Week 5 HW: Protein design II
Part A: SOD1 binder peptide design
Using the Uniprot database, here is the SOD1 Protein sequence that I managed to find:
sp|P00441|SODC_HUMAN Superoxide dismutase Cu-asdn OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
After using the PepMLM Colab I managed to obtain these 4 peptides and their perplexity. After that I added the known binder and computed its perplexity the same way, in the end having these 5 proteins:
WRYGVVAVAWWE 13.755085 WRYYVVALAHWE 18.777272 WRYPAAGVRLKK 13.841338 HRYPAAGVELWE 22.190305 FLYRWLPSRRGG 21.421776
Perplexity tells us how confident the model is that a peptide sequence fits (or binds) the given protein context. In this case we can see that the most likely peptides to bind to my human SOD1 protein are the first and third one. Something wierd that I notice is that the know binders perplexity is quite high which makes it less plausible, despite it being tested to work. I’ve tried multiple times but still got the same result, which is a little bit unexpected.
Next we had to navigate through the alphafold server and see how my peptides actually bind to the SOD1 protein. Here are the results, alongside their ipTM score:
FLYRWLPSRRGG (the know binder) ipTM = 0.29
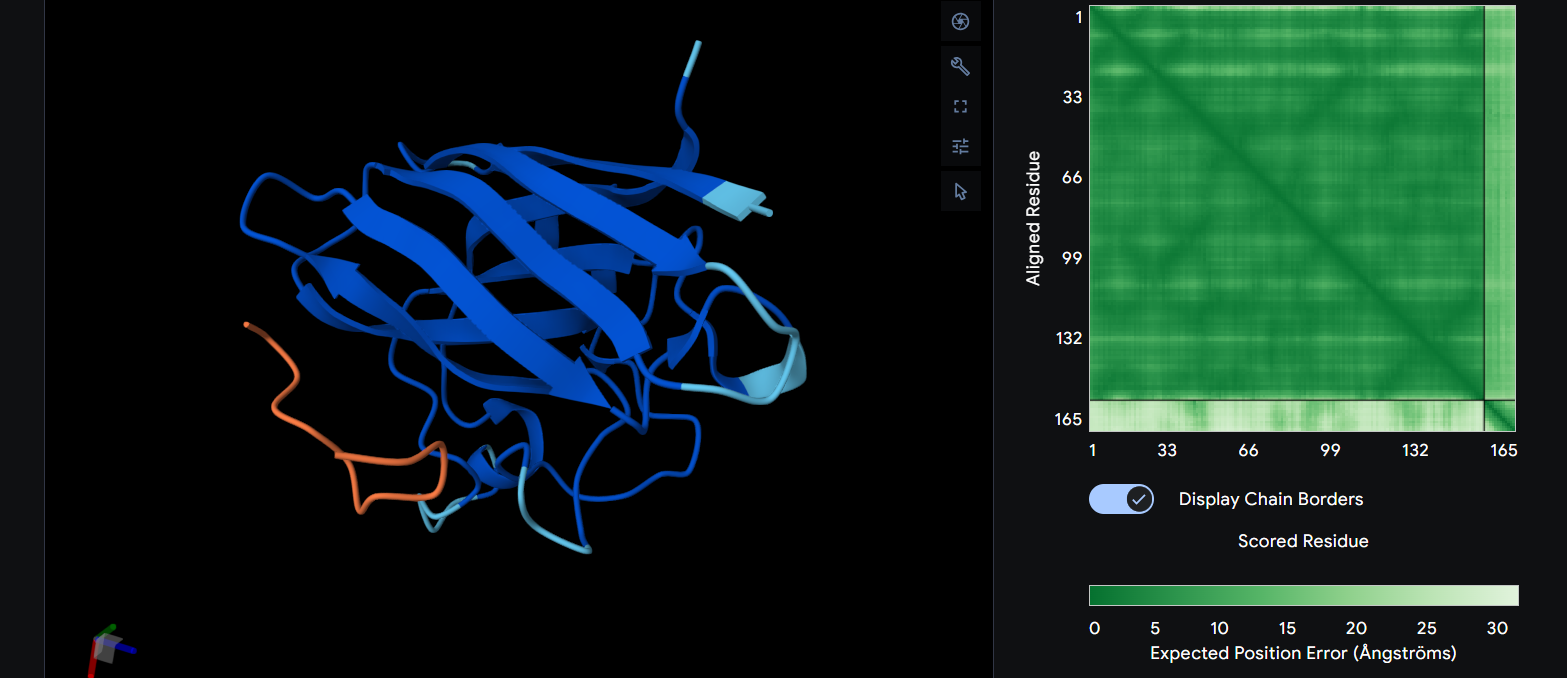
We can see that it has a β-barrel fold and it is completely surface bound, having only one part to bind with the protein. Also something wierd that I noticed aswell is the ipTM being relatively low on a protein that is known to bind to it. I can’t explain why but on my simulation it doesn’t bind near the N-terminus, rather it bind somewhere near the center.
WRYGVVAVAWWE ipTM = 0.34
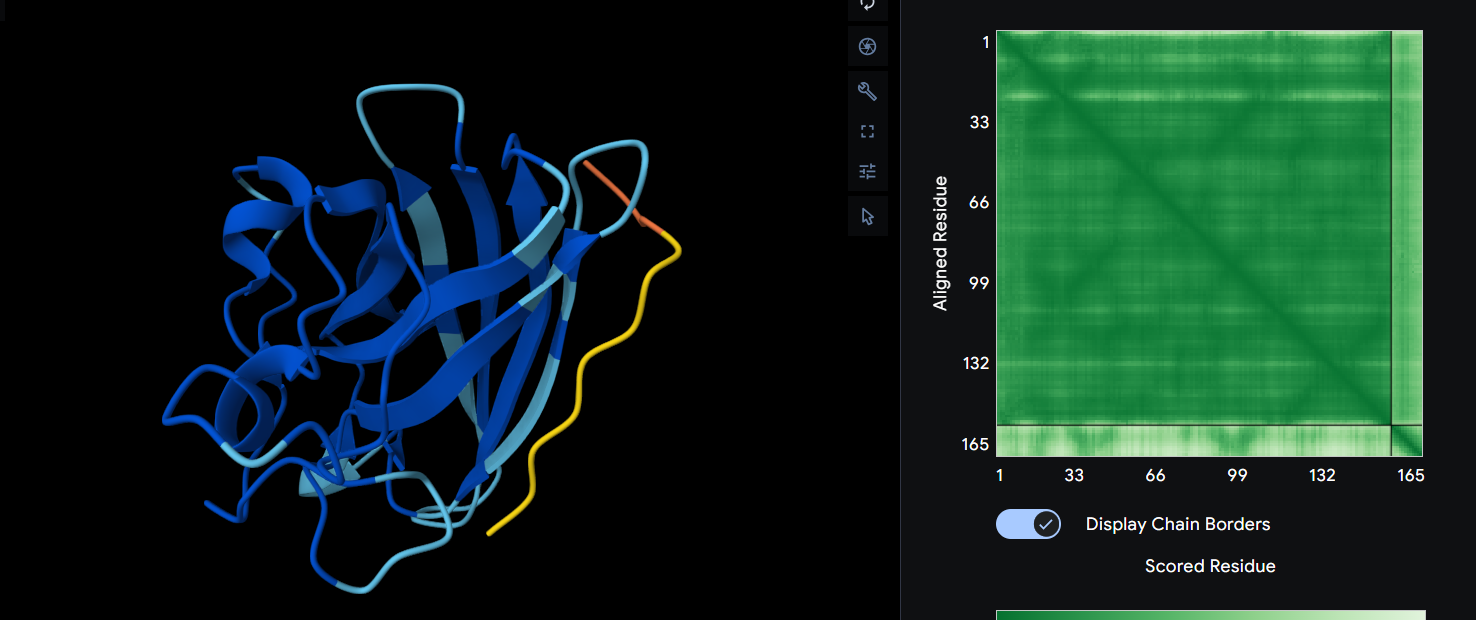
It is still has a β-barrel fold and completely surface bound. However what is different is the fact that the prediction says it binds both ways and we can see that for this peptide the alphafold tells us that it’s more likely to exist in that shape. On the other hand, this peptide does actually bind to the A4V mutation and it affects its structure.
WRYYVVALAHWE ipTM = 0.29
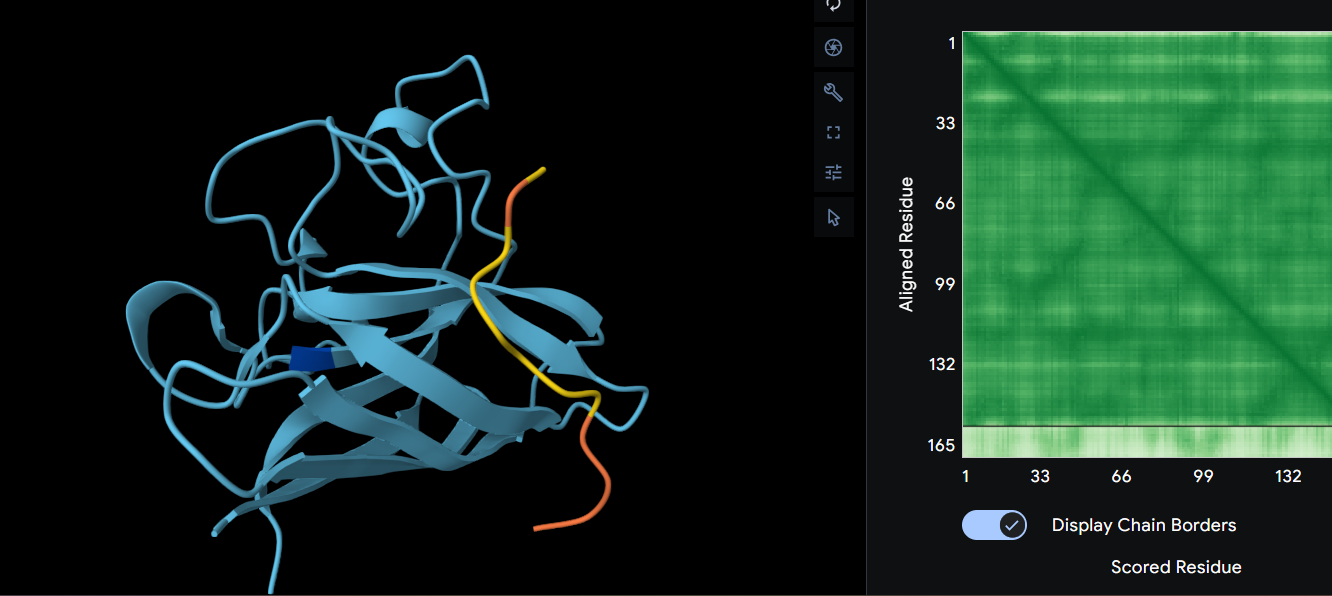
For this protein we’ll have another β-barrel fold and surface bound. This protein affects the whole proteins structure, meaning despite the peptide is more stable, it might affect the proteins structure more than the others did so far. This one doesn’t look like it’s binding to the N-terminus either, but it still affects the folding probability.
WRYPAAGVRLKK ipTM = 0.31
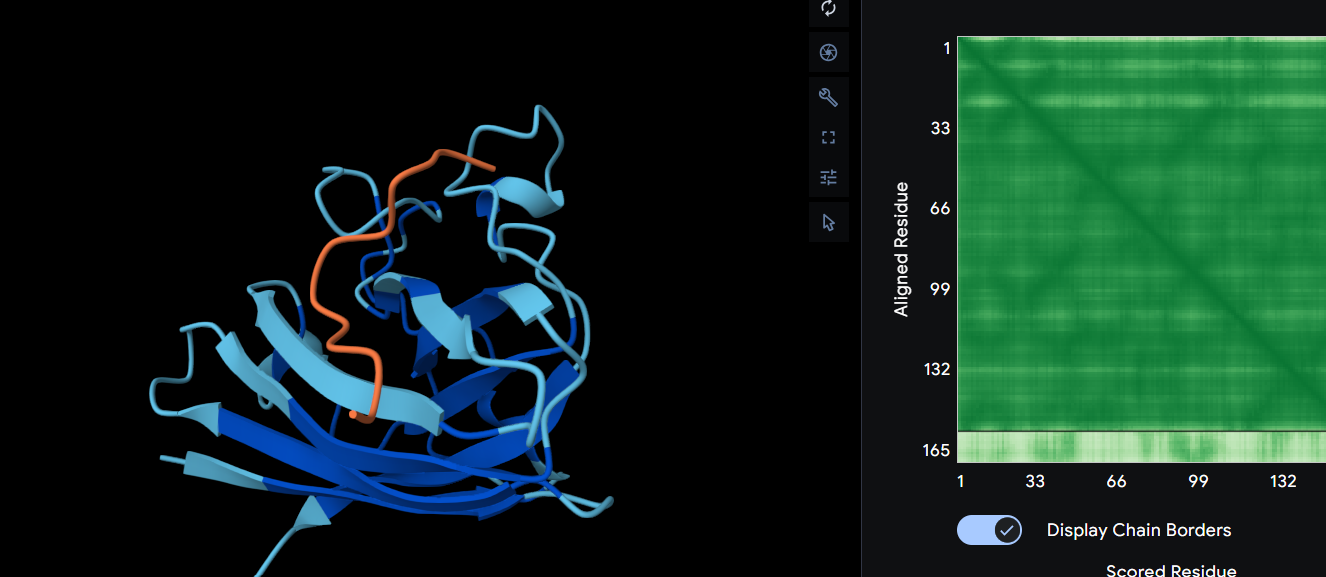
In this scenario we can observe what looks like a dimer interface since it’s a little bit shifted away from the β-barrel region, yet it is still surface bound. After its prediction, it binded on the completely other part of the protein so it doesn’t affect the A4V mutation. This peptide affects the structure and alphafold is also unsure about it’s own folding.
HRYPAAGVELWE ipTM = 0.3
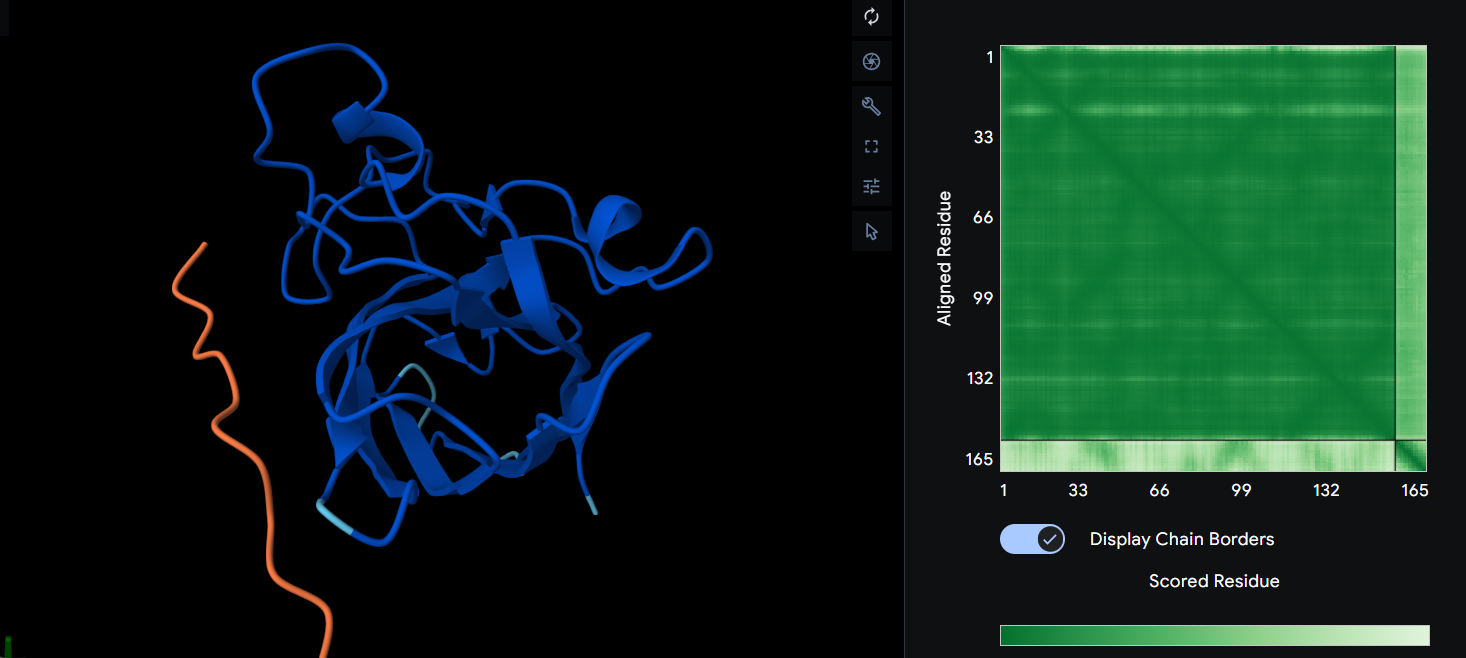
Yet again we have a dimer interface that is surface bounded. However, in this scenario the protein itself is more likely to fold the way alphafold predicts. It is nowhere near the N-terminus of the A4V mutation.
Unfortunately none binded to the A4V mutation.
Conclusions
Judgind by the ipTM values, we can say that almost all of them have a higher ipTM score than the one of the known binder. The best peptide generated is the ‘WRYGVVAVAWWE’, which was generated by PepMLM since it binds to the mutation directly and it also keeps the structure of the protein pretty much the same. The ipTM value is also higher, suggesting a potentially comparable interaction interface according to AlphaFold-Multimer predictions.
Using its exact format, here are the probabilities generated by Peptiverse for each peptide:
For FLYRWLPSRRGG
💧 Solubility => Soluble 1.000 🩸 Hemolysis => Non-hemolytic 0.047 🔗 Binding Affinity => Weak binding 5.962 ⚖️ Molecular Weight => 1507.7Da
For WRYGVVAVAWWE
💧 Solubility => Soluble 1.000 🩸 Hemolysis => Non-hemolytic 0.125 🔗 Binding Affinity => Medium binding 7.948 ⚖️ Molecular Weight => 1521.7Da
For WRYYVVALAHWE
💧 Solubility => Soluble 1.000 🩸 Hemolysis => Non-hemolytic 0.068 🔗 Binding Affinity => Weak binding 6.839 ⚖️ Molecular Weight => 1592.8Da
For WRYPAAGVRLKK
💧 Solubility => Soluble 1.000 🩸 Hemolysis => Non-hemolytic 0.014 🔗 Binding Affinity => Weak binding 6.139 ⚖️ Molecular Weight => 1444.7Da
For HRYPAAGVELWE
💧 Solubility => Soluble 1.000 🩸 Hemolysis => Non-hemolytic 0.028 🔗 Binding Affinity => Weak binding 5.372 ⚖️ Molecular Weight => 1427.6Da
One thing that we notice straight away is that the peptide with the highest ipTM is also has the highest predicted binding affinity, which is pretty much obvious since it actually targets the mutation. We also notice that not for every ipTM value that is higher than the one of the known binder means better affinity, like for ‘HRYPAAGVELWE’ (the last one). After the predictions, all peptides are soluble and for most cases, stronger binders usually mean a more hemolytic probability, despite still being non-hemolytic. The exception would be ‘WRYPAAGVRLKK’(the third one). In conclusion, I believe that the best generated peptide that balances binding affinity and therapeutic properties is ‘WRYYVVALAHWE’ (the second one). Personally I would still suggest that the protein we should continue is the one with the best binding affinity, due to it targetting the mutation and being the most likely one to bind. I believe that the hemolytic properties I don’t think it’s that much of a problem since it is still considered safe.
After the moPPit generation, I have found these 4 more peptides: ‘LTEKITQTLHTC’, ‘SKQYKCQLTGDI’, ‘STDQTCGKVKLM’, ‘DEKKFQRCTTTT’, and their respective Hemolysis, Solubility, Affinity (from the first 10 aminoacids) and Motif: [0.9491806849837303, 0.75, 5.967530727386475, 0.7040762305259705], [0.9336216971278191, 0.8333333134651184, 6.318202018737793, 0.7738364934921265], [0.9584326893091202, 0.75, 6.513046741485596, 0.7761058807373047], [0.9732194431126118, 0.9166666865348816, 5.4131293296813965, 0.7708268165588379]. First of all I see that for some reason they all have insanely large hemolytic properties so that is already a dangerous sign. Their binding affinity is also not as good as the pepMLM generated ones and the solubility is probably more precise which is a lpus but doesn’t help with all the disadvantages that the high hemolytic nature brings.
Part C: L-Protein Engineering. Random Mutagenis
Here is an example of code that can be used for this activity:
(I have absolutely no clue how to make it all beautifully alligned in a box for code)
import random
def generate_random_mutants(sequence, num_mutants=5, min_mutations=2): """ Generate random mutants of a protein sequence.
Parameters:
sequence (str): Original amino acid sequence
num_mutants (int): Number of random mutant sequences to generate
min_mutations (int): Minimum number of residues to mutate in each mutant
Returns:
list of tuples: [(mutant_sequence, mutations_list), ...]
"""
aa_list = list("ACDEFGHIKLMNPQRSTVWY") # 20 standard amino acids
mutants = []
for _ in range(num_mutants):
seq_list = list(sequence)
num_mut = random.randint(min_mutations, max(5, len(sequence)//5)) # random # mutations
positions = random.sample(range(len(seq_list)), num_mut)
mutations = []
for pos in positions:
original = seq_list[pos]
new_aa = random.choice([aa for aa in aa_list if aa != original])
seq_list[pos] = new_aa
mutations.append(f"{original}{pos+1}{new_aa}") # e.g., A4V
mutant_seq = "".join(seq_list)
mutants.append((mutant_seq, mutations))
return mutants
#Example usage:
original_seq = "MKTAYIAKQRQISFVKSHFSRQDILDLWIYHTQGYFP"
mutants = generate_random_mutants(original_seq, num_mutants=3)
for m, mut_list in mutants:
print(m, mut_list)