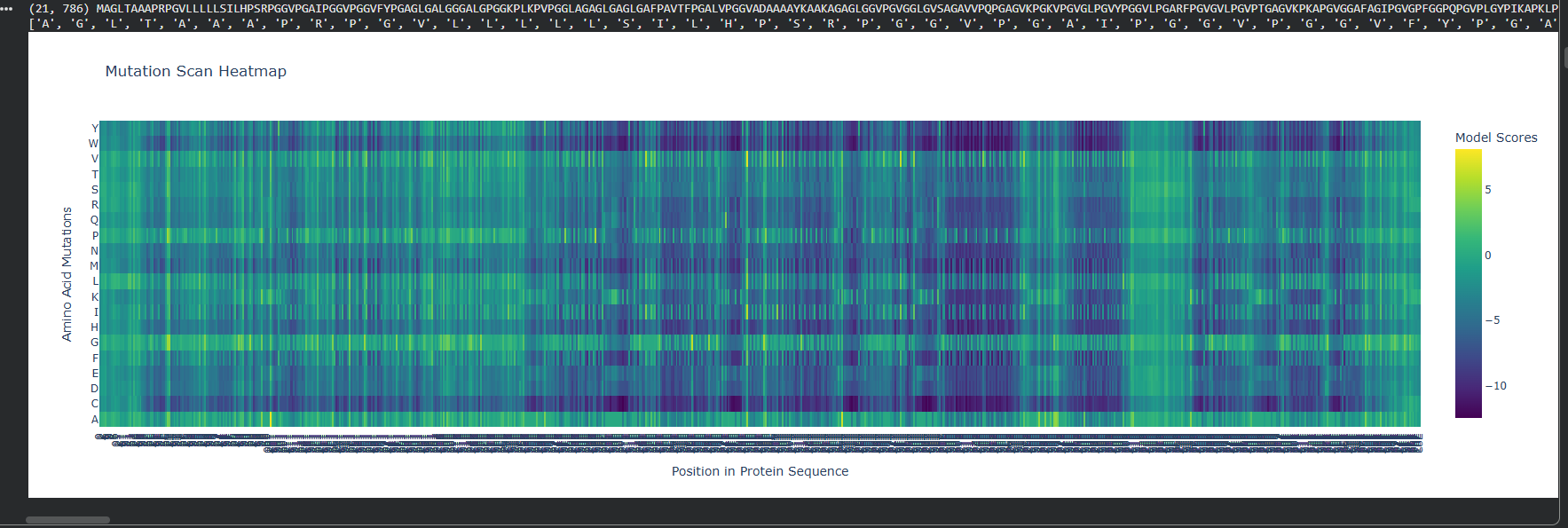
Relative DMS
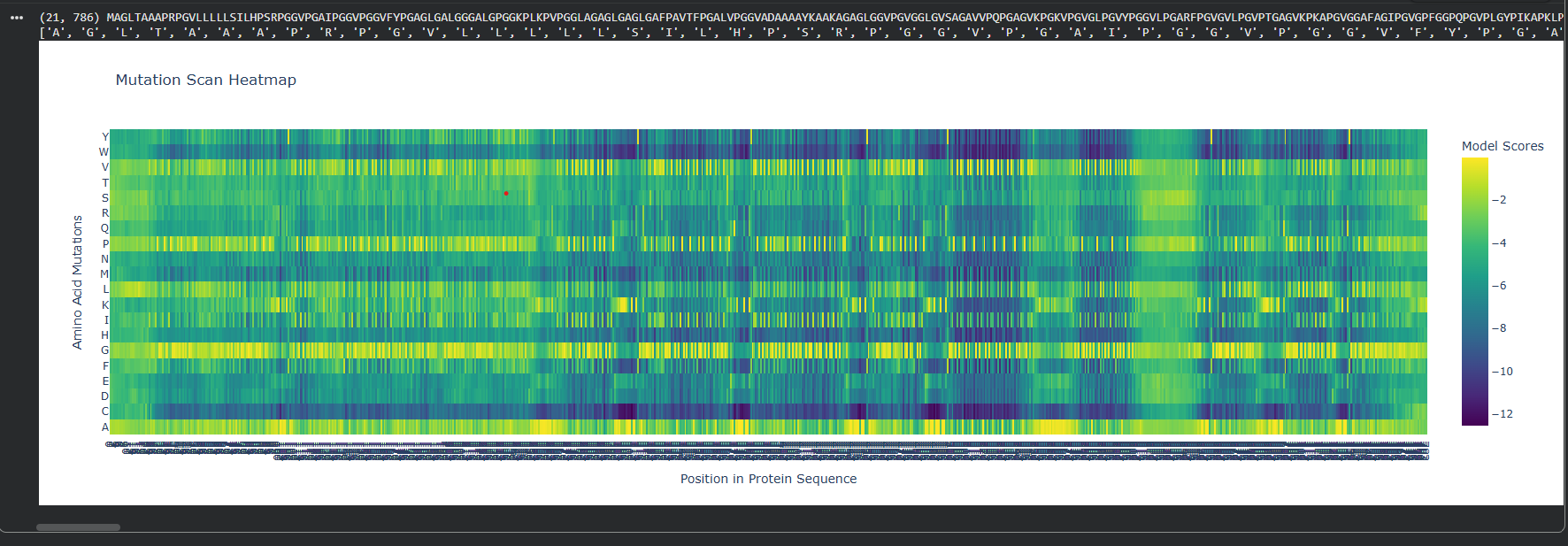
Absolute DMS
We can see that the Relative DMS shows that most Mutations don’t affect te function of the protein, unless it’s a mutation around the middle of the structure (where I assume is the secondary structure due to how the protein looks), where almost any mutation greatly affect the proteins functionality. In very few places we can even see that mutation can improve the functionality in relation to the strating population.
On the other hand, the Absolute DMS shows us that most mutations don’t change the enzyme activity, the binding affinity, the measured growth rate and the chance of survival. However no mutation can improve the proteins function.
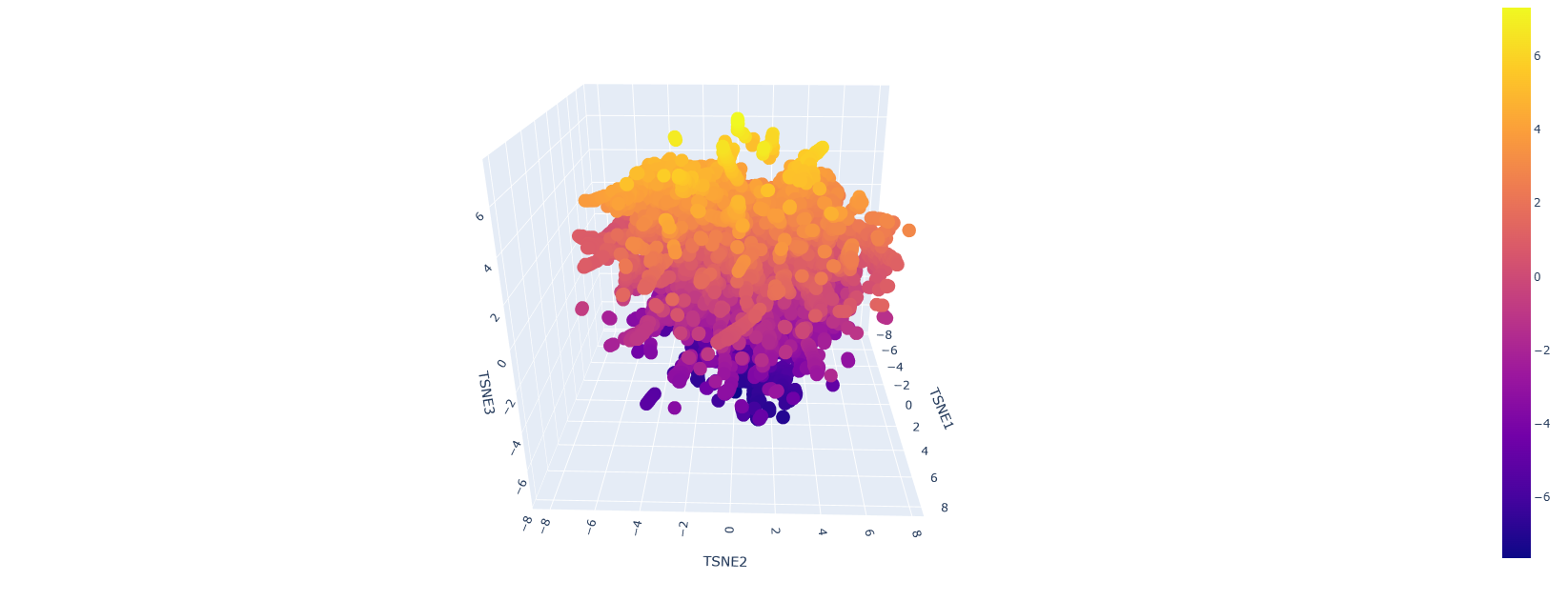
Here is how the LSA inspection looks like:
This is a sort of close-up look on it:
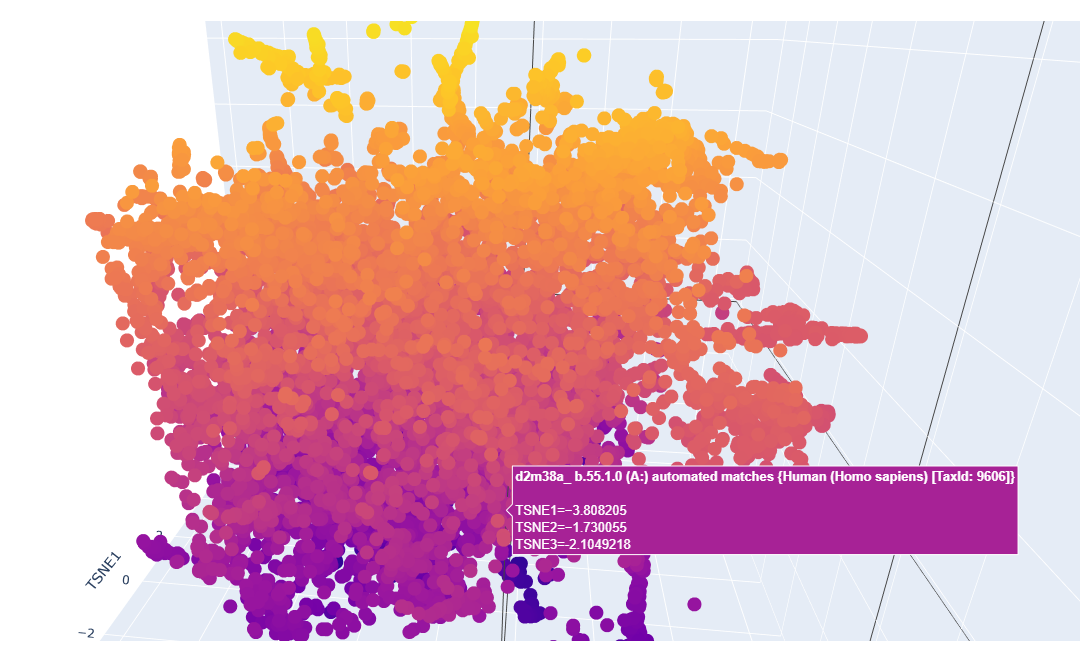
It looks like a mess but upon a closer inspection we can clearly say that we have similar protein designs. The yellow parts represent proteins that are similar and some even better, the dark blue or purple ones are the mutations that are not viable.
C2. Protein folding
Here is the folded protein model by the given colab code:
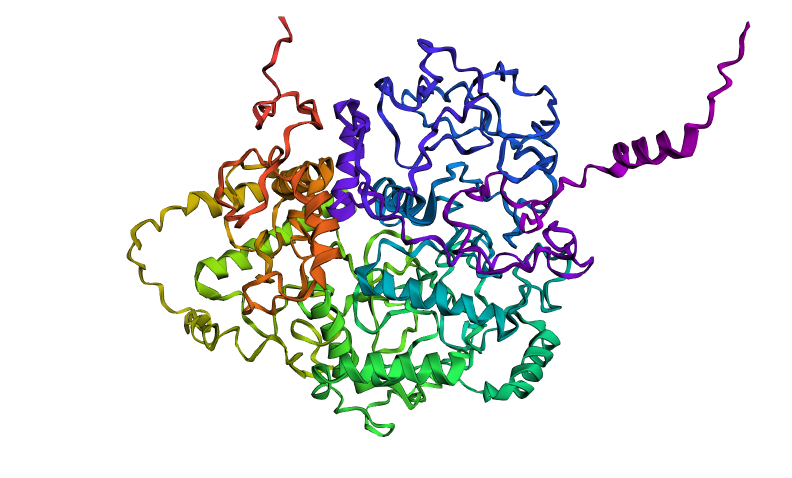
They are matching judgind by the fact that the one that I found is a pretty bad representations. We can see the helices in the middle being similar.
So my colab keeps on crashing, I will try to solve this issue as soon as possible so I can run the final tests on mutations and inverse folding
Here are some mutations I managed to do:
Working on the mutations we can see that
C3. Inverse Folding
Currently I’m experiencing some issues regarding the inverse folding…
Part D
Computational Design to Stabilize the MS2 Lysis Protein and Disrupt Its Interaction with E. coli DnaJ
Proposed Computational Approaches
- In Silico Mutagenesis and Stability Prediction
- Protein Language Models (PLMs):
- Use state-of-the-art PLMs (e.g., ESM-2, ProtT5, MutaPLM) for zero-shot and guided mutagenesis to predict stabilizing mutations across the L protein sequence.
- ΔΔG Calculations (measure the stability change of a protein upon mutation):
- Employ energy-based tools (FoldX, RosettaMP, and MD-based free energy estimators) to quantify the impact of mutations on protein stability, especially in the membrane context.
- Molecular Dynamics (MD) Simulations:
- Validate top mutations with MD and enhanced sampling (e.g., GaMD) to assess conformational stability and flexibility.
- Pipeline Management and Reproducibility
- Workflow Management:
- Implement the pipeline using Snakemake for reproducibility, scalability, and transparent documentation of all computational steps.
- Visualization and Analysis:
- Use ChimeraX and PyMOL for model inspection, interface mapping, and figure generation.
Justification for Tool Selection
- PLMs (e.g., ESM-2, MutaPLM, ProtT5):
- These models capture evolutionary and structural constraints, enabling accurate prediction of mutational effects, even in the absence of deep MSAs. MutaPLM, in particular, offers explicit modeling of mutation effects and supports explainable, engineerable protein design.
- AlphaFold-Multimer/AlphaFold3:
- These AI-based predictors provide high-accuracy models of protein complexes, crucial for mapping the L:DnaJ interface and guiding mutagenesis.
- Energy-Based Stability and Interface Tools (FoldX, RosettaMP, DockQ):
- These methods are validated for ΔΔG and interface scoring, allowing quantitative assessment of stability and PPI disruption.
- MD Simulations:
- MD provides atomistic insight into the dynamic consequences of mutations, complementing static predictions and revealing potential allosteric effects.
- Snakemake:
- Ensures reproducibility, modularity, and scalability of the computational workflow, facilitating transparent and collaborative research.
Potential Pitfalls and Limitations
- Modeling Limitations:
- AlphaFold-Multimer and PLMs may overpredict interface confidence or fail to capture dynamic, transient, or allosteric interactions, especially for small or flexible proteins like L and DnaJ.
- ΔΔG predictors (FoldX, RosettaMP) are sensitive to input structure quality and may not fully account for membrane context or oligomerization.
- Biological Relevance:
- In silico predictions may not translate directly to in vivo function due to cellular context, post-translational modifications, or compensatory mechanisms.
- Disrupting the L:DnaJ interface may inadvertently destabilize L or affect its oligomerization, requiring careful multi-objective optimization.