Biological Engineering Application: A Distributed, Low Cost Environmental Biosensing Platform For my project, I’ve been imagining a simple, low cost environmental biosensing system that anyone could use — something like disposable test strips with engineered microbes or cell free components that change color when they detect contaminants in water or soil. Think of it as an open source, modular “bio test strip” that communities could use to check for things like heavy metals, PFAS, or harmful bacteria without needing a lab. What draws me to this idea is how practical and empowering it could be. A lot of communities don’t have access to reliable water testing, and waiting for official reports can take weeks or months. A cheap, easy to use biosensor could help people catch problems early, respond faster, and feel more in control of their environment. It also has a nice educational angle — something schools, citizen science groups, or community labs could use to learn about biology while doing something meaningful. There are already tools in synthetic biology that point in this direction (like cell free biosensors or engineered yeast reporters), but I’m interested in pushing the idea toward something more distributed and democratized — something that doesn’t require a lab coat or a research budget to use. Governance / Policy Goals for an Ethical Future Because this kind of biosensor could end up in the hands of a lot of different people — community groups, teachers, students, DIYbio hobbyists, or folks in low resource settings — I want to think carefully about how to make sure it’s used safely and responsibly. That’s where my governance goals come in. Main Goal: Prevent Harm (Non Malfeasance) The first and most obvious goal is making sure the tool doesn’t accidentally cause harm — biologically, socially, or environmentally. If the biosensor uses engineered microbes, I need to think about the possibility of accidental release. Even “safe” strains can behave unpredictably in the wild, so containment and design safeguards matter. There’s also the social side. Environmental data can be sensitive. A false positive could cause unnecessary panic; a false negative could give people a false sense of security. So part of this goal is making sure the results are easy to interpret and hard to misuse. Side Goal 1: Promote Security and Prevent Malicious Use Because the platform is meant to be low cost, open source, and easy to distribute, it has all the qualities that make a tool empowering — but also potentially vulnerable. Someone could try to repurpose the biosensor to detect things it was never meant to detect, like human biomarkers or pathogens in ways that violate privacy. There’s also the supply chain angle: if the components can be tampered with, someone could alter the biosensor to give misleading results. So part of the governance plan is making sure the system can’t be easily weaponized or misused. Side Goal 2: Promote Equity and Autonomy This one feels especially important. Communities that deal with pollution often have complicated relationships with outside researchers or government agencies. They may lack access to testing, or they may distrust the people who usually control environmental data. I want this tool to do the opposite — to give communities more control, not less. That means thinking about access, affordability, and who gets to decide how the data is used. The goal is to support autonomy, not create new dependencies or power imbalances. Governance Actions Below are three governance actions, each analyzed through the required four aspects. Option 1: Community Biolab “Safety‑by‑Design” Toolkit Purpose Right now, community labs vary widely in safety practices. This toolkit standardizes risk assessment, training, and experiment planning. Design • Developed by community labs + biosafety experts • Includes checklists, training modules, and a risk‑flagging app • Incentives: badges, access to shared equipment, recognition Assumptions • Community members will voluntarily adopt the toolkit • Training can be made simple and engaging • Labs have enough resources to implement it Risks • Over‑reliance on checklists instead of real understanding • Labs may treat badges as performative rather than meaningful • Could create a divide between “certified” and “uncertified” labs Option 2: DNA Synthesis “Pre Check” Assistant Purpose Small labs often lack tools to screen DNA orders. This assistant helps them avoid ordering risky sequences. Design • Web app that screens sequences against risk lists • Built with input from synthesis companies and regulators • Provides explanations, not just warnings Assumptions • Users will run sequences through the tool • Risk lists are accurate and up to date • False positives won’t frustrate users Risks • False negatives could create a false sense of security • Malicious users could probe the system to find “safe” variants • Could burden small labs if too strict
Part 1 Here is the image that I managed to make using electrophoresis gel art: vs It’s not really the same but if you imagine the emoji having long hair you can sort of see it.
Part 1: Here is the reference image that I managed to make using the opentrons art GUI: I tried my best to recreate it on my own (no ai) in colab but it didn’t work quite well: I will most likely have fun with the python code later aswell! Maybe I’ll design an even better looking cat.
Part A How many molecules of amino acids do you take with a piece of 500 grams of meat? Let’s assume that meat is on average 20% protein. so 500g of meat would mean 100g of protein.
After calculations we can assume that 1Da/molecule = 1g/mol, so by having around 100 Da on a amino acid we can say that we have approximatelly 1 mol of amino acid residue in 500g of meat.
Part A: SOD1 binder peptide design Using the Uniprot database, here is the SOD1 Protein sequence that I managed to find:
sp|P00441|SODC_HUMAN Superoxide dismutase Cu-asdn OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
After using the PepMLM Colab I managed to obtain these 4 peptides and their perplexity. After that I added the known binder and computed its perplexity the same way, in the end having these 5 proteins:
Part A What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion HF PCR Master Mix is a ready-to-use mixture designed for high accuracy DNA amplification. It contains several component that make PCR efficient, like dNTPs (the building blocks of DNA which are incorporated by the polymerase in the newly synthesized DNA strand), Reaction buffer which can either help maintain the optimal chemical environment or a GC buffer that can amplify GC-rich DNA templates, and Mg²⁺ ions that are an essesntial cofactor for the DNA polymerase activity. The most notable component is the Phusion High-Fidelity DNA polymerase, which has proofreading activity between 3’-> 5’ exonuclease which corrects mistakes during replication, having a lower rate than the standard Taq polymerase. The Phusion HF PCR Master Mix can also contain Tracking Dye, allowing direct loading of PCR product on agarose gels without adding loading buffer.
Subsections of Homework
Week 1 HW: Principles and Practices
Biological Engineering Application: A Distributed, Low Cost Environmental Biosensing Platform
For my project, I’ve been imagining a simple, low cost environmental biosensing system that anyone could use — something like disposable test strips with engineered microbes or cell free components that change color when they detect contaminants in water or soil. Think of it as an open source, modular “bio test strip” that communities could use to check for things like heavy metals, PFAS, or harmful bacteria without needing a lab.
What draws me to this idea is how practical and empowering it could be. A lot of communities don’t have access to reliable water testing, and waiting for official reports can take weeks or months. A cheap, easy to use biosensor could help people catch problems early, respond faster, and feel more in control of their environment. It also has a nice educational angle — something schools, citizen science groups, or community labs could use to learn about biology while doing something meaningful.
There are already tools in synthetic biology that point in this direction (like cell free biosensors or engineered yeast reporters), but I’m interested in pushing the idea toward something more distributed and democratized — something that doesn’t require a lab coat or a research budget to use.
Governance / Policy Goals for an Ethical Future
Because this kind of biosensor could end up in the hands of a lot of different people — community groups, teachers, students, DIYbio hobbyists, or folks in low resource settings — I want to think carefully about how to make sure it’s used safely and responsibly. That’s where my governance goals come in.
Main Goal: Prevent Harm (Non Malfeasance)
The first and most obvious goal is making sure the tool doesn’t accidentally cause harm — biologically, socially, or environmentally. If the biosensor uses engineered microbes, I need to think about the possibility of accidental release. Even “safe” strains can behave unpredictably in the wild, so containment and design safeguards matter.
There’s also the social side. Environmental data can be sensitive. A false positive could cause unnecessary panic; a false negative could give people a false sense of security. So part of this goal is making sure the results are easy to interpret and hard to misuse.
Side Goal 1: Promote Security and Prevent Malicious Use
Because the platform is meant to be low cost, open source, and easy to distribute, it has all the qualities that make a tool empowering — but also potentially vulnerable. Someone could try to repurpose the biosensor to detect things it was never meant to detect, like human biomarkers or pathogens in ways that violate privacy.
There’s also the supply chain angle: if the components can be tampered with, someone could alter the biosensor to give misleading results. So part of the governance plan is making sure the system can’t be easily weaponized or misused.
Side Goal 2: Promote Equity and Autonomy
This one feels especially important. Communities that deal with pollution often have complicated relationships with outside researchers or government agencies. They may lack access to testing, or they may distrust the people who usually control environmental data.
I want this tool to do the opposite — to give communities more control, not less. That means thinking about access, affordability, and who gets to decide how the data is used. The goal is to support autonomy, not create new dependencies or power imbalances.
Governance Actions
Below are three governance actions, each analyzed through the required four aspects.
Option 1: Community Biolab “Safety‑by‑Design” Toolkit
Purpose
Right now, community labs vary widely in safety practices. This toolkit standardizes risk assessment, training, and experiment planning.
Design
• Developed by community labs + biosafety experts
• Includes checklists, training modules, and a risk‑flagging app
• Incentives: badges, access to shared equipment, recognition
Assumptions
• Community members will voluntarily adopt the toolkit
• Training can be made simple and engaging
• Labs have enough resources to implement it
Risks
• Over‑reliance on checklists instead of real understanding
• Labs may treat badges as performative rather than meaningful
• Could create a divide between “certified” and “uncertified” labs
Option 2: DNA Synthesis “Pre Check” Assistant
Purpose
Small labs often lack tools to screen DNA orders. This assistant helps them avoid ordering risky sequences.
Design
• Web app that screens sequences against risk lists
• Built with input from synthesis companies and regulators
• Provides explanations, not just warnings
Assumptions
• Users will run sequences through the tool
• Risk lists are accurate and up to date
• False positives won’t frustrate users
Risks
• False negatives could create a false sense of security
• Malicious users could probe the system to find “safe” variants
• Could burden small labs if too strict
Option 3: Genetic Kill Switch Design Library
Purpose
Provide standardized, vetted kill switch designs for engineered microbes.
Design
• Curated library with documentation
• Includes failure modes and testing guidelines
• Could be maintained by a consortium (iGEM, ABSA, NIH)
Assumptions
• Kill switches work reliably across contexts
• Users will implement them correctly
• Regulators will accept standardized designs
Risks
• Overtrust in kill switch reliability
• Dual use: knowledge could help people defeat containment
• False sense of safety could encourage riskier deployments
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
1
2
• By helping respond
2
1
3
Foster Lab Safety
• By preventing incident
1
2
3
• By helping respond
1
3
3
Protect the environment
• By preventing incidents
2
2
1
• By helping respond
2
3
2
Other considerations
• Minimizing costs and burdens to stakeholders
1
2
3
• Feasibility?
1
2
2
• Not impede research
1
2
2
• Promote constructive applications
1
2
1
Which governance combination works together?
Prioritization and Recommendation
Looking back at the scoring, the option that clearly rises to the top for me is Option 1: the Community Biolab Safety by Design Toolkit. It’s the most practical, the most immediately useful, and honestly the one that feels closest to the spirit of HTGAA — empowering people to do biology safely, confidently, and creatively. Community labs are incredibly diverse in experience and resources, so giving them a simple, friendly toolkit that makes safety feel approachable (rather than intimidating) seems like the biggest win with the least friction.
I would pair this with Option 3: the Genetic Kill Switch Design Library, especially for any projects that involve engineered organisms leaving the bench. Even if my own biosensing project ends up being cell free, I’ve realized how often people underestimate environmental risks. A well documented, vetted kill switch library gives people a starting point that’s safer than whatever they might cobble together on their own. It’s not perfect — kill switches can fail, and people can over trust them — but it’s still a meaningful step toward responsible design.
Option 2, the DNA Synthesis Pre Check Assistant, is valuable, but it feels more specialized. It’s great for labs that regularly order DNA, but it doesn’t help the broader community as much as the other two. It also introduces some workflow friction, and I can imagine small labs getting frustrated if the tool flags things too aggressively. So I see it as a useful add on rather than a core priority.
Trade offs, Assumptions, and Uncertainties
There are a few trade offs I had to think through:
• Option 1 relies on people actually using it. Tools and checklists only work if people adopt them, and community labs can be very independent. I’m assuming that if the toolkit is well designed, friendly, and not preachy, people will actually want to use it.
• Option 3 assumes kill switches are reliable enough to matter. In reality, biology is messy. Even the best kill switches can fail in unexpected environments. I’m assuming that a curated library with clear documentation will reduce misuse and overconfidence.
• Option 2 assumes people want to screen their own DNA orders. Some labs might see this as extra work or feel like they’re being policed. I’m assuming the tool can be designed in a way that feels helpful rather than punitive.
There’s also uncertainty around how these tools would be maintained long term. Community resources often start strong and then fade unless someone takes ownership.
Who I Would Recommend This To?
If I had to choose an audience, I’d direct this recommendation to leaders of community biology labs, DIYbio networks, and local makerspaces. These are the groups that would benefit the most from Option 1 and Option 3, and they’re also the ones who can adopt new norms quickly without waiting for national regulations.
I’d also share it with organizations like iGEM, ABSA, and the Global Community Bio Summit, since they already play a role in shaping norms and could help maintain shared resources like the kill switch library.
If you want, I can help you craft the final ethical reflection section in the same human tone, so your whole assignment feels cohesive and personal.
Homework Questions from Professor Jacobson
Cells copy their DNA using an enzyme called DNA polymerase. It does a pretty good job, but it’s not flawless—on average, it makes about one mistake per million base pairs. When you compare that to the size of the human genome (about 4.2 billion base pairs), you end up with roughly 3,200 potential errors every time a cell divides.
That sounds alarming, but cells have several built‑in proofreading and repair systems. These teams of proteins constantly scan the DNA, spot errors, and fix them. They dramatically cut down the number of mistakes, though they can’t eliminate them entirely.
Another topic is how many different DNA sequences could encode a typical human protein—and why some sequences work better than others. A “typical” protein is about 345 amino acids long, which corresponds to around 1,036 base pairs of DNA. Because the genetic code is redundant, multiple codons can specify the same amino acid. But in real organisms, not all codons are treated equally. Some are translated faster or more accurately, depending on the cell’s machinery. And translation speed matters: if the ribosome moves too quickly or too slowly, the protein may not fold correctly, which can affect its function.
Homework Questions from Dr. LeProust
The most widely used technique for making synthetic DNA fragments (oligonucleotides) is solid‑phase phosphoramidite synthesis. It works extremely well for short sequences, but the longer the oligo, the more problems arise. Once you get past about 200 nucleotides, errors start piling up and the chemistry becomes less efficient.
This is why you can’t just “chemically synthesize” a full 2,000‑base‑pair gene in one go—the error rate becomes too high over such a long stretch. Instead, long genes are usually built by assembling many shorter, more reliable pieces.
Homework Question from George Church
Animals need ten essential amino acids that they can’t make themselves and must obtain from their diet:
arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine.
This ties into the “Lysine Contingency” from Jurassic Park. In the story, the dinosaurs are engineered so they can’t produce lysine, meaning they would die without special supplements from the lab. The issue is that this wouldn’t actually work—no animals can make lysine, not even modern ones. They all get it from food.
So if the dinosaurs escaped, they could simply eat plants or animals that contain lysine and survive just fine. It’s a clever narrative idea, but biologically unrealistic. A more plausible failsafe would involve engineering a dependency on something that doesn’t already exist in nature or that only the lab can provide.
Professor Jacobson
DNA Polymerase Error Rate and How Cells Deal With It
DNA polymerase is the enzyme responsible for copying DNA, but it isn’t flawless. On its own, it makes about one mistake per million bases it copies. Considering the human genome is roughly 3 billion base pairs, that would add up to around 3,000 errors every time a cell divides if nothing else helped.
Fortunately, cells have several layers of quality control. DNA polymerase can actually proofread as it goes, using a 3′→5′ exonuclease activity to remove incorrectly added bases. This proofreading step alone improves accuracy dramatically—down to about one error per billion bases.
After replication finishes, additional mismatch repair systems sweep through the DNA to catch and fix leftover mistakes.
Another reason this error rate doesn’t cause constant problems is that most of the human genome doesn’t code for proteins. Many mutations land in non‑coding regions and have no effect. And if a cell does accumulate harmful mutations—something that often happens in cancer-prone cells—it can trigger apoptosis, a built‑in self‑destruct program that prevents damaged cells from continuing to divide.
How Many Ways You Can Code a Protein—and Why Most Sequences Fail
In theory, there are an astronomical number of DNA sequences that could encode a typical human protein. A protein of about 345 amino acids (roughly 1,036 base pairs) can be encoded in many different ways because the genetic code is redundant. When you do the math, you get around 10¹⁵⁷ possible DNA sequences that would all produce the same amino acid chain.
But in real cells, most of those sequences would perform poorly.
One major reason is codon bias. Different organisms prefer certain codons over others, and using rare codons can slow translation because the matching tRNAs are scarce. The DNA sequence also affects how the mRNA folds, and if the mRNA forms very stable structures, ribosomes can stall or stop prematurely. That can reduce protein production or even change how the protein folds, which affects its function.
Dr. LeProust – Oligonucleotide Synthesis
The Current Method
Today, the standard way to make synthetic DNA fragments (oligonucleotides) is solid‑phase phosphoramidite chemical synthesis. It’s reliable, efficient, and widely used.
Why Oligos Get Hard to Make Beyond ~200 Nucleotides
The challenge with long oligos is that each chemical step has a small chance of failing. Even with a high stepwise efficiency—say, around 99.5%—those tiny losses add up. As the sequence gets longer, the probability of ending up with a perfectly correct strand drops sharply. By the time you reach 200 nucleotides, the amount of full‑length, error‑free product becomes very small.
Other issues, like side reactions and physical crowding on the solid support, also make long sequences harder to synthesize cleanly.
Why You Can’t Directly Synthesize a 2,000 bp Gene
Trying to chemically synthesize a 2,000 base pair gene in one piece just isn’t practical. The accumulated errors would leave you with almost no usable product. Instead, scientists synthesize shorter oligos and then assemble them into full genes using enzymatic or biological methods.
George Church
The Ten Essential Amino Acids
Most animals—including humans—can’t make ten of the amino acids they need. These essential amino acids must come from the diet:
arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine.
Arginine is sometimes considered “semi‑essential,” especially during periods of growth.
Why the Jurassic Park “Lysine Contingency” Doesn’t Work
In Jurassic Park, the dinosaurs are engineered so they supposedly can’t make lysine, meaning they’d die without supplements from the lab. The problem is that no vertebrate can make lysine anyway—all animals already depend on dietary lysine.
And lysine is easy to find in nature. Plants, animals, insects—almost everything in a natural ecosystem contains lysine. So escaped dinosaurs could simply eat lysine‑rich food and survive.
It’s a fun plot device, but biologically it doesn’t hold up. A real failsafe would need to involve a dependency on something not found in the wild.
Week 2 HW: DNA read, write and edit
Part 1
Here is the image that I managed to make using electrophoresis gel art:
vs
It’s not really the same but if you imagine the emoji having long hair you can sort of see it.
A friend of mine said that it reminds it of Frisks face from undertale and ever since I’ve started seeing it aswell.
Part 3
I chose fibroin, which is one of the two main proteins found in silk. Silk in its raw state consists of two main proteins, sericin and fibroin, with a glue-like layer of sericin coating two singular filaments of fibroin called brins.
I used uniprot to find a sequence for fibroin that is produced by the Aliatypus thompsoni, also know as the trapdoor spider:
SQ SEQUENCE 193 AA; 18898 MW; 94DFC97AA25F9796 CRC64;
ASSASGASSS IGVASSKGVA SSSKTATKAR ISAGSSGSST STKSSSSAST AVPTNLSGSR
SHALSSSNSG QDNTVGDDFG LGYISGGILP VNTPALNFPS DLGSLTSGLL SSLDGPVLPS
VEYRITSLTS SVLSLLSTSG GAFNYSSFAK NLAILAYQIS VSNPGLSVSQ VVSETLLESV
GALIHILVSS QVG
After I reverse translated, the protein looks like this (using https://www.genecorner.ugent.be/rev_trans.html):
GCGAGCAGCGCGAGCGGCGCGAGCAGCAGCATTGGCGTGGCGAGCAGCAAAGGCGTGGCG
AGCAGCAGCAAAACCGCGACCAAAGCGCGCATTAGCGCGGGCAGCAGCGGCAGCAGCACC
AGCACCAAAAGCAGCAGCAGCGCGAGCACCGCGGTGCCGACCAACCTGAGCGGCAGCCGC
AGCCATGCGCTGAGCAGCAGCAACAGCGGCCAGGATAACACCGTGGGCGATGATTTTGGC
CTGGGCTATATTAGCGGCGGCATTCTGCCGGTGAACACCCCGGCGCTGAACTTTCCGAGC
GATCTGGGCAGCCTGACCAGCGGCCTGCTGAGCAGCCTGGATGGCCCGGTGCTGCCGAGC
GTGGAATATCGCATTACCAGCCTGACCAGCAGCGTGCTGAGCCTGCTGAGCACCAGCGGC
GGCGCGTTTAACTATAGCAGCTTTGCGAAAAACCTGGCGATTCTGGCGTATCAGATTAGC
GTGAGCAACCCGGGCCTGAGCGTGAGCCAGGTGGTGAGCGAAACCCTGCTGGAAAGCGTG
GGCGCGCTGATTCATATTCTGGTGAGCAGCCAGGTGGGC
Afterwards I had to optimize the codon sequence, which results in this:
GCATCTAGCGCGTCTGGTGCGAGCAGCTCTATTGGTGTGGCAAGCTCTAAGGGCGTTGCGTCTAGCAGCAAAACCGCGACCAAAGCGCGTATTAGCGCGGGTAGCAGCGGCAGCTCTACCAGCACCAAGTCTAGCAGCTCTGCAAGCACTGCGGTTCCGACCAACTTATCTGGCTCTCGTAGCCATGCATTAAGCTCTTCTAACAGCGGCCAGGACAACACTGTTGGTGATGATTTTGGCCTGGGCTATATTAGCGGTGGCATCCTGCCAGTTAACACCCCAGCGCTGAATTTTCCAAGCGATTTAGGCTCTTTAACTAGCGGCCTGCTGAGCTCTTTAGATGGCCCAGTGTTACCGTCTGTTGAGTATCGTATCACTTCTTTAACTTCTTCTGTTTTAAGCCTGCTGAGCACTAGCGGCGGTGCGTTTAACTACTCTAGCTTCGCGAAAAACCTGGCAATCCTGGCGTACCAAATCAGCGTTAGCAACCCGGGTCTGAGCGTGTCTCAGGTGGTTTCTGAGACCCTGTTAGAATCTGTTGGCGCACTGATCCATATCCTGGTGAGCAGCCAGGTGGGT
The question still stands… How can I produce this protein using my DNA?
Well obvioulsy I can’t make the sequence straight from my DNA since my body doesn’t produce silk. What I can do instead is use my DNA as a backbone (which I can just extract from my cells) and edit it so it has the fibroin sequence using CRIPS-based editing or PCR-based gene assembly. After I have the right sequence for fibroin production, it will be insedrted into a plasmid and then transported into E-coli cells. Inside a cell, RNA polymerase reads the DNA sequence and it produces mRNA copy of it. Ribosomes translates it into the polypeptide chain which is then folded into it’s desired shape (in our case silk fibers).
Part 4
4.2
This is the given example with the E-coli glowing phosphorescent green under UV light.
4.3-6
Part 5
5.1 DNA READ
i) Since I’m a computer engineer at base, I would love to be able to make a synthetic DNA data storage. Other than my personal preference for this conjucture of amazing subjects, DNA data storage is the most efficient way of storing information since it doesn’t need any energy after creation and can store insane ammounts of information (~215 petabytes in 1 gram of DNA) for long periods of time in minimal spaces. However sequencing is critical due to the errors that can occur during encoding. Since DNA stores information passively, sequencing is crucial when decoding aswell.
ii) I would most likely need the Third-generation sequencing mostly because the DNA sequences that are supposed to be stored are long. Not only that, but since ONT can read continious sequences of DNA molecules and give me real-time answers clearly makes it the superior option. In addition, it does not require PCR amplification to create a signal, meaning it bypasses the “vicious” cycle of amplification biases that can sometimes lead to errors in GC-rich regions of the genome.
5.2 DNA write
i) In my opinion, it would be a dream to be able to produce a synthetic genetic construct that allows a host organism to efficintly produce spider silk proteins. The production of silk should most likely be regulated so I know exactly the length of each strand, or why not make them arbitrarily. Why you may ask? Silk is one of the strongest biodegradable materials known to man, and it also has a lot of uses in medicine, textile industry and engineering. For some kids out there maybe the “I want to become Spiderman” wish might come true one day.
ii) So what technologies would I need?
If I wanted to synthesize a spider‑silk gene, I’d start by ordering commercially made oligonucleotides and then assemble them using methods like Gibson Assembly or overlap‑extension PCR. These approaches are well‑suited for building long, repetitive genes and let me incorporate codon optimization along with any regulatory elements I need, such as promoters, ribosome‑binding sites, and terminators.
To verify the construct, I’d rely on a combination of Sanger sequencing and long‑read next‑generation sequencing (like PacBio or Oxford Nanopore). Sanger is extremely accurate (around 99.99%) but it’s slow and not ideal for very long or highly repetitive sequences, so I’d use it mainly to confirm individual repeat units. Long‑read NGS, on the other hand, can capture the entire gene in one go and handles repetitive regions well, though each read is a bit less accurate and requires some bioinformatic assembly.
Using both methods together gives a high level of confidence that the full‑length gene was synthesized correctly, striking a good balance between accuracy, speed, and scalability.
5.3 DNA edit
i) When I think about valuable DNA edits, I’m most interested in changes that improve health, resilience, and environmental sustainability, rather than enhancements for their own sake. In humans, this could mean correcting disease-causing mutations, like inherited metabolic disorders or certain forms of blindness, where fixing a single faulty gene could prevent suffering without changing a person’s identity. Another promising area is reducing age-related decline, helping people stay healthier as they get older rather than trying to extend lifespan indefinitely. To me, the worst part about aging isn’t my appearance changing, but me losing my senses (poorer eyesight and hearing), so something of great interest to me would be to manage to slow down something like eyesight loss.
I’m cautious about human enhancement, such as boosting intelligence or physical ability, because of ethical concerns around fairness and consent. But using DNA editing to prevent disease, strengthen ecosystems, and support sustainable agriculture seems like a direction with clear benefits and manageable risks.
ii) What technology or technologies would you use to perform these DNA edits and why?
If the goal is to reduce eyesight degradation, particularly conditions driven by inherited mutations or age‑related cellular decline—the most appropriate technologies would be those that offer high precision and minimal disruption to the genome. In this context, CRISPR‑based tools remain the leading candidates, but not the traditional cut‑and‑repair versions. Instead, I would look toward base editing and prime editing, which are designed to make extremely targeted changes without creating double‑strand breaks. That matters because retinal cells are delicate, slow‑dividing, and difficult to replace, so any editing approach needs to be as gentle and predictable as possible.
Base editors are useful when eyesight degradation stems from a single incorrect DNA letter, because they can convert one nucleotide into another with high specificity. Prime editors go a step further, allowing slightly larger corrections or small insertions and deletions, which could be valuable for conditions where the underlying mutation is more complex. Both technologies aim to correct the genetic cause of degeneration before it leads to irreversible damage.
For age‑related vision loss, where the problem isn’t a single mutation but a gradual decline in cellular function, the focus would shift toward editing pathways that support cellular resilience, mitochondrial health, or protective responses to oxidative stress. Again, precision tools like prime editing would be preferable, because they allow subtle adjustments to gene regulation without fundamentally altering the identity or behavior of retinal cells.
Across all of these possibilities, the priority is safety: technologies that minimize off‑target effects, avoid unnecessary DNA breaks, and allow careful control over the extent of the edit. The retina is one of the most sensitive tissues in the body, so any intervention must be both scientifically justified and ethically grounded
Week 3 HW: Lab automation
Part 1:
Here is the reference image that I managed to make using the opentrons art GUI:
I tried my best to recreate it on my own (no ai) in colab but it didn’t work quite well:
I will most likely have fun with the python code later aswell! Maybe I’ll design an even better looking cat.
This paper looks at how a research team used the Opentrons OT-2—an affordable liquid-handling robot—to take over one of the most tedious parts of protein research: setting up crystallization experiments. Growing protein crystals is famously slow, delicate work, and when scientists need lots of crystals that all behave the same way—for studying molecular structures or making biomaterials—the manual effort quickly becomes exhausting. The researchers wanted to know if a general-purpose robot like the OT-2 could handle this large-scale setup reliably.
A difficult part about protein crystalization automation comes from the preparation of the samples. To speed up the process, they programmed the robot in Python and modified it to work with larger 24-well crystallization plates. This wasn’t plug-and-play—they had to design and 3D-print a custom adapter and create a matching software definition so the robot would know how to handle the plates. They then put the system through three tests: mixing colored liquids to check accuracy, growing crystals of lysozyme, and crystallizing a more complex protein from Campylobacter jejuni that their lab studies as a biomaterial.
The results were better than expected. The robot could mix solutions, fill reservoirs, and set up crystallization drops accurately and consistently. When compared with plates prepared by human researchers, the robot’s plates actually produced crystals more reliably—although the robot worked a bit more slowly. Just as importantly, it spared scientists from hours of repetitive pipetting, reduced experiment-to-experiment variation, and managed most tricky steps with only small tweaks for thicker liquids or mixing needs.
In short, the study shows that a relatively inexpensive, flexible robot can take over a big chunk of protein crystallization work. That makes experiments more consistent and far less labor-intensive. For labs that don’t have the budget for specialized crystallization machines, this approach could be a practical and accessible alternative.
I’m not yet 100% sure what my final project, so for the sake of answering the question regarding lab automation in my project let’s take the fungal-material growth project. The project consists of a small automated setup for growing fungal mycelium in a controlled environment. Lab automation is important because it reduces the ammount of work keeping the conditions (like humidity, temperature, airflow, and moisture) of a certain test ideal, that would be absolute hell to do by hand. Mycellium is very sensitive, automating them should make the growth process more consistent and easier to reproduce.
The setup will include sensors that track humidity and temperature, along with a microcontroller that adjusts a humidifier, heater, and small fans to keep conditions stable. I also want to automate substrate hydration using a simple pump system, since moisture levels strongly influence how the material develops. A camera will record the growth over time, allowing me to monitor progress and analyze how different conditions affect the final material. The camera is very important since it can help me detect contamination and also measure the are covered by the fungi
All data from the sensors and the camera will be logged automatically, giving me a clear picture of how the fungus responds to different environments. By combining these elements, the project becomes a small “smart” fungal farm that can grow mycelium materials in a controlled, repeatable way.
Week 4 HW: Protein Design I
Part A
How many molecules of amino acids do you take with a piece of 500 grams of meat?
Let’s assume that meat is on average 20% protein. so 500g of meat would mean 100g of protein.
After calculations we can assume that 1Da/molecule = 1g/mol, so by having around 100 Da on a amino acid we can say that we have approximatelly 1 mol of amino acid residue in 500g of meat.
Therefore in total we have 6.022*10^23 amino acid molecules.
Why are there only 20 natural animo acids?
DNA uses triplet codons so there are 64 possible codons. Out of these, 3 are stop codons so there remain only 61 available code for amino acids. These 61 codons map redundantly to 20 core amino acids. Early life likely used fewer than 20. As new amino acids were gradually incorporated, the code expanded—then “froze.” Once the translation machinery (ribosomes, tRNAs, enzymes) became complex, changing the code would have been catastrophic. So evolution locked in the set. It’s just like a code that wants to become more and more complex but instead of adding new data types, they are forced to use already existing data types to form something else which would most likely be useless and would just become more difficult to use.
Also each amino acid uses a specific tRNA and a matching animoacyl-tRNA synthetase enzyme, so adding new ones require an even more complex and risky path to not accidentally cause cross-reactions between them.
The already existing 20 amino acids can achieve hydrophobic cores, charged interactions, structural rigidity, a stable 3D structure after folding.
Can you make other non-natural amino acids? Design some new amino acids.
Yes you can design non-natural amino acids and scientist have already done so. We can design new ones by modifying the side chain while keeping the core amino acid backbone: NH₂–CH(R)–COOH. Instead of R I can place something else.
For example, by replacing one single methyl group with a trifluoromethyl group in Leucine (R = –CH₂–C(CH₃)(CF₃)) we will obtain a superhydrophonic core builder that is more resitant to oxidation.
Another example would be an nnAA that changes shape when exposed to light allows light-controlled protein folding. We can obtain this by replacing the side chain with an anzobenzene group (R = –CH₂–C₆H₄–N=N–C₆H₄).
Where did amino acids come from before enzymes that make them, and before life started?
The first thing we need to understand is that amino acids can form abiotically, they can be synthesized through chemical reactions under certain conditions, whithout needing enzymes or life. Some of the key ways scientists think amino acids could have formed before life include Miller-Urey type synthesis (simulated early Earth’s atmosphere and electric sparks and managed to obtain several amino acids), hydrothermal vents (Dee-sea hydrothermal vents release hot, mineral-rich water that can catalyze chemical reactions to produce amino acids from simpler molecules like CO₂, H₂, and NH₃) and extraterrestrial delivery (amino acids found in meteorites).
Enzymes are proteins that can catalyze reactions in life, but this doesn’t mean that reactions don’t work without them. They still do, slower and less specific, but under the right conditions they can still happen.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A D-amino acid is the mirrored version of an L-amino acid, so the α-helix headedness of a D-amino acid will be the opposite of one of a L-amino acid.
Since almost all amino acids in natural protein are L-amino acids we can understand easier how they form.
The α-helix is defined by specific torsion angles (ϕ and ψ) along the peptide backbone:ϕ (phi) = rotation around N–Cα bond and ψ (psi) = rotation around Cα–C bond. For a helix to form, the backbone must adopt a set of angles that allows regular hydrogen bonding between the carbonyl oxygen of residue i and the amide hydrogen of residue i+4. The specific geometry of L-amino acids makes this hydrogen-bonding network more favorable in a right-handed twist.
L-amino acids have the Cα in the S configuration (except glycine). When you try to twist the backbone into a helix a right-handed twist positions the side chains away from the helix axis without clashing with the backbone. On the other hand, a left-handed twist would force side chains into steric clashes with the backbone or the other side chains. A left-handed helix is possible but it will require a lot more energy to form.
So L-amino acids form right-handed helices, so if we mirror it we can say that R-amino acids form left-handed helices
Can you discover additional helices in proteins?
Yes you can discover additional helices. An α-helix is a secondary structure element characterized by: Backbone hyrogen bonds (between carbonyl oxygen of residue i and the amide hydrogen of residue i+4), Specific φ and ψ angles, and right-handed twist. If a stretch of amino acids satisfies these geometric and hydrogen-bonding criteria, it can be classified as an α-helix.
There are several ways to identify α-helices in proteins:
X-ray crystallography & cryo-EM
These methods give atomic-resolution structures
You can look at the backbone position and see repeating helical patterns.
Software and algorithms
Like DSSP, STRIDE and PyMol
Sequence prediction
Certain amino acids have high helix-forming propensity
Predictive algorithms like PSIPRED or JPred can guess where helices are based on sequence alone.
There also exist other types of helices like 3₁₀ helices (tighter, 3 residues per turn) and π-helices (looser, ~4.4 residues per turn)
Why are most molecular helices right-handed?
Almost all natural amino acids are L-amino acids. I’ve talked a little bit earlier about how do L-amino acids make right-handed helices. Shortly, when the peptide chain coils to form an α-helix, the side chains naturally point outward, and the backbone hydrogen-bonding pattern is energetically most favorable when the helix is right-handed.
Most molecular helices are right-handed because the chirality of their building blocks makes right-handed twisting energetically favorable, avoiding steric clashes and stabilizing hydrogen bonds.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
First of all let’s see what a β-sheet. A β-sheet is a common secondary structure in proteins, formed by β-strands linked by hydrogen bonds. Some characteristics of β-sheet are Backbone hydrogen bonding (Between the C=O of one strand and the NH of another strand), side chain orientation (Side chains alternate above and below the sheet) and its arrangement (parallel β-sheet strands run in the same N→C direction, antiparallel β-sheet strands run in opposite directions).
β-sheets have an inherent tendency to stack into extended, layered structures, and this is driven by several forces:
Hydrogen bonding:
Exposed backbone NH and C=O groups at the edges of β-sheets can form additional hydrogen bonds with other β-sheets. This allows β-sheets to “zipper” together, forming extended sheets.
Hydrophobic interactions:
Side chains alternate above and below the sheet.
Hydrophobic side chains on one face can pack against hydrophobic side chains of another sheet.
This minimizes exposure to water, driving aggregation.
Van der Waals forces:
Close packing of side chains in stacked β-sheets leads to favorable van der Waals contacts, further stabilizing aggregates.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloid disease involve proteins that misfold and aggregate into β-sheet–rich fibrils.
The backbone always wants to form hydrogen bond. Every protein backbone has repeating carbonyl and amide groups. If these groups are not satisfied by internal folding, they seek hydrogen bonds elsewhere.
When a protein partially unfolds, its backbone becomes exposed and neighbouring molecules can form hydrogen bonds with it and it forms a cross-β structure (since the easiest repeating pattern for intermolecular H-bonding is a β-sheet alignment), the hallmark of amyloid fibrils.
Another factor could be the fact that β-sheets allow infinite extension. Unlike α-helices, that are stabilized within 1 molecule, β-sheets cam form between different molecules, in an extended reapeting way, without needing complex folding. This means they can stack and grow into long fibrils.
Also, amyloid β-sheets have high thermodynamic stability and are often more stable than the original folded protein. So under stress, mutation or aging, proteins can fall into deep energy minimum.
Amyloid β-sheets can be used as materials due to their mechanical strength and self-assembly mechanisms. There are some applications being explored right now, such as tissue engineering scaffolds, drug delivery systems and sustainable biomaterials.
Part B
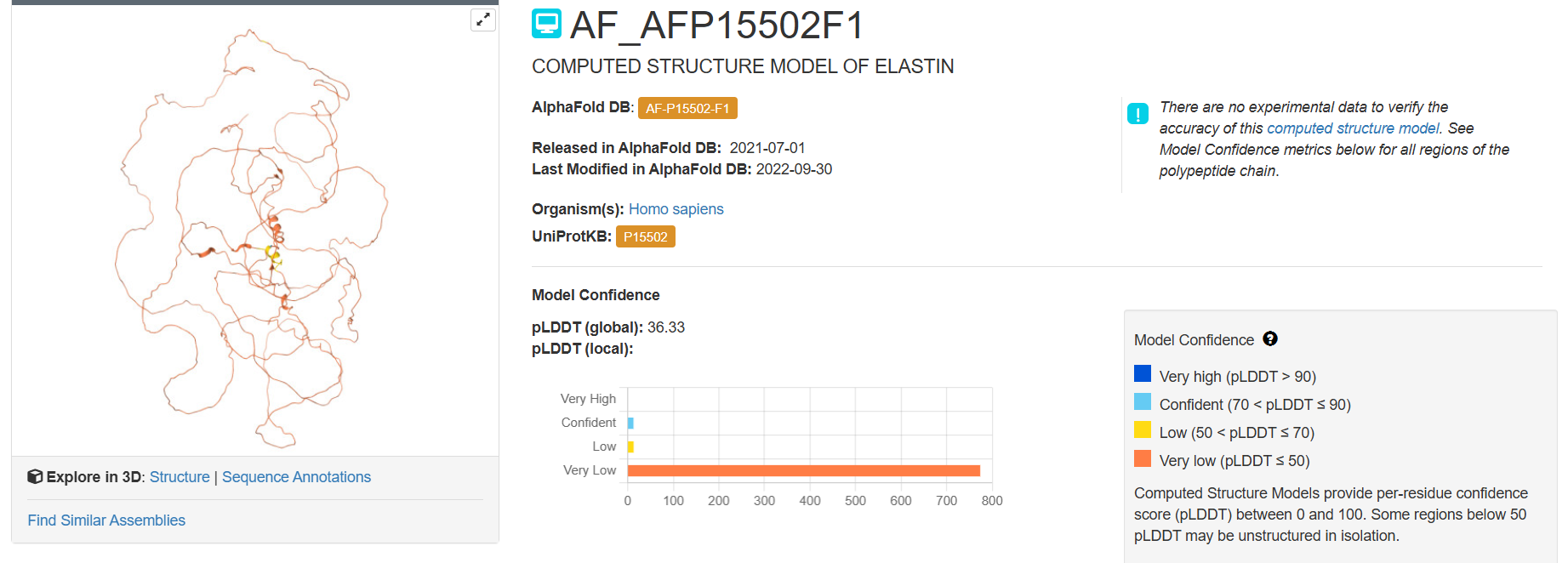
chosen protein: Elastin
Elastin is a protein encoded by the ELN gene in humans and several other animals. Elastin is a key component in the extracellular matrix of jawed vertebrates. It is highly elastic and present in connective tissue of the body to resume its shape after stretching or contracting. Elastin helps skin return to its original position whence poked or pinched. Elastin is also in important load-bearing tissue of vertebrates and used in places where storage of mechanical energy is required.
I was done with fibroin and silk, but I wanted to keep a sort of continuity between my proteins so I chose Elastin due to its stretching functionality.
Here is the elastin sequence of the human body found on Uniprot:
It has 786 amino acids, the most frequent one being G, which appears 225 times.
There are 250 homologs for my protein.
Elastin belongs to the elastic fiber protein family, alongside elaunin and oxytalan.
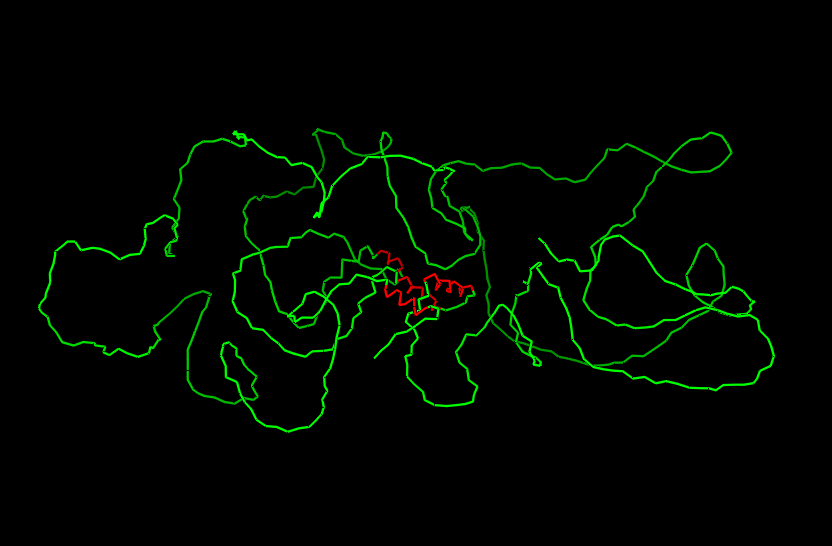
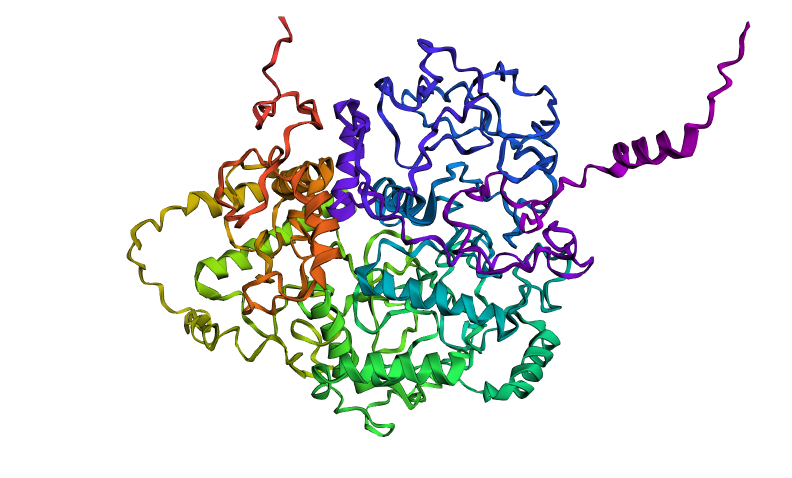
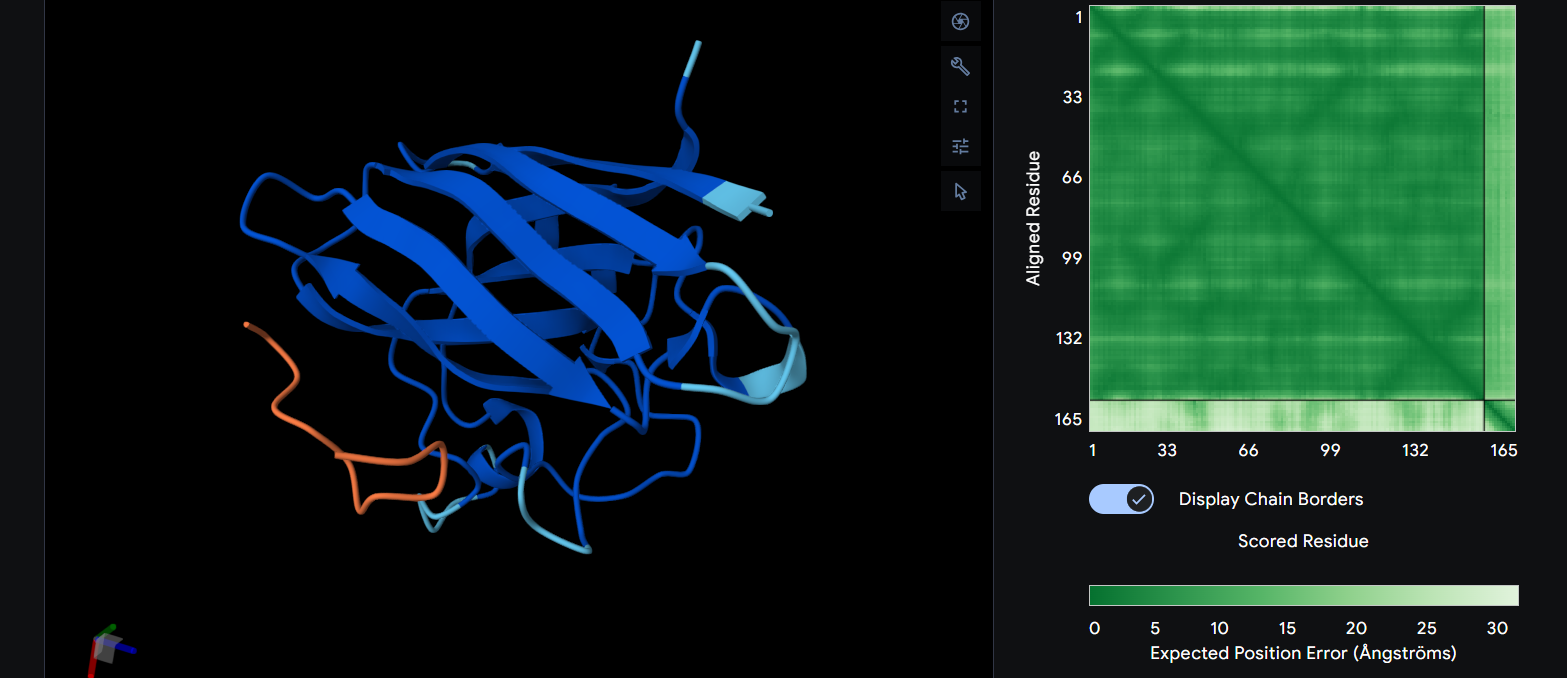
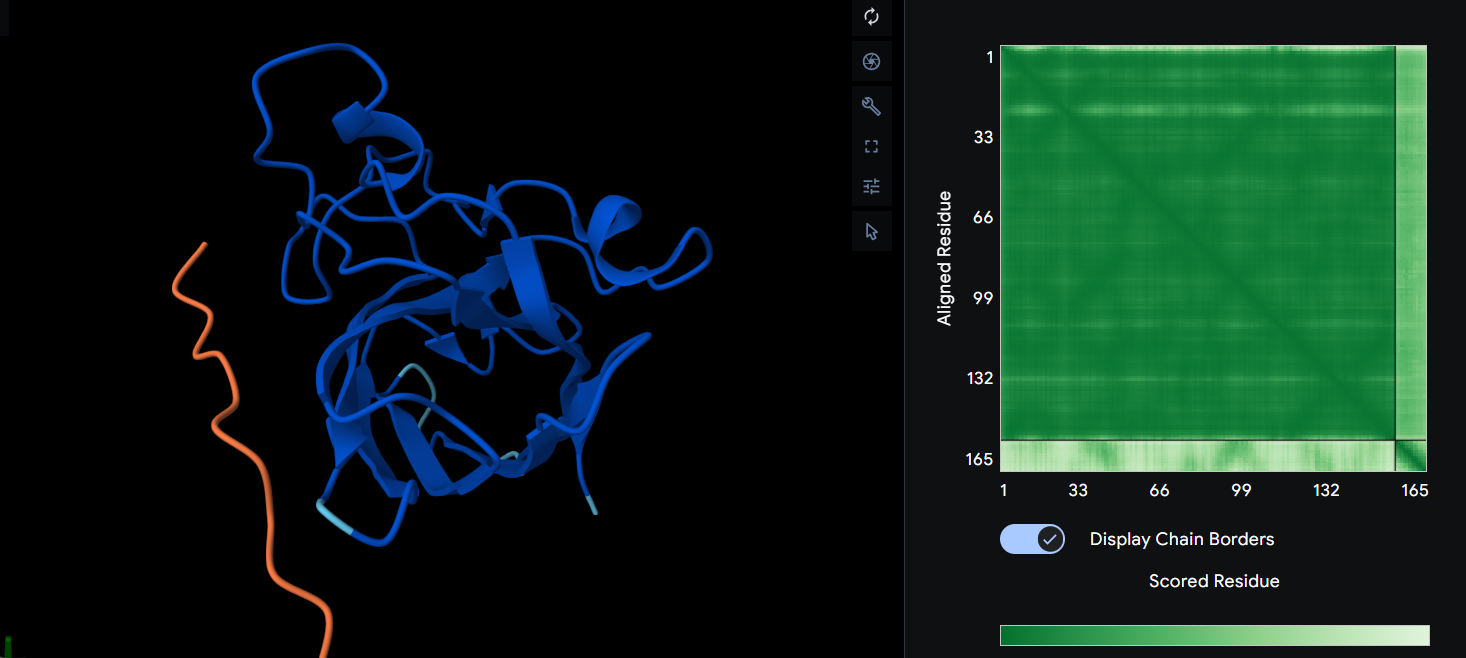
Here you can see the 3D representation I found on RCSB of Elastin found in human body:
It was release firstly in 2021.07.01 and lastly modified in 2022.09.30. as you can see on the bottom right, the confidence score is very low. There aren’t any other molecules in the solved structure apart from my protein.
Elastin belongs to the fibrous protein family and is structurally classified as a structural extracellular matrix (ECM) protein.
For the next part I couldn’t find from RCSB the PDB file needed so I searched for it and found the exact protein on Alphafolds protein structure database.
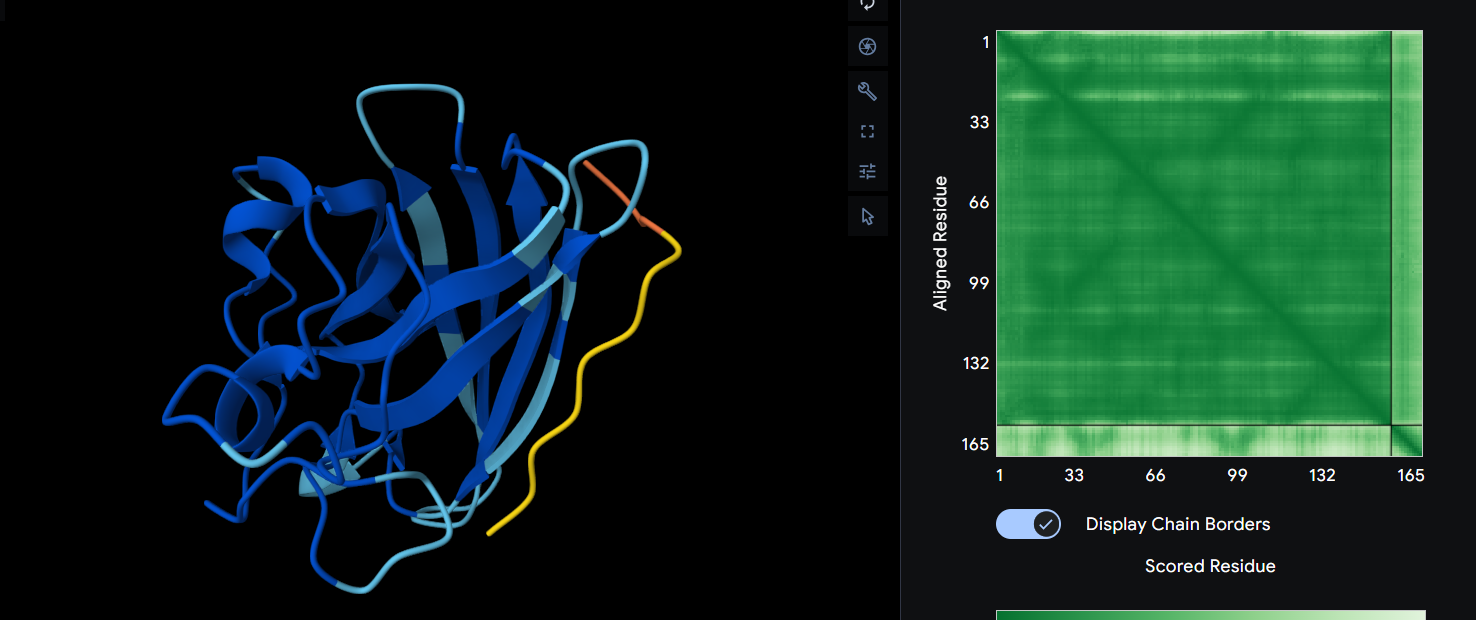
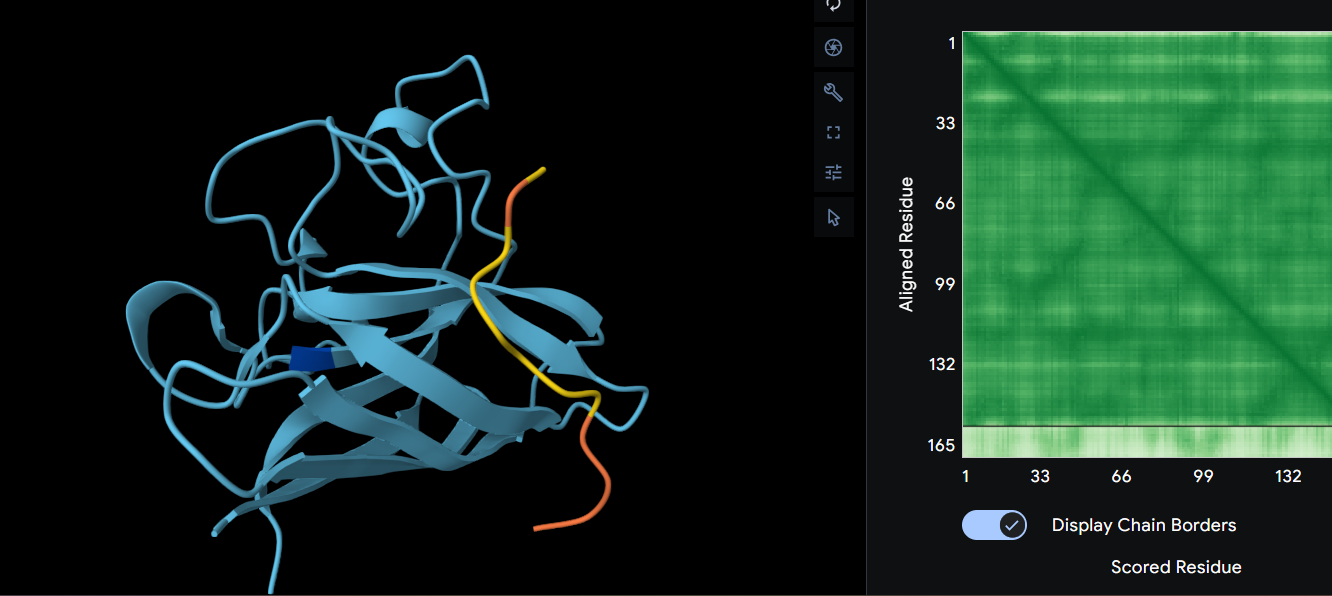
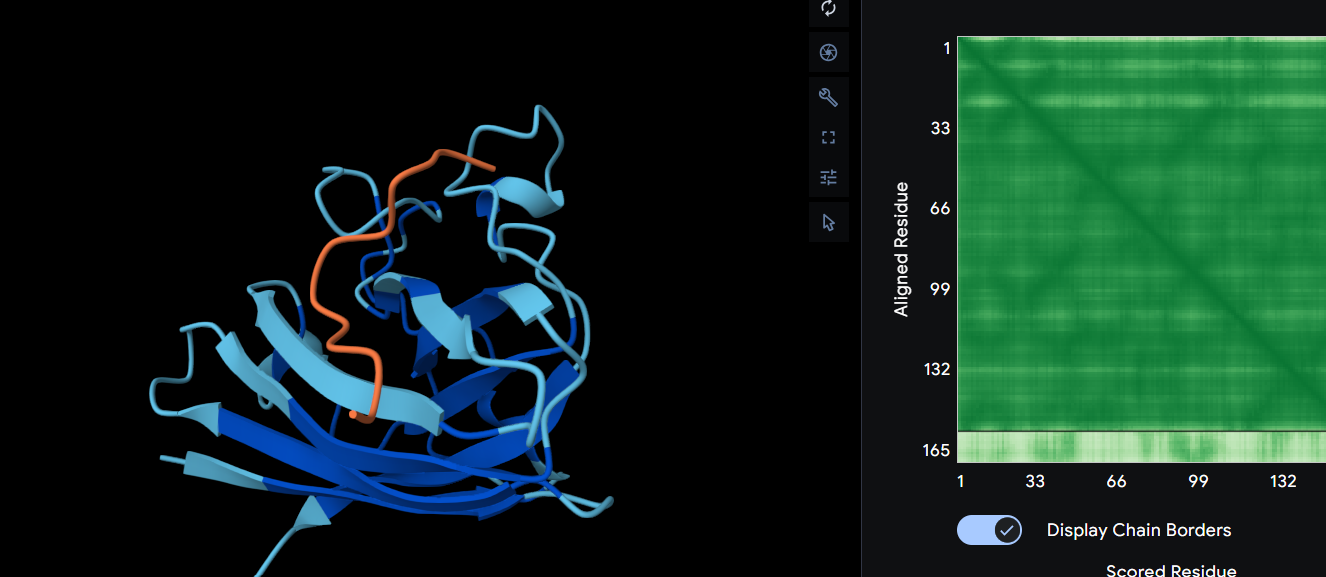
Ribbon
Cartoon
Ball and Sticks
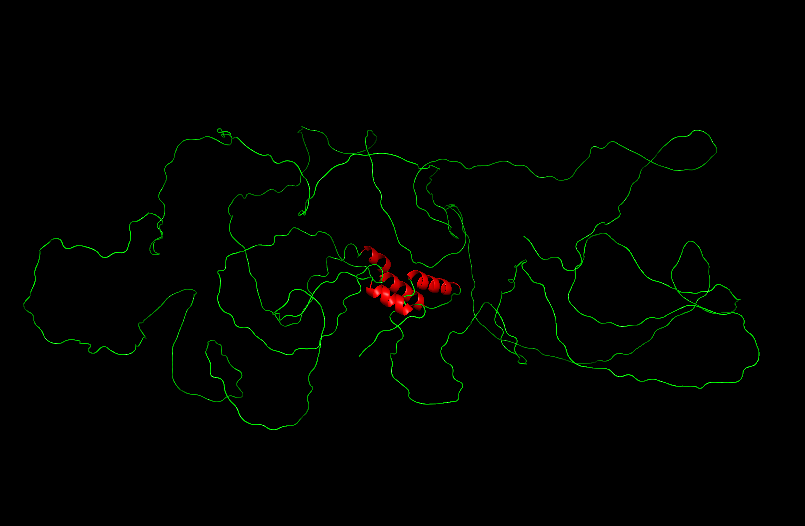
After coloring the secondary structure (colored in cyan and purple) we can clearly see that it has more helices than sheets.
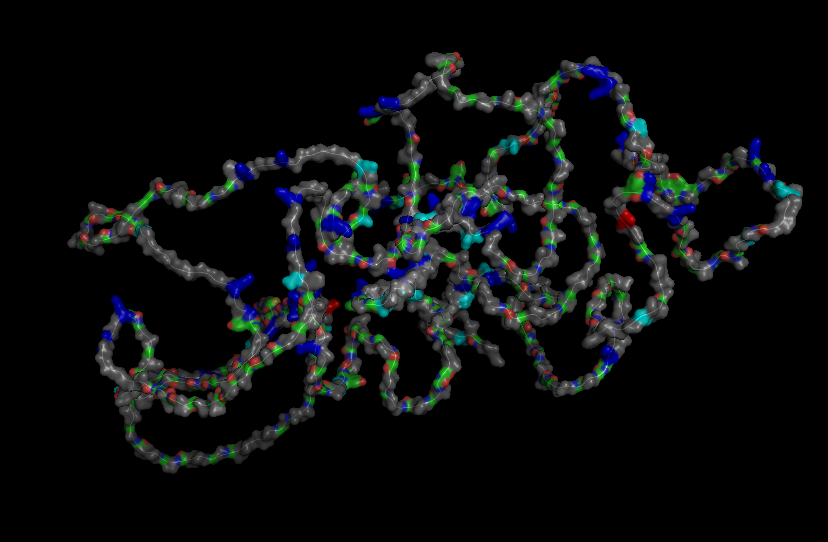
Here is the Protein after highlighting the residues:
With grey I have the Hydrophobic residue and with the everything else is the Hydrophilic residue. The hydrophilic residue is divided into Polar uncharged (green), positively charged (blue) and negatively charged (red) residues. We can see that Elastin is highly hydrophobic due to how many grey regions are spread across the whole protein. There are very few hydrophilic parts as we can see and they are mostly positively charged around the edges. Inside the helixes of the secondary structures we can see yet again how most of it are hydrophobic which is understandable, due to Elastins ability to coacervate and form elastic fibers.
The Elastin has little to no binding pockets which is perfect due to its nature. Elastin is an intrinsically disordered protein (IDP). It doesn’t fold into a compact globular shape like enzymes or receptors, instead, it forms long, flexible, coiled chains that assemble into elastic fibers. This is also a reason for the Hydrophobic core.
Part C
C1. Protein Language modelling
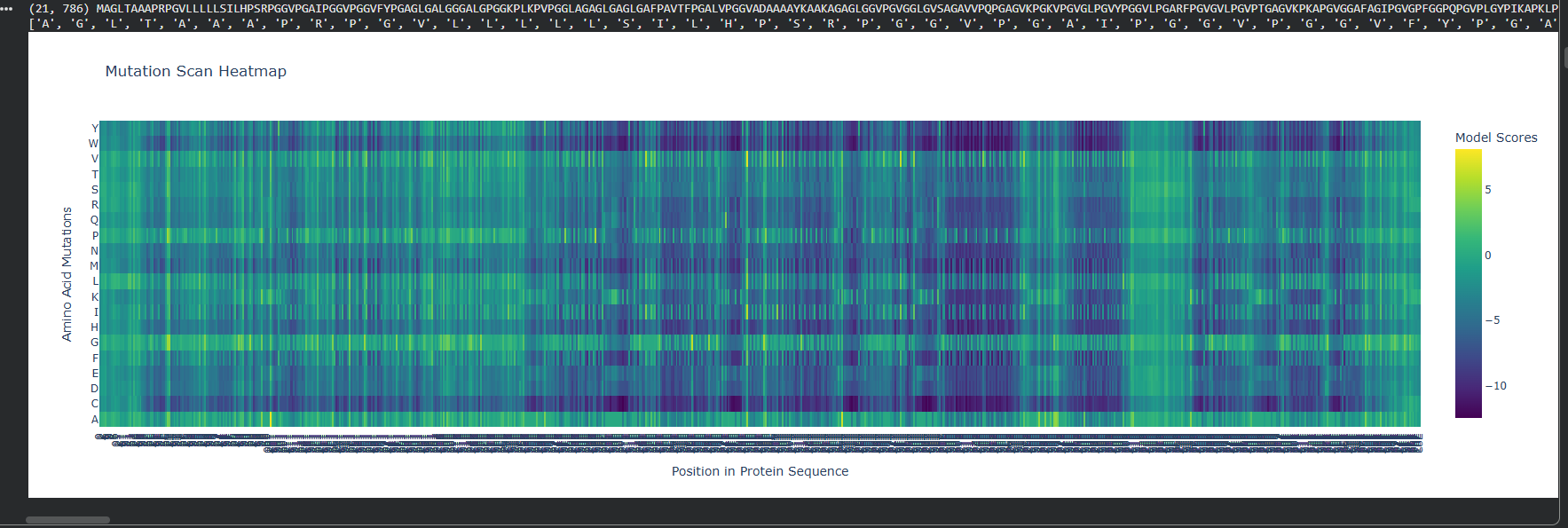
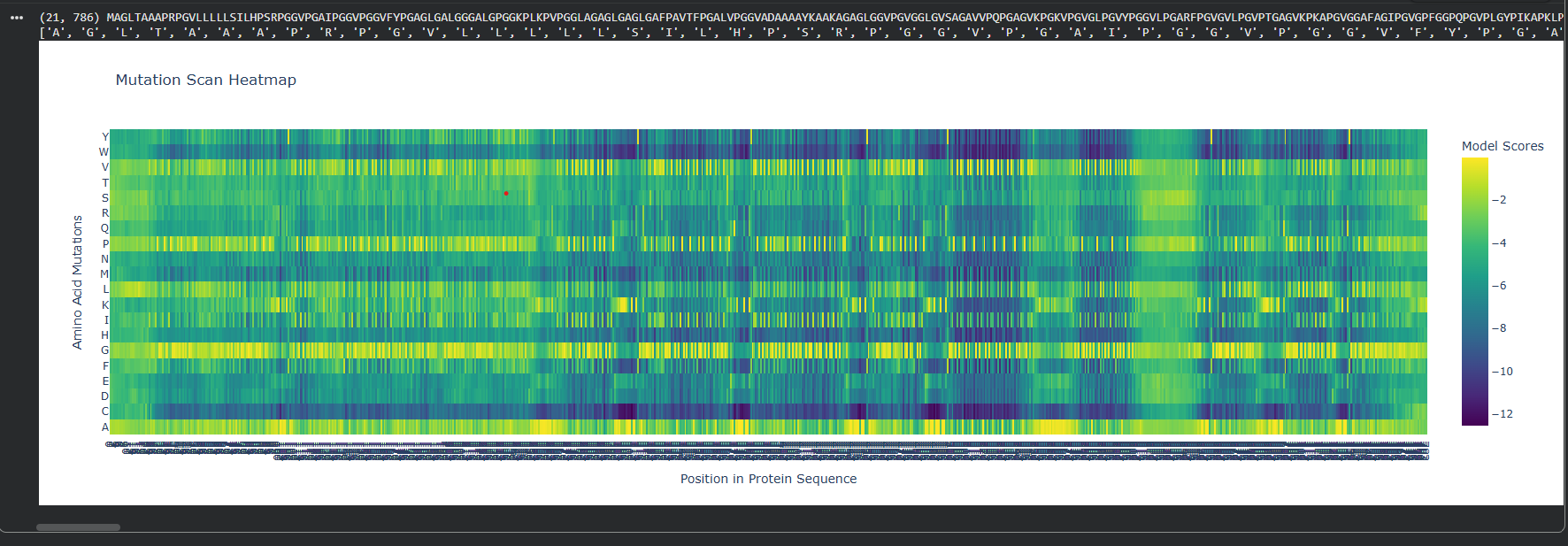
At first I ran two DMS (deep mutational scans) with the given options of relative and absolute to also analyze the difference between them.
Relative DMS
Absolute DMS
We can see that the Relative DMS shows that most Mutations don’t affect te function of the protein, unless it’s a mutation around the middle of the structure (where I assume is the secondary structure due to how the protein looks), where almost any mutation greatly affect the proteins functionality. In very few places we can even see that mutation can improve the functionality in relation to the strating population.
On the other hand, the Absolute DMS shows us that most mutations don’t change the enzyme activity, the binding affinity, the measured growth rate and the chance of survival. However no mutation can improve the proteins function.
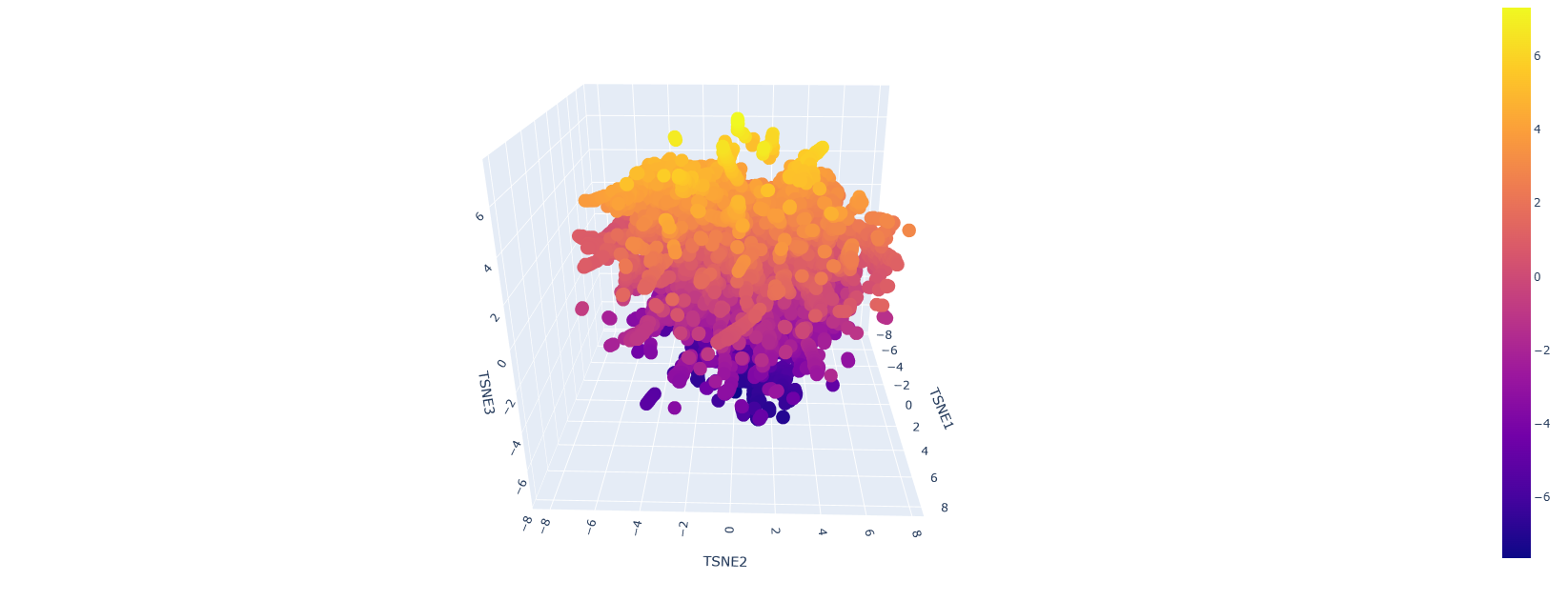
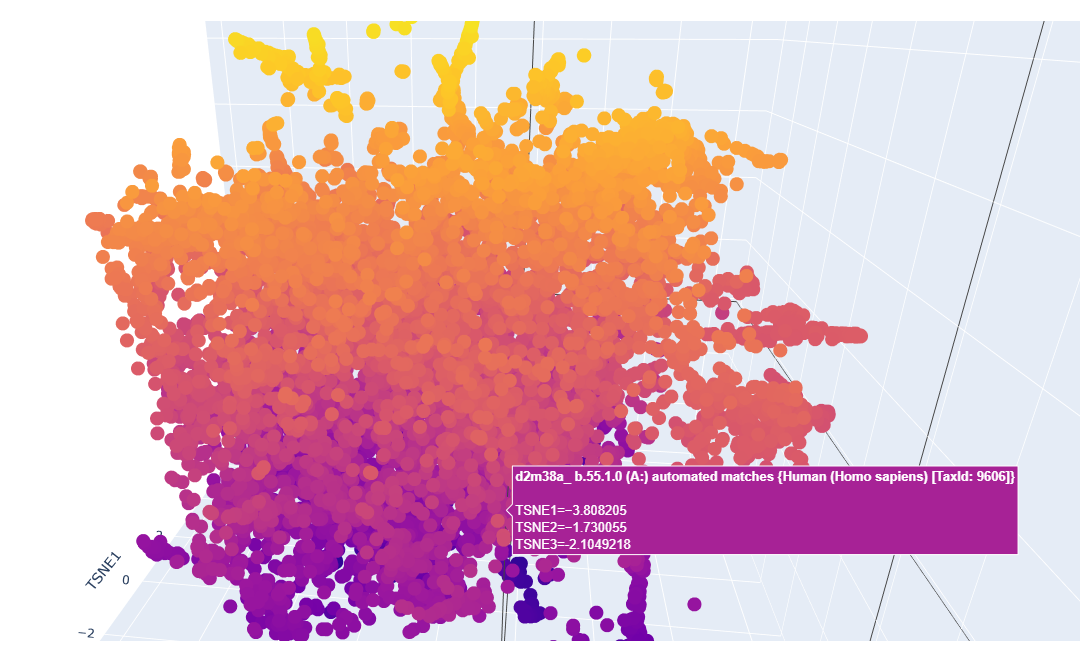
Here is how the LSA inspection looks like:
This is a sort of close-up look on it:
It looks like a mess but upon a closer inspection we can clearly say that we have similar protein designs. The yellow parts represent proteins that are similar and some even better, the dark blue or purple ones are the mutations that are not viable.
C2. Protein folding
Here is the folded protein model by the given colab code:
They are matching judgind by the fact that the one that I found is a pretty bad representations. We can see the helices in the middle being similar.
So my colab keeps on crashing, I will try to solve this issue as soon as possible so I can run the final tests on mutations and inverse folding
Here are some mutations I managed to do:
Working on the mutations we can see that
C3. Inverse Folding
Currently I’m experiencing some issues regarding the inverse folding…
Part D
Computational Design to Stabilize the MS2 Lysis Protein and Disrupt Its Interaction with E. coli DnaJ
Proposed Computational Approaches
In Silico Mutagenesis and Stability Prediction
Protein Language Models (PLMs):
Use state-of-the-art PLMs (e.g., ESM-2, ProtT5, MutaPLM) for zero-shot and guided mutagenesis to predict stabilizing mutations across the L protein sequence.
ΔΔG Calculations (measure the stability change of a protein upon mutation):
Employ energy-based tools (FoldX, RosettaMP, and MD-based free energy estimators) to quantify the impact of mutations on protein stability, especially in the membrane context.
Molecular Dynamics (MD) Simulations:
Validate top mutations with MD and enhanced sampling (e.g., GaMD) to assess conformational stability and flexibility.
Pipeline Management and Reproducibility
Workflow Management:
Implement the pipeline using Snakemake for reproducibility, scalability, and transparent documentation of all computational steps.
Visualization and Analysis:
Use ChimeraX and PyMOL for model inspection, interface mapping, and figure generation.
Justification for Tool Selection
PLMs (e.g., ESM-2, MutaPLM, ProtT5):
These models capture evolutionary and structural constraints, enabling accurate prediction of mutational effects, even in the absence of deep MSAs. MutaPLM, in particular, offers explicit modeling of mutation effects and supports explainable, engineerable protein design.
AlphaFold-Multimer/AlphaFold3:
These AI-based predictors provide high-accuracy models of protein complexes, crucial for mapping the L:DnaJ interface and guiding mutagenesis.
Energy-Based Stability and Interface Tools (FoldX, RosettaMP, DockQ):
These methods are validated for ΔΔG and interface scoring, allowing quantitative assessment of stability and PPI disruption.
MD Simulations:
MD provides atomistic insight into the dynamic consequences of mutations, complementing static predictions and revealing potential allosteric effects.
Snakemake:
Ensures reproducibility, modularity, and scalability of the computational workflow, facilitating transparent and collaborative research.
Potential Pitfalls and Limitations
Modeling Limitations:
AlphaFold-Multimer and PLMs may overpredict interface confidence or fail to capture dynamic, transient, or allosteric interactions, especially for small or flexible proteins like L and DnaJ.
ΔΔG predictors (FoldX, RosettaMP) are sensitive to input structure quality and may not fully account for membrane context or oligomerization.
Biological Relevance:
In silico predictions may not translate directly to in vivo function due to cellular context, post-translational modifications, or compensatory mechanisms.
Disrupting the L:DnaJ interface may inadvertently destabilize L or affect its oligomerization, requiring careful multi-objective optimization.
Week 5 HW: Protein design II
Part A: SOD1 binder peptide design
Using the Uniprot database, here is the SOD1 Protein sequence that I managed to find:
After using the PepMLM Colab I managed to obtain these 4 peptides and their perplexity. After that I added the known binder and computed its perplexity the same way, in the end having these 5 proteins:
Perplexity tells us how confident the model is that a peptide sequence fits (or binds) the given protein context. In this case we can see that the most likely peptides to bind to my human SOD1 protein are the first and third one. Something wierd that I notice is that the know binders perplexity is quite high which makes it less plausible, despite it being tested to work. I’ve tried multiple times but still got the same result, which is a little bit unexpected.
Next we had to navigate through the alphafold server and see how my peptides actually bind to the SOD1 protein. Here are the results, alongside their ipTM score:
FLYRWLPSRRGG (the know binder) ipTM = 0.29
We can see that it has a β-barrel fold and it is completely surface bound, having only one part to bind with the protein. Also something wierd that I noticed aswell is the ipTM being relatively low on a protein that is known to bind to it. I can’t explain why but on my simulation it doesn’t bind near the N-terminus, rather it bind somewhere near the center.
WRYGVVAVAWWE ipTM = 0.34
It is still has a β-barrel fold and completely surface bound. However what is different is the fact that the prediction says it binds both ways and we can see that for this peptide the alphafold tells us that it’s more likely to exist in that shape. On the other hand, this peptide does actually bind to the A4V mutation and it affects its structure.
WRYYVVALAHWE ipTM = 0.29
For this protein we’ll have another β-barrel fold and surface bound. This protein affects the whole proteins structure, meaning despite the peptide is more stable, it might affect the proteins structure more than the others did so far. This one doesn’t look like it’s binding to the N-terminus either, but it still affects the folding probability.
WRYPAAGVRLKK ipTM = 0.31
In this scenario we can observe what looks like a dimer interface since it’s a little bit shifted away from the β-barrel region, yet it is still surface bound. After its prediction, it binded on the completely other part of the protein so it doesn’t affect the A4V mutation. This peptide affects the structure and alphafold is also unsure about it’s own folding.
HRYPAAGVELWE ipTM = 0.3
Yet again we have a dimer interface that is surface bounded. However, in this scenario the protein itself is more likely to fold the way alphafold predicts. It is nowhere near the N-terminus of the A4V mutation.
Unfortunately none binded to the A4V mutation.
Conclusions
Judgind by the ipTM values, we can say that almost all of them have a higher ipTM score than the one of the known binder. The best peptide generated is the ‘WRYGVVAVAWWE’, which was generated by PepMLM since it binds to the mutation directly and it also keeps the structure of the protein pretty much the same. The ipTM value is also higher, suggesting a potentially comparable interaction interface according to AlphaFold-Multimer predictions.
Using its exact format, here are the probabilities generated by Peptiverse for each peptide:
One thing that we notice straight away is that the peptide with the highest ipTM is also has the highest predicted binding affinity, which is pretty much obvious since it actually targets the mutation. We also notice that not for every ipTM value that is higher than the one of the known binder means better affinity, like for ‘HRYPAAGVELWE’ (the last one). After the predictions, all peptides are soluble and for most cases, stronger binders usually mean a more hemolytic probability, despite still being non-hemolytic. The exception would be ‘WRYPAAGVRLKK’(the third one). In conclusion, I believe that the best generated peptide that balances binding affinity and therapeutic properties is ‘WRYYVVALAHWE’ (the second one). Personally I would still suggest that the protein we should continue is the one with the best binding affinity, due to it targetting the mutation and being the most likely one to bind. I believe that the hemolytic properties I don’t think it’s that much of a problem since it is still considered safe.
After the moPPit generation, I have found these 4 more peptides: ‘LTEKITQTLHTC’, ‘SKQYKCQLTGDI’, ‘STDQTCGKVKLM’, ‘DEKKFQRCTTTT’, and their respective
Hemolysis, Solubility, Affinity (from the first 10 aminoacids) and Motif: [0.9491806849837303, 0.75, 5.967530727386475, 0.7040762305259705], [0.9336216971278191, 0.8333333134651184, 6.318202018737793, 0.7738364934921265], [0.9584326893091202, 0.75, 6.513046741485596, 0.7761058807373047], [0.9732194431126118, 0.9166666865348816, 5.4131293296813965, 0.7708268165588379]. First of all I see that for some reason they all have insanely large hemolytic properties so that is already a dangerous sign. Their binding affinity is also not as good as the pepMLM generated ones and the solubility is probably more precise which is a lpus but doesn’t help with all the disadvantages that the high hemolytic nature brings.
Part C: L-Protein Engineering. Random Mutagenis
Here is an example of code that can be used for this activity:
(I have absolutely no clue how to make it all beautifully alligned in a box for code)
import random
def generate_random_mutants(sequence, num_mutants=5, min_mutations=2):
"""
Generate random mutants of a protein sequence.
Parameters:
sequence (str): Original amino acid sequence
num_mutants (int): Number of random mutant sequences to generate
min_mutations (int): Minimum number of residues to mutate in each mutant
Returns:
list of tuples: [(mutant_sequence, mutations_list), ...]
"""
aa_list = list("ACDEFGHIKLMNPQRSTVWY") # 20 standard amino acids
mutants = []
for _ in range(num_mutants):
seq_list = list(sequence)
num_mut = random.randint(min_mutations, max(5, len(sequence)//5)) # random # mutations
positions = random.sample(range(len(seq_list)), num_mut)
mutations = []
for pos in positions:
original = seq_list[pos]
new_aa = random.choice([aa for aa in aa_list if aa != original])
seq_list[pos] = new_aa
mutations.append(f"{original}{pos+1}{new_aa}") # e.g., A4V
mutant_seq = "".join(seq_list)
mutants.append((mutant_seq, mutations))
return mutants
#Example usage:
original_seq = "MKTAYIAKQRQISFVKSHFSRQDILDLWIYHTQGYFP"
mutants = generate_random_mutants(original_seq, num_mutants=3)
for m, mut_list in mutants:
print(m, mut_list)
Week 6 HW: Genetic circuits I
Part A
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion HF PCR Master Mix is a ready-to-use mixture designed for high accuracy DNA amplification. It contains several component that make PCR efficient, like dNTPs (the building blocks of DNA which are incorporated by the polymerase in the newly synthesized DNA strand), Reaction buffer which can either help maintain the optimal chemical environment or a GC buffer that can amplify GC-rich DNA templates, and Mg²⁺ ions that are an essesntial cofactor for the DNA polymerase activity. The most notable component is the Phusion High-Fidelity DNA polymerase, which has proofreading activity between 3’-> 5’ exonuclease which corrects mistakes during replication, having a lower rate than the standard Taq polymerase. The Phusion HF PCR Master Mix can also contain Tracking Dye, allowing direct loading of PCR product on agarose gels without adding loading buffer.
What are some factors that determine primer annealing temperature during PCR?
The primer melting temperature is the estimate of the DNA-DNA hybrid stability and critical in determining the annealing temperature. The primer annealing temperature (Ta) in PCR is the temperature at which primers bind to complementary DNA template. Too high Ta will produce insufficient primer-template hybridization resulting in low PCR product yield. Too low Ta may possibly lead to non-specific products caused by a high number of base pair mismatches.
Some factors that influence the Ta are the primer melting temperature (Ta = 0.3 x Tm(primer) + 0.7 Tm (product) – 14.9, best primers have melting temperature at 52-58 °C), the primer length (Longer primers form more hydrogen bonds => higher Tm => higher Ta, typical primers are between 18-22 nucleotides long), GC content (since G-C base pairs have 3 hydrogen bonds, higher GC content mean higher Tm, best primers have around 40-60% GC content), GC clamps (more than 3 G’s or C’s should be avoided in the last 5 bases at the 3’ end of the primer because they can help promote specific binding at the 3’ end) and primer secondary structures (strong secondary structures can reduce effective binding to the template => adjustments to Ta)
Compare and contrast PCR and Restriction Enzymes Digest
PCR
amplifies a specific DNA segment using primers and DNA polymerase;
produces many copies of the target DNA region;
They need a DNA template, forwards and reverse primers, dNTPs, reactions buffer and DNA polymerase;
Require thermal cycling;
Produces copies of a specific sequence (with variable size given by the primers positions);
Preferred for studying a specific gene.
Restriction Enzyme Digest
uses restriction endonucleases to cut DNA at specific recognition sequences;
Produce DNA fragments with defined ends;
They need DNA substrate, restriction enzymes, restriction buffer and stabilizers;
Require constant temperature;
Produce predictable fragments depending on the location of restrictions sites in the DNA;
Preferred for cutting DNA for cloning into vectors.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson Assembly requires that all fragments share designed overlapping ends, without relying on restriction sites.
We can ensure that the DNA is suitable following the next steps:
1. Design overlapping sequences
Each adjacent DNA fragment must share ~20–40 bp of identical sequence at their ends. Typically, the PCR primer design add at the 5’ end sequences homologous to the neighboring fragment or vector. These overlaps determine the order and orientation of assembly. If the fragments don’t overlap, then they will not assemble in Gibson.
2. PCR product design consideration
Standard Taq polymerase may cause occasional errors. To prevent that we can use high-fidelity polymerase to minimize mutations and ensure correct overlapping and clean single-band amplification. Like this we can avoid unwanted sequences between overlaps and inserts.
3. Preparing Restriction-Digested DNA
For Gibson, restriction enzymes are not required, but we can still use them to linearize a vector and ensure the cut region exposed ends match the designed overlaps.
4. End Compatibility
Gibson Assembly requires double-stranded DNA with overlapping ends so we must ensure that the Gibson mix overlaps are exact matches (no mismatches or frame shifts). The Gibson mix uses exonuclease to chew back 5’ ends, allow complementary overlaps to anneal, fill gaps and anneal.
5. Fragment quality
DNA needs to be purified of primers, enzymes and nonspecific products.
6. Avoid internal problems
We need to check for secondary structures disruptions, mis-annealing caused by repetitive sequences and unintended homology elsewhere.
7. Sequence verification
In silico design tools we can confirm overlaps.
How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA doesn’t normally pass through the membrane of E.coli, so we need to “shock” the membrane, making it temporarily permeable. The most common methods are:
Heat Shock Transformation
Cells are first made “competent” using cold salts like CaCl₂, to help neutralize negative charges on both DNA and the cell membrane. DNA is added and cells are kept on ice, the a short heat shock (~42°C for ~ 30-60 seconds) is applied. Like this we can cause temporary pores or increased fluidity in the membrane, pulling the DNA into the cell.
Electroporation
It acts with a similar purpose: to create pores in the membrane of the cells. Cells are washed to remove salts and placed in a special buffer where DNA is added and a brief high-voltage pulse is applied, causing transient pores in the cells membrane.
Describe another assembly method in detail
Another interesting method that I’ve learned about is the traditional restriction-ligation cloning.
Restriction–ligation cloning is a molecular biology method used to insert a specific DNA fragment into a plasmid vector. It begins by cutting both the vector and the DNA insert with restriction enzymes such as EcoRI or XhoI, which recognize specific DNA sequences. These enzymes often generate “sticky ends,” short single-stranded overhangs that can base-pair with complementary sequences. When the vector and insert have compatible ends, they can anneal through hydrogen bonding. The enzyme T4 DNA Ligase is then used to covalently join the DNA backbone by forming phosphodiester bonds. This creates a stable recombinant plasmid containing the inserted DNA fragment. The ligated plasmid is introduced into competent cells of Escherichia coli through transformation. Inside the bacteria, the plasmid is replicated, allowing amplification of the inserted gene. The cells are grown on selective media to identify those that have successfully taken up the plasmid. Finally, colonies are screened and verified to confirm that the correct DNA insert is present.