I am a graduate student at Utah State University, studying biological engineering on the PhD track. I am passionate about biotechnology and hope to have my own startup one day that helps produce products that today are made from petrochemicals but tomorrow will be made from biosystems! I also work for a non-profit/startup studio called the Climate Foundation that pioneers marine permaculture and other innovative technologies to fight climate change.
I’m excited to take part in this course and improve my website!
Homework Week 1 Biological Engineering Application First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Homework Week 1, Lecture Prep Homework Questions from Professor Jacobson: Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Part 1: Benchling & In-silico Gel Art
Here is a run of all the enzymes we were told to use in our virtual digest on benchling.
Here is an ‘art run’ of the virtual digest, I think this looks like an elephant.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Lab Automation Design links:
http://opentrons-art.rcdonovan.com/?id=14ylm4e9u480k3whttp://opentrons-art.rcdonovan.com/?id=jsx1tq762tv3s6q
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications. Malcı, K., Meng, F., Galez, H., Franja Da Silva, A., Caro-Astorga, J., Batt, G. and Ellis, T., 2026. Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT-2 and Flex. ACS Synthetic Biology.
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
6.6 x 1023 molecules of amino acids approximately
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The digestive system breaks down the components of food (proteins, fats, carbohydrates) into their consitutitive parts. So proteins become amino acids which our body then uses for its own cells, we don’t just assimilate cow proteins or cells, only the nutrients.
Subsections of Homework
Week 1 HW: Principles and Practices
Homework Week 1
Biological Engineering Application
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
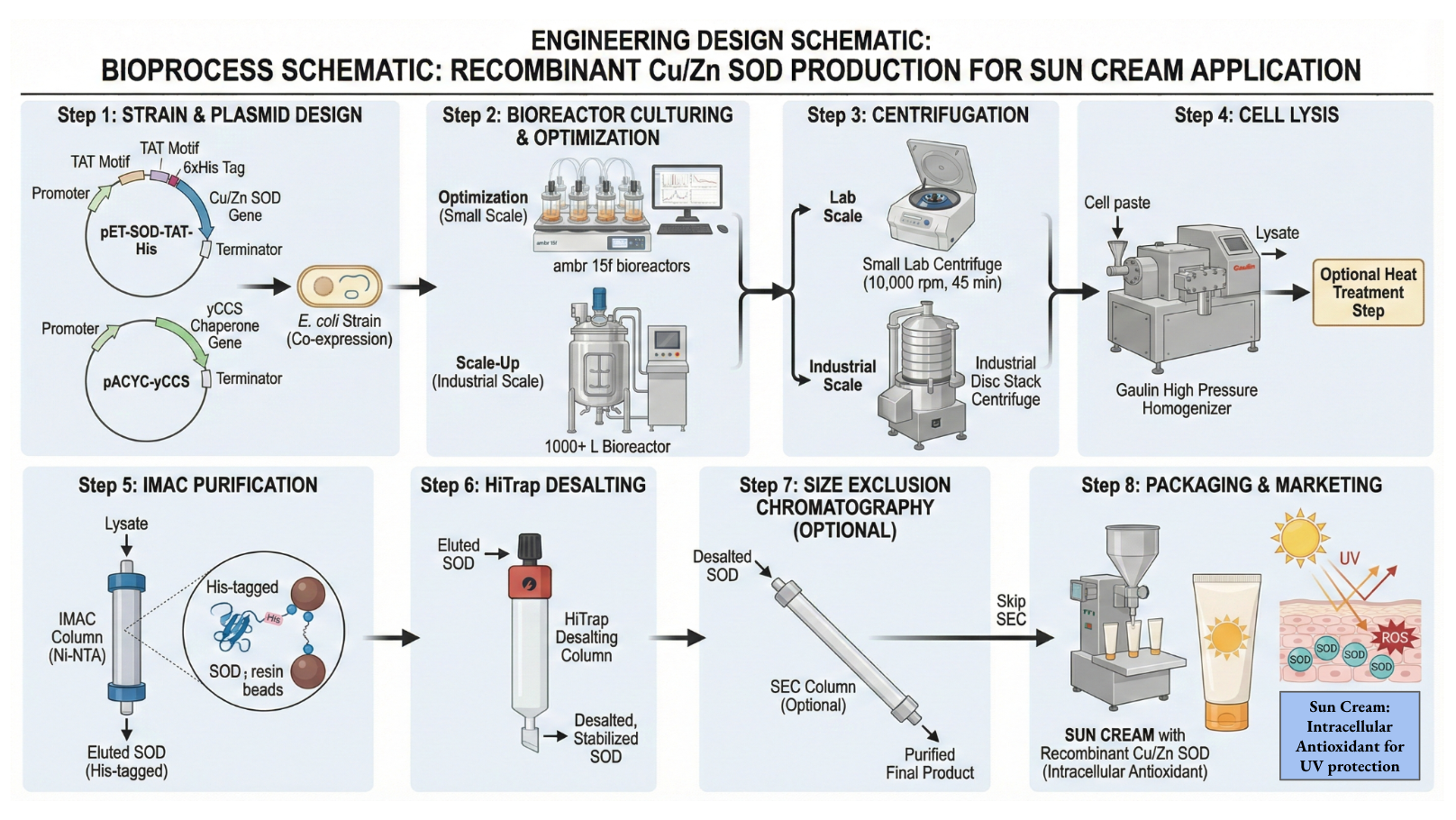
A biological engineering application I want to develop is a bioactive skin‑repair sunscreen that not only blocks ultraviolet (UV) radiation but also repairs UV‑induced intracellular damage in skin cells. The goal is to prevent skin cancer more effectively than traditional sunscreens, which mainly work by absorbing or reflecting UV light rather than actively reversing oxidative damage. I am particularly motivated by living in high‑altitude, high‑UV environments (Colorado and Utah), where cumulative sun exposure and sunburn risk are significant. Traditional sunscreens can be underused, improperly applied, or raise environmental concerns, especially regarding some organic UV filters and marine ecosystems. A bioactive formulation that incorporates antioxidant enzymes and intracellular delivery peptides could help address these limitations by scavenging reactive oxygen species (ROS) generated after UV exposure. Implementing this vision would require advances in protein engineering, biomanufacturing, formulation science, and automated biodesign.
Reference: Chen, X. et al. (2016) “Topical application of superoxide dismutase mediated by HIV-TAT peptide attenuates UVB-induced damages in human skin,” European Journal of Pharmaceutics and Biopharmaceutics, 107, pp. 286–294. Available at: https://doi.org/10.1016/j.ejpb.2016.07.023.
Here is a potential biomanufacturing workflow I have designed for a simplistic version of this bioactive sunscreen.
Briefly, the workflow involves: recombinant expression of a therapeutic enzyme such as superoxide dismutase (SOD), purification and quality control, fusion or formulation with a cell‑penetrating peptide (for example, TAT), and incorporation into a stable topical vehicle compatible with large‑scale manufacturing and regulatory requirements.
Governance and policy goals
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
My overarching governance goal is to ensure that a bioactive sunscreen is safe, trusted, and equitably accessible, rather than becoming a niche or risky cosmetic for a privileged few. In particular, I would prioritize:
Bioliteracy and trust: The public should understand, at a basic level, how the product works (e.g., enzyme‑mediated ROS scavenging and intracellular delivery) and why it is considered safe, to avoid both hype and backlash.
Robust safety and regulatory oversight: Regulatory review (e.g., FDA or equivalent) and clinical trials should evaluate not only acute irritation and erythema, but also systemic exposure, long‑term effects, and off‑label use (eyes, mucous membranes, damaged skin).
Equitable access and affordability: Pricing and distribution policies should ensure that high‑risk populations (rural communities, outdoor workers, high‑UV regions) can obtain the product at cost parity with or below conventional sunscreens.
Governance Actions
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Traditional sunscreens are regulated primarily on their ability to block UV and their short‑term safety, yet there are still gaps in evidence for cancer prevention and long‑term systemic safety. For a bioactive sunscreen, I propose strengthening peer‑reviewed evidence and clinical trials that specifically evaluate reduction in UVB‑induced skin damage, biomarker changes, and long‑term cancer risk proxies.
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Preclinical and formulation phase
Academic and industrial labs characterize enzyme stability, activity after formulation, skin penetration, and reduction of UV‑induced oxidative damage in ex vivo and small in vivo models.
Formulation scientists optimize the topical vehicle to control enzyme release, minimize systemic absorption, and ensure compatibility with existing UV filters.
Research funders (public agencies, foundations) support comparative studies that benchmark bioactive sunscreen against high‑quality conventional products, not substandard controls.
Early‑stage human studies
Clinical research centers conduct small randomized trials that measure biomarkers such as erythema, sunburn cells, DNA damage indicators, and local inflammation in human volunteers, similar in scale to existing sunscreen and nanoparticle‑based sunscreen studies.
These trials include participants with a range of skin types and ages, and track short‑term tolerability, sensitization, and signs of systemic exposure.
Independent ethics boards oversee study design, with clear informed consent that explains the bioengineered components and remaining uncertainties.
Pivotal trials and regulatory review
Larger, longer trials follow people over months or seasons of real‑world use, combining controlled UV‑challenge tests with everyday conditions.
Regulators treat the bioactive sunscreen as a drug‑like product rather than a simple cosmetic, requiring data on chronic use, systemic exposure, and environmental release, in line with recent calls to modernize sunscreen regulation.
Results and protocols are published and deposited in open repositories, with pre‑registration of trials to reduce publication bias and allow independent reanalysis.
Public acceptance and uptake
Honest media and marketing that explain mechanism, benefits, risks, and limits
Coordination with clinicians and public‑health agencies so it is framed as a preventive health tool
Take lessons from GLP‑1 drugs: strong evidence plus clear communication and clinician endorsement to build trust and awareness
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
This plan assumes that the bioactive sunscreen will ultimately prove cost‑effective enough that payers and governments are willing to fund subsidies, which depends on strong trial data and realistic cost projections. It assumes that manufacturers can scale production without dramatic price spikes or supply bottlenecks. It also assumes that providing the product at low or no cost will translate into consistent use, which may not hold without ongoing education and cultural alignment. The effectiveness and mechanisms of action, outlined in Chen et al. 2016, could be found to be not accurate and overestimated in different skin types or groups of people. This product could always fail in clinical trials or be found unsafe. Public acceptance could be tricky, the TAT peptides being sourced from HIV could create public backlash and getting ahead of such a media storm and sourcing another TAT peptide that is not associated with a disease would be ideal.
Reference: Chen, X. et al. (2016) “Topical application of superoxide dismutase mediated by HIV-TAT peptide attenuates UVB-induced damages in human skin,” European Journal of Pharmaceutics and Biopharmaceutics, 107, pp. 286–294. Available at: https://doi.org/10.1016/j.ejpb.2016.07.023.
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Failures:
This product could always fail in clinical trials or be found unsafe. If it’s not actually feasible to generate a bioactive and protective sunscreen that outperforms traditional sunscreens and is cost competitive that could cause development of this product to fail or hit roadblocks.
Consequences:
There could be long term health consequences for such a product not caught in clinical trials. Public acceptance of such a biotechnology product could lead to the acceptance of other more dangerous products.
Ideally, the product would work and a bioactive sunscreen would reduce skin cancer rates drastically and equitably.
3 governance actions:
Action 1: Peer review and clinical trials
Purpose:
Traditional sunscreens are regulated primarily on their ability to block UV and their short‑term safety, yet there are still gaps in evidence for cancer prevention and long‑term systemic safety. For a bioactive sunscreen, I propose strengthening peer‑reviewed evidence and clinical trials that specifically evaluate reduction in UVB‑induced skin damage, biomarker changes, and long‑term cancer risk proxies.
Design:
Academic and industrial researchers design studies that characterize enzyme stability, skin penetration, ROS reduction, and DNA damage markers in human skin.
Clinical investigators run randomized controlled trials to compare the bioactive sunscreen with standard SPF‑matched sunscreens across diverse skin types and age groups.
Regulators (e.g., FDA or international counterparts) require data on systemic absorption, endocrine disruption potential, and chronic exposure, consistent with current concerns about conventional actives.
Independent review boards and journals enforce transparency, data sharing, and conflict‑of‑interest disclosures.
Assumptions:
This action assumes that mechanisms seen in early clinical and ex vivo studies (e.g., TAT‑SOD decreasing UVB‑induced damage) will translate to large, diverse populations and real‑world use. It also assumes that trial endpoints (e.g., sunburn cells, oxidative stress biomarkers) are valid proxies for long‑term cancer risk.
Risks of failure and “success”:
Failure risks: Trials could reveal limited benefit over high‑quality traditional sunscreens, unacceptable safety issues, or strong inter‑individual variability.
Success risks: Demonstrating strong protection might create risk compensation, where users rely solely on the product and ignore complementary behaviors like shade or protective clothing.
Action 2: Media, marketing, and science communication
Purpose:
I propose a coordinated science communication strategy to build informed trust rather than hype, ensuring the product is understood as one part of a broader sun‑safety toolkit.
Design (actors and implementation):
Science communicators, dermatologists, and public health agencies jointly develop clear, accessible messaging about how the bioactive sunscreen works, what is known, and what remains uncertain.
Marketing campaigns emphasize evidence‑based benefits, appropriate use, and limitations, avoiding sensational claims about being “sunburn‑proof” or “cancer‑proof.”
Community health workers and local organizations co‑design outreach in high‑risk communities, emphasizing culturally relevant framing and local representation, echoing the importance of trust described by George Shultz.
Assumptions:
This action assumes that transparent, co‑created communication can overcome distrust linked to “biotech,” “engineered enzymes,” or associations with HIV‑derived TAT peptides. It also assumes that industry actors will accept constraints on exaggerated marketing.
Risks of failure and “success”:
Failure risks: Poorly framed messages could trigger fear (e.g., focus on “HIV‑TAT”) or be dismissed as corporate spin.
Success risks: Extremely successful marketing might oversell benefits and normalize dependence on a proprietary product instead of promoting broader sun‑safety and environmental protections.
Action 3: Equitable distribution and subsidization
Purpose:
I propose policies to treat bioactive sunscreen as a preventive health tool rather than a luxury cosmetic, aligning distribution with skin‑cancer prevention goals.
Design (actors and implementation):
Public health agencies and insurers consider covering the product for high‑risk groups (e.g., outdoor workers, immunocompromised patients, very fair skin types) as a preventive intervention.
Governments and payers negotiate pricing or volume guarantees to keep retail costs comparable to or lower than common sunscreens in pharmacies and supermarkets.
Community clinics and rural health programs distribute the product alongside educational materials on sun safety and skin‑cancer screening.
Assumptions:
This action assumes that strong cost‑effectiveness arguments can be made (e.g., reduced future skin‑cancer treatment costs) and that manufacturing can scale without driving prices beyond what subsidies can realistically cover.
Risks of failure and “success”:
Failure risks: If manufacturing remains expensive, subsidies could be politically unpopular or unsustainable, reinforcing inequities.
Success risks: If access is expanded without strong safety and literacy measures, misuse or overreliance could spread broadly rather than being confined to early adopters.
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Peer Review/Clinical Trials
Media and Marketing
Equitable Distribution
Enhance Biomedicine
1
n/a
2
• By preventing disease
1
n/a
2
• By helping treat illness
1
n/a
2
• Ensure safe bioproduct
1
n/a
n/a
Enhance Bioliteracy
1
2
3
• By educating the public
2
1
3
• By fostering trust in biotechnologies
1
3
2
Protect the environment
3
2
1
• By reducing other sunscreens
n/a
1
2
Other considerations
• Minimizing costs and burdens to stakeholders
1
2
3
• Feasibility?
1
2
3
• Promote constructive applications
3
1
2
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
I would prioritize a combined strategy that begins with rigorous peer review and clinical trials (Action 1), then layers on responsible science communication (Action 2), and finally builds toward equitable distribution (Action 3). Robust evidence generation is non‑negotiable; without it, any marketing or subsidization risks amplifying harms or wasting resources. Once a strong safety and efficacy case exists, coordinated communication can promote informed uptake while explicitly acknowledging uncertainties and avoiding overclaiming. Only after those foundations are in place would I advocate for large‑scale subsidization and integration into public health programs, given the political, economic, and logistical complexity of equitable distribution.
For a target audience, I would imagine this recommendation addressed to a national public‑health agency (a federal department of health or an NIH‑like funding body) that can coordinate research funding, regulatory pathways, and outreach partnerships. A key trade‑off is time: prioritizing careful trials and communication may slow wide deployment, but it helps preserve long‑term public trust and reduces the risk that an early failure poisons attitudes toward future bioactive preventives. My largest uncertainties involve real‑world cost‑effectiveness, long‑term systemic safety, environmental impact compared with current sunscreens, and how public attitudes toward engineered biologics will evolve.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues.
In class, the discussion of ethics, safety, and governance highlighted how quickly powerful biotechnologies can move from lab to market and how unevenly risks and benefits are distributed across communities. George P. Shultz’s essay “Trust Is the Coin of the Realm” reinforced that durable trust is built when communities see their own members involved in decision‑making, rather than having technologies imposed on them. For bioactive sunscreen and similar interventions, this suggests that governance should not only involve regulators and companies but also community representatives in high‑UV, high‑risk areas, including rural, low‑income, and historically marginalized groups.
Governance actions I see as appropriate include: funding community‑based participatory research around sun‑safety tools; requiring public comment and local advisory boards when introducing novel preventive biotechnologies; and supporting training programs that help people from affected communities become scientists, clinicians, and communicators. These steps could align the development of bioactive sunscreens with the broader ethical goal of biotechnology that is responsive to, and shaped by, the communities it is meant to serve
Week 2 Lecture Prep
Homework Week 1, Lecture Prep
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error rate = 1x104 from Slide 15
Length: If the human genome is 3 billion bp, then that would be approximately 3x105 errors.
Discrepancy: This is dealt with by repair mechanisms
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Different ways to code: With 61 codons coding for 21 amino acids there are a large number of ways to code for the average protein.
In practice why doesn’t this work: tRNA availablity, mRNA secondary structure, co-translation folding errors.
Citation: Gemini Ai prompt: ‘why do alternate codes for protein coding fail, list the most often reasons and provide scientific backing’
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
The most common method of oligo syntesis is solid phase synthesis with the phosphoramidite method.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Errors accumulate and the sequence becomes inaccurate.
Why can’t you make a 2000bp gene via direct oligo synthesis?
The error rate of such a process is too high to be feasible. Typically you would use smaller fragments and join them with Gibson or another method of nucleotide assembly.
Homework Question from George Church:
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Phenylalanine, Valine, Threonine - PVT
Tryptophan, Isoleucine, Methionine - TIM
Histidine, Arginine, Leucine, Lysine - HALL
The lysine contingency, from the film jurassic park, is not a good contingency considering all animals have to get these 10 amino acids from their diet anyways. It’s not like the parks managers were very good with any of their ethical considerations or failsafe systems though. Life finds a way.
Citations: I just directly Google searched the question ‘What are the 10 essential amino acids in all animals’ and ‘Lysine Contingency’
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
n/a
Here is a run of all the enzymes we were told to use in our virtual digest on benchling.
Here is an ‘art run’ of the virtual digest, I think this looks like an elephant.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
I did not participate physically to produce this. (N/A)
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
CATHL3, also known as cathelicidin antimicrobial peptide is the protein I have chosed. I chose it because antimicrobial peptides have the potential to be used as medicines or novel tools for microbial biotechnologies.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Codon usage needs to be optmized to match up the GC content and frequency of amino acids used to the specific organism you want to insert your gene of interest into. This is important because different organisms use amino acids at different frequencies and have preferences. This is know as codon usage bias. I chose E. coli because I want to test what happens when you put an anitmicrobial peptide in a microbe. How will this impact its growth and intracellular resource pool distribution? This is the question I want to answer.
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
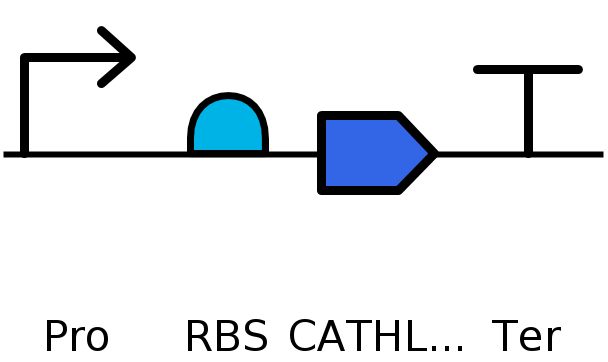
I would use cell-dependent protein production methods such as E. coli. I would use plasmid insertion rather than genome editing for simplicity and use a high yield industrial strain optmized for protein production. This would use a cell dependent system of transcription and translation. Transcription occurs with RNA polymerase and create an mRNA sequence based off the DNA template. This is then translated by ribosomes using matching tRNAs to produce an amino acid chain from the mRNA. The amino acid chain folds as it is produced and becomes the functional protein, depending on any posttranslational modifications it may need. A sample circuit I would use would look like this:
Fully assembled plasmid using pTwist Amp High Copy : pTwist Amp vector.
CATHL3 assembly
Part 5: DNA Read/Write/Edit
5.1 DNA READ
What DNA would you want to sequence (e.g., read) and why?
For this project I would sequence the nuclear genome, the organelle genomes, and specifically the regions encoding peroxisome biogenesis and metabolism in Asparagopsis taxiformis. I want to capture the full set of genes that build and traffic proteins to the novel peroxisome‑like organelle my previous research has discovered, along with enzymes involved in halogenated metabolite and methane‑mitigation chemistry, so I can link this organelle’s structure to its potential function in reducing ruminant methane emissions when Asparagopsis taxiformis is used as a feed additive.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use a combination of Illumina short‑read sequencing and long‑read sequencing such as PacBio HiFi or Oxford Nanopore. Illumina gives me highly accurate, deep coverage for polishing assemblies and doing fine‑scale variant calling, while long‑read platforms let me span repeats and complex loci where peroxisome‑associated pathways and specialized metabolic gene clusters often reside. Together, they give me both the clean base calls and the genome contiguity I need to understand this new organelle genetically.
Is your method first-, second- or third-generation or other? How so?
In terms of classification, Illumina is a second‑generation, short‑read technology because it uses massively parallel sequencing‑by‑synthesis to generate large numbers of relatively short reads. PacBio HiFi and Oxford Nanopore are third‑generation methods because they read single DNA molecules directly as long continuous sequences, using real‑time polymerase movies or nanopore current signals instead of clonal clusters.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
My input is high‑molecular‑weight genomic DNA from A. taxiformis tissues that are enriched for the peroxisome‑containing gland‑like cells, plus RNA if I include transcriptomics. I would gently extract and purify the DNA to preserve long fragments, then fragment it to a few hundred base pairs for Illumina while keeping it long (10–20 kb or more) for PacBio or Nanopore. After that, I would repair ends, ligate platform‑specific adapters, optionally perform limited PCR amplification for Illumina libraries, carry out size selection and quality control, and finally load the prepared libraries onto the respective flow cells or chips.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
For Illumina, the key steps are cluster generation on a flow cell followed by sequencing‑by‑synthesis, where fluorescently labeled, reversible terminator nucleotides are added one at a time and imaged each cycle so the instrument can call bases from the color patterns over time. For PacBio HiFi, circular SMRTbell templates are read multiple times by a single polymerase in tiny wells, and the fluorescent signals from repeated passes are combined into a high‑accuracy consensus read. For Nanopore, individual DNA molecules are driven through nanopores, the changes in ionic current are measured continuously, and a neural network base caller converts those current traces into base sequences.
What is the output of your chosen sequencing technology?
The output I get is primarily FASTQ files containing reads and their quality scores, with Illumina producing massive numbers of accurate short reads and PacBio or Nanopore producing long reads that span complex regions. After assembly and polishing, I end up with contigs or scaffolds representing the nuclear and organelle genomes, plus expression or full‑length transcript datasets if I sequence RNA, which I can then mine for peroxisome biogenesis genes, targeting signals, and specialized metabolic pathways linked to methane‑reducing activity.
5.2 DNA WRITE
What DNA would you want to synthesize (e.g., write) and why?
For this section I would want to build on my CuZn SOD intracellular bioactive sunscreen project described in HW1, I would synthesize a human or optimized CuZn SOD1 coding sequence fused to an appropriate targeting or delivery module, such as a cell‑penetrating peptide, secretion signal, or organelle‑targeting sequence, all codon‑optimized for my chosen expression host. I want the DNA to encode a version of CuZn SOD that retains high catalytic activity while being efficiently expressed, folded, and localized in skin cells, so I can maximize intracellular ROS scavenging and UV protection without causing toxicity or mislocalization.
What technology or technologies would you use to perform this DNA synthesis and why?
To make this DNA, I would use commercial gene synthesis based on high‑fidelity oligonucleotide synthesis and assembly (e.g., array‑ or column‑synthesized oligos assembled by Gibson assembly or similar chemistries, as offered by providers like Twist or IDT). These platforms can rapidly deliver error‑checked, sequence‑verified genes in the 1–3 kb range, which easily covers a CuZn SOD fusion construct plus regulatory elements, and they save me the time and error‑prone work of stitching the gene together manually.
What are the essential steps of your chosen sequencing methods?
The essential steps of this DNA writing process start with de novo synthesis of many short oligonucleotides that together span my designed CuZn SOD construct, usually up to about 150–200 bases each. Those oligos are then assembled into the full‑length gene using overlap‑based assembly methods such as PCR assembly or Gibson assembly, followed by cloning into a plasmid, transformation into E. coli, and sequencing of individual clones to identify a correct, error‑free construct that matches my design exactly
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The main limitations of this synthesis approach for my project are not accuracy but sequence length, turnaround time, and some sequence constraints. Chemically synthesized oligos have finite length and error rates, so long or highly repetitive or GC‑rich constructs can be harder to assemble and may require more screening and verification, and typical commercial turnaround is days to a couple of weeks rather than real‑time. For a single CuZn SOD sunscreen construct in the 1–3 kb range, though, the method is highly accurate and quite scalable, and the practical bottleneck becomes how many design variants and expression systems I want to test in parallel rather than the DNA writing itself.
5.2 DNA EDIT
What DNA would you want to edit and why?
Seaweeds function as an “orphan crop” because they are under‑domesticated compared with major terrestrial crops, yet they can be cultivated at scale without using arable land or freshwater and can draw down carbon while providing habitat. For this section I would look at a seaweed domestication project, I would edit nuclear genes in key macroalgal species that control traits like growth rate, nutrient and CO₂ uptake, temperature and salinity tolerance, disease resistance, and morphology that suits offshore farms. I would also target genes in metabolic pathways that produce valuable compounds for food ingredients, animal feed, fertilizers and biostimulants, biomaterials, and biofuels, because tuning these pathways can turn seaweed into a versatile biotechnology platform that does not rely on arable land or freshwater while feeding into multiple product streams.
What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR‑based genome editing as my primary technology, focusing on CRISPR‑Cas9 or Cas12 for knockouts and cis‑regulatory edits, and base editors or prime editors where I need precise nucleotide changes. CRISPR has already been demonstrated in several macroalgae and microalgae, often via ribonucleoprotein delivery or transient vectors, and it offers a flexible way to generate both loss‑of‑function alleles (for traits like reduced fouling or altered life cycle) and fine‑tuned alleles that enhance stress tolerance and product profiles for marine permaculture systems.
How does your technology of choice edit DNA? What are the essential steps?
CRISPR technologies edit DNA by directing a programmable nuclease to a specific genomic site via a guide RNA and then relying on the cell’s endogenous repair pathways. In a typical Cas9 workflow, the Cas9–guide complex induces a double‑strand break at the target sequence, which the cell repairs either by error‑prone non‑homologous end joining, creating insertions or deletions that knock out gene function, or by homology‑directed repair if I supply a repair template encoding a desired allele. Base editors fuse a deaminase to a catalytically impaired Cas protein so they can convert specific bases (like C to T or A to G) in a narrow “editing window” without making a full double‑strand break, which can be useful for subtle tuning of enzymes involved in carbon capture or metabolite production in seaweeds.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Before editing, I need to design guide RNAs targeting seaweed genes associated with domestication traits (e.g., cell wall properties, stress signaling, carbon and nitrogen metabolism, and biosynthetic enzymes for high‑value products) and check off‑target likelihood against available genomes or de novo assemblies. My inputs include purified Cas protein (for RNP delivery), synthetic guide RNAs, and sometimes plasmid vectors expressing Cas and gRNAs, plus donor DNA templates if I want precise knock‑ins or promoter swaps; the biological input is seaweed cells, spores, or gametes that I can electroporate. In practice, I would generate or use existing genomic data for my chosen species, design and synthesize guides and donors, prepare RNPs or vectors, deliver them into the algal cells using species‑appropriate methods, regenerate edited thalli, and then phenotype lines under marine permaculture‑like conditions for growth and bioproduct yields.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The main limitations of these editing methods in seaweeds are efficiency, precision in non‑model species, and regeneration bottlenecks. Editing efficiencies in macroalgae can be lower and more variable than in classic model organisms because transformation, cell wall penetration, and tissue regeneration systems are still being optimized, and off‑target risks can be harder to quantify when reference genomes are incomplete. It can also be challenging to stack multiple edits or apply large‑scale multiplexing at industrial scale, so even though CRISPR is conceptually powerful, the practical rate at which we can generate and field‑test elite cultivars for marine permaculture and multi‑product seaweed biorefineries is still constrained by biology, culture methods, and regulatory frameworks.
When editing seaweed genomes for domestication, I need to consider ecological impacts, including the risk that modified strains might escape farms, outcompete wild populations, or alter marine food webs in unforeseen ways. I also have to weigh questions of ownership, access, and benefit sharing, especially for coastal communities and Indigenous groups who rely on local seaweeds and may hold traditional knowledge about their uses. Finally, I should be transparent about the purposes of editing and ensure that the push for climate and bioproduct benefits does not override careful biosafety assessment and robust regulatory oversight.
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Malcı, K., Meng, F., Galez, H., Franja Da Silva, A., Caro-Astorga, J., Batt, G. and Ellis, T., 2026. Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT-2 and Flex. ACS Synthetic Biology.
This publication outlines a golden gate cloning workflow automation process with Opentrons that would greatly improve my current labs workflow. I think the automation of such work would increase our output by 1-2 orders of magnitude. I think combining such a tool with more robust golden gate kits for assembling more complex genetic devices would greatly speed up synthetic biology work more broadly.
The paper presents Slowpoke, a low cost, modular automation workflow for Golden Gate cloning on Opentrons OT 2 and Flex, aimed at making routine synthetic biology cloning faster and more reproducible while remaining accessible through open source protocols and a simple graphical interface. It automates key steps of the cloning pipeline, including assembly setup, transformation, plating, and colony PCR, so that users mainly handle plate movements and colony picking rather than complex protocol programming.
Using the MoClo Yeast Toolkit and the SubtiToolKit on both OT 2 and Flex, the authors show that Slowpoke achieves high assembly efficiency in basic transcription unit builds and maintains strong performance in more complex, multi part combinatorial assemblies. By releasing all code, templates, and documentation for community use and adaptation, the work positions Slowpoke as a generalizable platform for robust, everyday Golden Gate cloning in synthetic biology labs.
Slowpoke implements an end to end Golden Gate automation pipeline on Opentrons OT 2 and Flex that covers assembly setup, transformation, plating, and colony PCR, with users mainly intervening for plate transfers and colony picking. A free graphical interface at https://slowpoke.streamlit.app generates robot protocols from simple file uploads, making protocol design largely point and click and enabling non specialists to automate cloning workflows. Using the MoClo Yeast Toolkit on OT 2, the workflow achieved 17 of 17 correct colonies for basic transcription unit assemblies, while on Flex 11 of 12 colonies were correct, demonstrating high efficiency on both platforms. With the SubtiToolKit on OT 2, 8 of 13 colonies were correct for basic assemblies, showing that the same pipeline can be ported across Golden Gate toolkits with good success rates. In a higher throughput test on Flex using YTK compatible parts, 55 out of 57 multi part combinations yielded correct constructs, indicating strong reliability for combinatorial six part assemblies. The authors release all code, templates, and documentation in the Slowpoke GitHub repository, providing an open, modular foundation that laboratories can adapt to their own Golden Gate toolkits and construct designs.
Write a description about what you intend to do with automation tools for your final project.
Project Ideas
SOD Sunscreen Evolution Screen
In this project, I’m evolving CuZn SOD variants as intracellular “bioactive sunscreens” that protect cells from reactive oxygen species generated during UV exposure. I start from a baseline SOD1 (human or consensus) fused to a trafficking module such as a cell‑penetrating peptide or organelle‑targeting signal, and then build a focused mutant library around catalytic and stability‑relevant residues. I use E. coli as the initial expression and screening chassis, where I can score variants by enzymatic activity and survival under oxidative stress, and later port top candidates into a mammalian context for proof‑of‑concept protection assays.
My experimental workflow begins with designing the SOD library using error‑prone PCR plus targeted mutations near the active site and key stability positions. I clone these variants into a standardized expression vector and express them in 96‑ or 384‑well format. I then quantify SOD activity in lysates using a plate‑based assay that measures inhibition of a superoxide‑driven colorimetric reaction, and I complement that with growth assays under superoxide‑generating compounds like paraquat. By ranking variants across both enzymatic and functional stress assays, I can identify sequence features that enhance activity and robustness in oxidative environments that resemble UV‑induced stress in skin cells.
This project is designed from the ground up for lab automation. I use automated liquid handling to set up PCR and assembly reactions for the library, perform transformations, and distribute transformants into multiwell plates. Induction, lysis, and reagent addition for the SOD activity assay are all scripted and executed in parallel, and a plate reader continuously records absorbance or fluorescence to generate kinetic activity curves. I also integrate an automated incubator/shaker for consistent growth and induction conditions and use simple analysis scripts to automatically flag top‑performing wells for re‑arraying and sequencing. In effect, I’m building a small, closed‑loop directed‑evolution platform for ROS‑protective enzymes.
CO₂ Co-Culture Toggle Project
In this project, I’m engineering a cyanobacteria–E. coli co-culture where the phototroph fixes CO₂ and secretes organic carbon that E. coli converts into a storable product such as PHB. I’m designing a bistable genetic toggle in E. coli that switches between a “carbon uptake and storage” state and a “carbon burn” state, controlled by small-molecule inducers or light. The cyanobacterium supplies fixed carbon (for example sucrose or lactate), while E. coli’s state determines whether that carbon is hoarded as product or consumed for growth, letting me explicitly test how dynamic carbon partitioning shapes community stability and overall CO₂ fixation.
Experimentally, I first validate the toggle in E. coli alone, using mutually repressing transcription factors that each drive a fluorescent reporter plus either a “storage” or “burn” enzyme module. I then work on a cyanobacterial partner engineered to secrete a defined carbon source at tunable rates. Finally, I assemble defined co-cultures in microplates or small bioreactors and measure CO₂ uptake, product formation, and community composition across different toggle states. My goal is to build a model platform for programmable carbon flux in microbial consortia that can later be generalized to other substrates and host pairs.
For this project, I’m leaning heavily on lab automation. I use automated DNA assembly to build toggle variants and transporter constructs in 96‑well format, followed by automated colony picking and growth in deep‑well plates. A liquid handler sets up a matrix of co‑culture conditions by varying initial species ratios, inducer concentrations, and light regimes. Plate readers then track OD and fluorescence over time for both partners, and automated sampling feeds into simple assays for secreted metabolites and storage products. The result is a high‑density dataset of co‑culture and induction conditions that lets me map how the toggle controls carbon flux in a fully programmable, automation‑friendly way.
Gut Microbiome for Mood and Health Regulation
In this project, I’m engineering a “mood‑ and craving‑aware” probiotic chassis—such as E. coli Nissle or a GRAS Lactobacillus—to modulate multiple neuroactive pathways linked to anxiety, mood, and addiction. One module produces GABA and/or 5‑HTP to support anxiolysis and prosocial mood; a second module modulates dopamine‑related metabolism to smooth extreme reward spikes associated with craving; and a third module expresses a short oxytocin‑mimetic peptide to reinforce bonding and compassionate affect. Each module is controlled by gut‑relevant sensors (for example bile salts, lactate, or inflammatory markers), so the system is most active under physiological states associated with stress, post‑exercise recovery, or post‑use withdrawal.
At the bench, I start by building and characterizing each pathway independently. I place GABA and 5‑HTP biosynthesis pathways under inducible or sensor‑driven promoters and quantify their outputs with metabolite assays. In parallel, I design circuits that either consume dopamine precursors or express mild inhibitors of dopamine biosynthesis, and I validate those using in vitro assays or bioreporters. For the oxytocin‑mimetic module, I fuse a short peptide to an appropriate signal peptide to ensure secretion and verify production and export. Once I’m confident in the individual modules, I combine them in one or more strains using orthogonal sensors and simple logic to activate different outputs under distinct input profiles that approximate real gut states.
Automation is central to how I explore this design space. I use a liquid handler to assemble promoter–RBS–gene combinations for each module and to transform and array the resulting clones. I then set up microtiter plate assays with controlled input conditions such as gradients of pH, lactate, bile salts, and carbon sources. Automated sampling into metabolite assays or bioreporter strains gives me dose–response curves across hundreds of construct–condition pairs. With basic scripting, I automatically identify designs that show high production under “desired” states (for example high lactate and bile salts) and minimal leakiness at baseline. As an extension, I can connect the system to a simple gut‑on‑a‑chip or microfluidic device to automate media switching and simulate feeding, exercise, or withdrawal cycles, turning this into a small but realistic testbed for programmable neuroactive probiotics.
Protein Design part 1 Week 4
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
6.6 x 1023 molecules of amino acids approximately
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The digestive system breaks down the components of food (proteins, fats, carbohydrates) into their consitutitive parts. So proteins become amino acids which our body then uses for its own cells, we don’t just assimilate cow proteins or cells, only the nutrients.
Why are there only 20 natural amino acids?
Technically there could be more but evolution got set with ~20 very early on in life’s history so those became the standard. The chemical diversity of those 20 is enough to support life.
Where did amino acids come from before enzymes that make them, and before life started?
Before life as we know it started (on Earth), amino acids could be found in primoridal geochemical or extraterrestrial reactions. Amino acids and their chemical precursors are often found naturally occuring on meteorites, hydrothermal vents, and other such geochemically active sites. It is important to consider that without living organisms to degrade them, any geochemical means of synthesis would accumulate these relatively stable molecules.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left handed. Biology is uses the L-amino acids but these create right handed α-helixes.
Can you discover additional helices in proteins?
rare π-helices often misidentified as α-bulges and 311-helices
Why are most molecular helices right-handed?
They are more thermodynacmically and energetically stable
Why do β-sheets tend to aggregate?
Hydrogen bonding occurs between the ends of the sheets allowing the sheets attach to each other and stack
What is the driving force for β-sheet aggregation?
Hydrophobic effects
Why do many amyloid diseases form β-sheets?
Protein aggregation in the form of sheets can cause disease but it is a thermodynamically favorable structure.
Can you use amyloid β-sheets as materials?
Yes they can be used for tissue scaffold, hydrogels, or other biological materials
Part B: Protein Analysis and Visualization
Human Cu/Zn superoxide dismutase (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals to oxygen and hydrogen peroxide, protecting cells from oxidative stress. I selected it because it is a classic, well‑characterized enzyme that we’ve already worked with in my recombinant CuZn SOD production project (bioactive sunscreen).
Cu-ZN is 154 aa long and its most frequent residue is Glycine (G).
Using UniProt’s BLAST on P00441 returns thousands of homologous SOD1/CuZnSOD sequences from many species (order of 10³–10⁴ hits, depending on database and thresholds).
SOD1 belongs to the Cu-Zn superoxide dismutase family and contains the Sod_Cu domain (Pfam PF00080)
Resolution: 1.07 Å, and an atomic structure that was solved in 2005
Sample visualizations:
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359
Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model.
Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model.
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101
Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Goal 1: Stabilize MS2 L so it remains correctly folded and membrane-competent under a wider range of conditions (temperature, expression levels), which should support more robust lysis and higher effective titers
Goal 2: Increase intrinsic toxicity by promoting oligomerization and pore formation of L in the bacterial envelope.
We’ll focus on increasing stability and increasing toxicity of the MS2 L lysis protein, using a purely computational design-and-screen pipeline before any wet-lab work.
Our first goal is increased stability: we want MS2 L to remain correctly folded and membrane-competent across a broad range of expression and environmental conditions, so that lysis is robust and supports higher effective titers.
Our second goal is increased toxicity: we aim to boost the intrinsic lytic potency of L by promoting its oligomerization and pore formation in the bacterial envelope.
To do this, we will use protein language models (such as ESM or ProtT5) as in silico mutagenesis engines, scoring single and selected double mutants of L, then ranking substitutions predicted to increase stability or fitness.
We will combine this with structure prediction and modeling, using AlphaFold on full-length and truncated L variants to obtain consistent 3D models of the soluble and transmembrane domains, and AlphaFold-Multimer or docking tools to model L oligomers within a membrane-like context.
Membrane-aware stability analysis will help constrain our designs: TMHMM-like predictors will define transmembrane boundaries and disorder regions, while Rosetta- or FoldX-style ΔΔG calculations, accessed through web servers, will refine a shortlist of promising positions around the LS motif and transmembrane helix.
We will also integrate coevolution and functional hotspot information by overlaying published near-saturating mutagenesis data for L, so that residues known to be essential for function are explicitly locked and excluded from substitution.
These tools are well suited to our problem because mutational studies show that even conservative changes near the LS motif can abolish function, meaning that we need a guided way to search sequence space that respects both evolutionary constraints and biophysical stability.
Language models provide a rapid, global scan of mutation space, while ΔΔG calculations and structure predictions help us reject variants that appear incompatible with correct helix packing or overall folding, yielding a concise list of testable stabilizing mutants.
For increased toxicity, MS2 L’s activity depends on efficient membrane insertion and formation of higher-order oligomers by its C-terminal transmembrane segment, so modeling and optimizing transmembrane helix–helix interfaces should directly affect pore stability and lytic potency.
By favoring small or helix-packing residues at predicted contact positions and avoiding unfavorable charges within the bilayer, we can design variants that, in silico, form more stable oligomers without disrupting the LS motif or essential hydrophobic patterning.
In single-gene lysis systems, lysis timing and efficiency emerge from a balance of expression level, folding, and toxicity, so improving stability should also reduce misfolding and escape mutants, enabling higher functional L levels and improved titers under comparable induction conditions.
Our overall objective is to computationally design MS2 L variants that combine improved structural stability in their membrane-inserted forms with increased