Week 2 HW: DNA Read Write and Edit
Part 1: Benchling & In-silico Gel Art
Here is a run of all the enzymes we were told to use in our virtual digest on benchling.

Here is an ‘art run’ of the virtual digest, I think this looks like an elephant.

Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
I did not participate physically to produce this. (N/A)
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
CATHL3, also known as cathelicidin antimicrobial peptide is the protein I have chosed. I chose it because antimicrobial peptides have the potential to be used as medicines or novel tools for microbial biotechnologies.
It’s amino acid sequence is:
sp|P19661|CTHL3_BOVIN Cathelicidin-3 OS=Bos taurus OX=9913 GN=CATHL3 PE=1 SV=2 METQRASLSLGRWSLWLLLLGLVLPSASAQALSYREAVLRAVDRINERSSEANLYRLLEL DPPPKDVEDRGARKPTSFTVKETVCPRTSPQPPEQCDFKENGLVKQCVGTITLDQSDDLF DLNCNELQSVRRIRPRPPRLPRPRPRPLPFPRPGPRPIPRPLPFPRPGPRPIPRPLPFPR PGPRPIPRPL
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
sp|P19661|CTHL3_BOVIN Cathelicidin-3 OS=Bos taurus OX=9913 GN=CATHL3 PE=1 SV=2 (reverse translated) atggaaacccagcgcgcgagcctgagcctgggccgctggagcctgtggctgctgctgctgggcctggtgctgccgagcgcgagcgcgcaggcgctgagctatcgcgaagcggtgctgcgc gcggtggatcgcattaacgaacgcagcagcgaagcgaacctgtatcgcctgctggaactggatccgccgccgaaagatgtggaagatcgcggcgcgcgcaaaccgaccagctttaccgtg aaagaaaccgtgtgcccgcgcaccagcccgcagccgccggaacagtgcgattttaaagaaaacggcctggtgaaacagtgcgtgggcaccattaccctggatcagagcgatgatctgttt gatctgaactgcaacgaactgcagagcgtgcgccgcattcgcccgcgcccgccgcgcctgccgcgcccgcgcccgcgcccgctgccgtttccgcgcccgggcccgcgcccgattccgcgc ccgctgccgtttccgcgcccgggcccgcgcccgattccgcgcccgctgccgtttccgcgcccgggcccgcgcccgattccgcgcccgctg
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Codon usage needs to be optmized to match up the GC content and frequency of amino acids used to the specific organism you want to insert your gene of interest into. This is important because different organisms use amino acids at different frequencies and have preferences. This is know as codon usage bias. I chose E. coli because I want to test what happens when you put an anitmicrobial peptide in a microbe. How will this impact its growth and intracellular resource pool distribution? This is the question I want to answer.
sp|P19661|CTHL3_BOVIN Cathelicidin-3 OS=Bos taurus OX=9913 GN=CATHL3 PE=1 SV=2 (codon optimized) ATGGAAACCCAGCGCGCAAGCCTGAGCCTGGGTCGTTGGTCGCTGTGGCTGCTGCTGCTGGGTCTGGTTCTGCCGAGTGCGAGCGCGCAGGCATTAAGCTATCGTGAAGCGGTGTTACGCGCGGTTGATCGCATTAATGAACGTAGCTCAGAAGCGAACCTGTACCGTCTGTTAGAACTGGATCCGCCGCCGAAAGATGTGGAAGATCGCGGTGCCCGTAAACCTACCTCGTTCACCGTGAAAGAAACCGTCTGCCCGCGCACGAGCCCGCAGCCGCCTGAACAGTGTGATTTCAAAGAAAATGGCCTGGTGAAGCAGTGTGTTGGCACCATTACCCTGGATCAGTCAGATGATCTGTTTGACCTGAACTGCAACGAACTGCAAAGCGTGCGTCGTATTCGTCCGCGTCCGCCGCGCCTGCCGCGTCCGCGTCCGCGTCCGCTGCCGTTTCCGCGTCCGGGCCCGCGCCCGATCCCGCGTCCGCTGCCGTTTCCGCGCCCGGGCCCGCGCCCGATTCCGCGTCCGCTGCCTTTTCCGCGTCCGGGCCCGCGTCCTATTCCGCGCCCGCTG
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
I would use cell-dependent protein production methods such as E. coli. I would use plasmid insertion rather than genome editing for simplicity and use a high yield industrial strain optmized for protein production. This would use a cell dependent system of transcription and translation. Transcription occurs with RNA polymerase and create an mRNA sequence based off the DNA template. This is then translated by ribosomes using matching tRNAs to produce an amino acid chain from the mRNA. The amino acid chain folds as it is produced and becomes the functional protein, depending on any posttranslational modifications it may need. A sample circuit I would use would look like this:
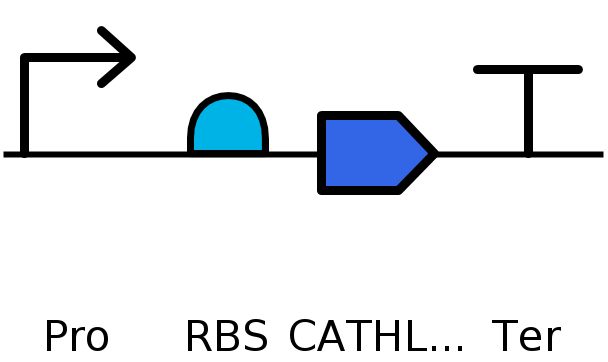
Part 4: Prepare a Twist DNA Synthesis Order
TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC - Promoter CATTAAAGAGGAGAAAGGTACC - RBS ATGGAAACCCAGCGCGCAAGCCTGAGCCTGGGTCGTTGGTCGCTGTGGCTGCTGCTGCTGGGTCTGGTTCTGCCGAGTGCGAGCGCGCAGGCATTAAGCTATCGTGAAGCGGTGTTACGCGCGGTTGATCGCATTAATGAACGTAGCTCAGAAGCGAACCTGTACCGTCTGTTAGAACTGGATCCGCCGCCGAAAGATGTGGAAGATCGCGGTGCCCGTAAACCTACCTCGTTCACCGTGAAAGAAACCGTCTGCCCGCGCACGAGCCCGCAGCCGCCTGAACAGTGTGATTTCAAAGAAAATGGCCTGGTGAAGCAGTGTGTTGGCACCATTACCCTGGATCAGTCAGATGATCTGTTTGACCTGAACTGCAACGAACTGCAAAGCGTGCGTCGTATTCGTCCGCGTCCGCCGCGCCTGCCGCGTCCGCGTCCGCGTCCGCTGCCGTTTCCGCGTCCGGGCCCGCGCCCGATCCCGCGTCCGCTGCCGTTTCCGCGCCCGGGCCCGCGCCCGATTCCGCGTCCGCTGCCTTTTCCGCGTCCGGGCCCGCGTCCTATTCCGCGCCCGCTGCATCACCATCACCATCATCACTAA - CDS CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA - Terminator
Annotated sequence: CATHL3 circuit
Fully assembled plasmid using pTwist Amp High Copy : pTwist Amp vector. CATHL3 assembly
Part 5: DNA Read/Write/Edit
5.1 DNA READ
What DNA would you want to sequence (e.g., read) and why?
For this project I would sequence the nuclear genome, the organelle genomes, and specifically the regions encoding peroxisome biogenesis and metabolism in Asparagopsis taxiformis. I want to capture the full set of genes that build and traffic proteins to the novel peroxisome‑like organelle my previous research has discovered, along with enzymes involved in halogenated metabolite and methane‑mitigation chemistry, so I can link this organelle’s structure to its potential function in reducing ruminant methane emissions when Asparagopsis taxiformis is used as a feed additive.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use a combination of Illumina short‑read sequencing and long‑read sequencing such as PacBio HiFi or Oxford Nanopore. Illumina gives me highly accurate, deep coverage for polishing assemblies and doing fine‑scale variant calling, while long‑read platforms let me span repeats and complex loci where peroxisome‑associated pathways and specialized metabolic gene clusters often reside. Together, they give me both the clean base calls and the genome contiguity I need to understand this new organelle genetically.
Is your method first-, second- or third-generation or other? How so?
In terms of classification, Illumina is a second‑generation, short‑read technology because it uses massively parallel sequencing‑by‑synthesis to generate large numbers of relatively short reads. PacBio HiFi and Oxford Nanopore are third‑generation methods because they read single DNA molecules directly as long continuous sequences, using real‑time polymerase movies or nanopore current signals instead of clonal clusters.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
My input is high‑molecular‑weight genomic DNA from A. taxiformis tissues that are enriched for the peroxisome‑containing gland‑like cells, plus RNA if I include transcriptomics. I would gently extract and purify the DNA to preserve long fragments, then fragment it to a few hundred base pairs for Illumina while keeping it long (10–20 kb or more) for PacBio or Nanopore. After that, I would repair ends, ligate platform‑specific adapters, optionally perform limited PCR amplification for Illumina libraries, carry out size selection and quality control, and finally load the prepared libraries onto the respective flow cells or chips.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
For Illumina, the key steps are cluster generation on a flow cell followed by sequencing‑by‑synthesis, where fluorescently labeled, reversible terminator nucleotides are added one at a time and imaged each cycle so the instrument can call bases from the color patterns over time. For PacBio HiFi, circular SMRTbell templates are read multiple times by a single polymerase in tiny wells, and the fluorescent signals from repeated passes are combined into a high‑accuracy consensus read. For Nanopore, individual DNA molecules are driven through nanopores, the changes in ionic current are measured continuously, and a neural network base caller converts those current traces into base sequences.
What is the output of your chosen sequencing technology?
The output I get is primarily FASTQ files containing reads and their quality scores, with Illumina producing massive numbers of accurate short reads and PacBio or Nanopore producing long reads that span complex regions. After assembly and polishing, I end up with contigs or scaffolds representing the nuclear and organelle genomes, plus expression or full‑length transcript datasets if I sequence RNA, which I can then mine for peroxisome biogenesis genes, targeting signals, and specialized metabolic pathways linked to methane‑reducing activity.
5.2 DNA WRITE
What DNA would you want to synthesize (e.g., write) and why?
For this section I would want to build on my CuZn SOD intracellular bioactive sunscreen project described in HW1, I would synthesize a human or optimized CuZn SOD1 coding sequence fused to an appropriate targeting or delivery module, such as a cell‑penetrating peptide, secretion signal, or organelle‑targeting sequence, all codon‑optimized for my chosen expression host. I want the DNA to encode a version of CuZn SOD that retains high catalytic activity while being efficiently expressed, folded, and localized in skin cells, so I can maximize intracellular ROS scavenging and UV protection without causing toxicity or mislocalization.
What technology or technologies would you use to perform this DNA synthesis and why?
To make this DNA, I would use commercial gene synthesis based on high‑fidelity oligonucleotide synthesis and assembly (e.g., array‑ or column‑synthesized oligos assembled by Gibson assembly or similar chemistries, as offered by providers like Twist or IDT). These platforms can rapidly deliver error‑checked, sequence‑verified genes in the 1–3 kb range, which easily covers a CuZn SOD fusion construct plus regulatory elements, and they save me the time and error‑prone work of stitching the gene together manually.
What are the essential steps of your chosen sequencing methods?
The essential steps of this DNA writing process start with de novo synthesis of many short oligonucleotides that together span my designed CuZn SOD construct, usually up to about 150–200 bases each. Those oligos are then assembled into the full‑length gene using overlap‑based assembly methods such as PCR assembly or Gibson assembly, followed by cloning into a plasmid, transformation into E. coli, and sequencing of individual clones to identify a correct, error‑free construct that matches my design exactly
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The main limitations of this synthesis approach for my project are not accuracy but sequence length, turnaround time, and some sequence constraints. Chemically synthesized oligos have finite length and error rates, so long or highly repetitive or GC‑rich constructs can be harder to assemble and may require more screening and verification, and typical commercial turnaround is days to a couple of weeks rather than real‑time. For a single CuZn SOD sunscreen construct in the 1–3 kb range, though, the method is highly accurate and quite scalable, and the practical bottleneck becomes how many design variants and expression systems I want to test in parallel rather than the DNA writing itself.
5.2 DNA EDIT
What DNA would you want to edit and why?
Seaweeds function as an “orphan crop” because they are under‑domesticated compared with major terrestrial crops, yet they can be cultivated at scale without using arable land or freshwater and can draw down carbon while providing habitat. For this section I would look at a seaweed domestication project, I would edit nuclear genes in key macroalgal species that control traits like growth rate, nutrient and CO₂ uptake, temperature and salinity tolerance, disease resistance, and morphology that suits offshore farms. I would also target genes in metabolic pathways that produce valuable compounds for food ingredients, animal feed, fertilizers and biostimulants, biomaterials, and biofuels, because tuning these pathways can turn seaweed into a versatile biotechnology platform that does not rely on arable land or freshwater while feeding into multiple product streams.
What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR‑based genome editing as my primary technology, focusing on CRISPR‑Cas9 or Cas12 for knockouts and cis‑regulatory edits, and base editors or prime editors where I need precise nucleotide changes. CRISPR has already been demonstrated in several macroalgae and microalgae, often via ribonucleoprotein delivery or transient vectors, and it offers a flexible way to generate both loss‑of‑function alleles (for traits like reduced fouling or altered life cycle) and fine‑tuned alleles that enhance stress tolerance and product profiles for marine permaculture systems.
How does your technology of choice edit DNA? What are the essential steps?
CRISPR technologies edit DNA by directing a programmable nuclease to a specific genomic site via a guide RNA and then relying on the cell’s endogenous repair pathways. In a typical Cas9 workflow, the Cas9–guide complex induces a double‑strand break at the target sequence, which the cell repairs either by error‑prone non‑homologous end joining, creating insertions or deletions that knock out gene function, or by homology‑directed repair if I supply a repair template encoding a desired allele. Base editors fuse a deaminase to a catalytically impaired Cas protein so they can convert specific bases (like C to T or A to G) in a narrow “editing window” without making a full double‑strand break, which can be useful for subtle tuning of enzymes involved in carbon capture or metabolite production in seaweeds.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Before editing, I need to design guide RNAs targeting seaweed genes associated with domestication traits (e.g., cell wall properties, stress signaling, carbon and nitrogen metabolism, and biosynthetic enzymes for high‑value products) and check off‑target likelihood against available genomes or de novo assemblies. My inputs include purified Cas protein (for RNP delivery), synthetic guide RNAs, and sometimes plasmid vectors expressing Cas and gRNAs, plus donor DNA templates if I want precise knock‑ins or promoter swaps; the biological input is seaweed cells, spores, or gametes that I can electroporate. In practice, I would generate or use existing genomic data for my chosen species, design and synthesize guides and donors, prepare RNPs or vectors, deliver them into the algal cells using species‑appropriate methods, regenerate edited thalli, and then phenotype lines under marine permaculture‑like conditions for growth and bioproduct yields.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The main limitations of these editing methods in seaweeds are efficiency, precision in non‑model species, and regeneration bottlenecks. Editing efficiencies in macroalgae can be lower and more variable than in classic model organisms because transformation, cell wall penetration, and tissue regeneration systems are still being optimized, and off‑target risks can be harder to quantify when reference genomes are incomplete. It can also be challenging to stack multiple edits or apply large‑scale multiplexing at industrial scale, so even though CRISPR is conceptually powerful, the practical rate at which we can generate and field‑test elite cultivars for marine permaculture and multi‑product seaweed biorefineries is still constrained by biology, culture methods, and regulatory frameworks.
When editing seaweed genomes for domestication, I need to consider ecological impacts, including the risk that modified strains might escape farms, outcompete wild populations, or alter marine food webs in unforeseen ways. I also have to weigh questions of ownership, access, and benefit sharing, especially for coastal communities and Indigenous groups who rely on local seaweeds and may hold traditional knowledge about their uses. Finally, I should be transparent about the purposes of editing and ensure that the push for climate and bioproduct benefits does not override careful biosafety assessment and robust regulatory oversight.