Group Final Project
Computational Engineering of the MS2 Lysis Protein to Improve Stability, Titers, and Toxicity
After reviewing the provided literature on the MS2 lysis protein (L) and discussing the project aims, our group has decided to focus on three interconnected goals:
Goal 1: Increase the stability of the L protein
As the “easiest” goal, it is the most computationally tractable. A stabilized protein is less prone to degradation and misfolding, which could directly lead to higher functional titers and serve as a robust starting point for any subsequent engineering.
Goal 2: Increase bacteriophage titers through improved lysis efficiency.
Phage therapy relies on high phage titers for effective bacterial killing and scalable manufacturing, but phage production can be limited by inefficient lysis or poor coordination between phage replication and host destruction. Improving the efficiency and timing of host cell lysis can therefore directly increase the number of phage particles released per infected cell.
The MS2 L protein is a small 75–amino acid membrane protein that triggers bacterial lysis and is essential for the release of new phage particles. In the paper Mutational analysis of the MS2 lysis protein L, it is described how MS2 L functions as a single-gene lysis protein that disrupts bacterial cell envelope integrity without classical enzymatic activity. Additionally, L interacts with the host chaperone DnaJ, which modulates its activity and timing of lysis. In MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ it is shown that lysis timing strongly affects the number of virions produced before the host cell bursts, meaning that engineering improved L variants may increase overall phage titers.
Goal 3: Increase the toxicity of the lysis protein.
This proposal addresses the subproblem of increasing the toxicity of the L lysis protein from Bacteriophage MS2. Instead of random mutagenesis, toxicity will be approached as a multi-factor optimization problem involving structural stability, membrane insertion, oligomerization efficiency, and expression kinetics in Escherichia coli. The objective is to design L variants that enhance membrane disruption while maintaining proper folding and stability.
E. coli chaperone DnaJ.
Additionally, we will explore disrupting the interaction between the L protein and the E. coli chaperone DnaJ.
The reading “Identification MS2 lysis protein dependency on DnaJ” establishes this interaction as critical for function. By computationally predicting and then disrupting this interface, we can test its necessity and potentially create a DnaJ-independent lysis mechanism, offering a new avenue for controlling lysis timing.
Together, these three goals form a coherent strategy: stabilizing the L protein may improve its folding and expression, which can increase functional titers, while further engineering of membrane disruption and host interactions may increase toxicity and lysis efficiency.
Proposed Computational Tools and Approaches
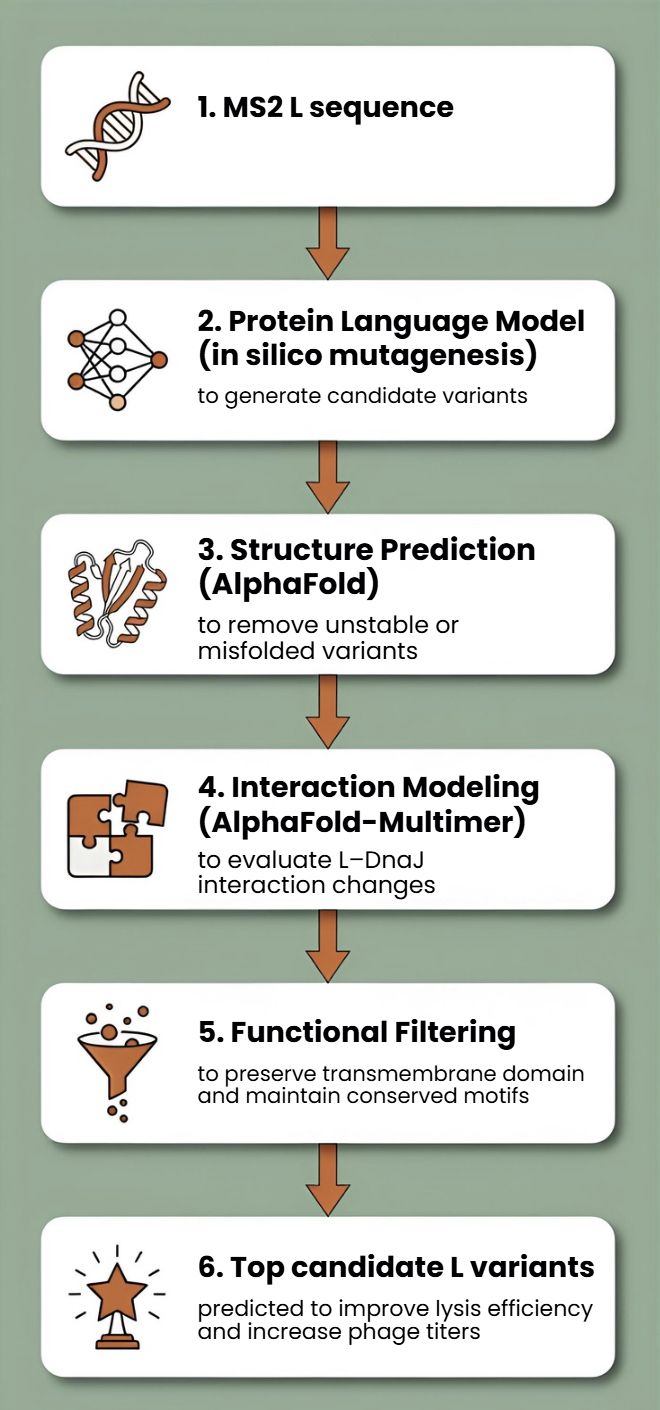
Proposed Tools and Approaches We will build a computational pipeline using the tools introduced in recitation and the provided resources. The key steps and tools are:
Step 1: Structural Modeling of the L Protein
Tool: AlphaFold2 (via ColabFold for ease of use).
Why: No high-resolution experimental structure of the full-length MS2 L protein exists. A reliable 3D model is the absolute foundation for all downstream analysis, allowing us to visualize which parts are structured vs. disordered.
Step 2: Modeling the L-DnaJ Complex
Tool: AlphaFold-Multimer.
Why: To disrupt the interaction, we first need to know where it occurs. AlphaFold-Multimer is the current state-of-the-art for predicting protein-protein complexes and will generate a testable model of the L protein bound to E. coli DnaJ.
Step 3: In Silico Mutagenesis for Stability
Tool: Rosetta (or FoldX). Specifically, the ddg_monomer application for predicting changes in folding free energy (ΔΔG).
Why: These tools are parameterized using vast amounts of experimental data on protein stability. They can systematically mutate each residue in our L protein model and predict whether the change (e.g., A->V) makes the protein more stable (negative ΔΔG) or less stable (positive ΔΔG).
Step 4: Visualizing and Selecting Interface Mutations
Tool: PyMOL and the HTGAA Protein Engineering Tools spreadsheet.
Why: We will use PyMOL to visually inspect the predicted L-DnaJ complex from Step 2 and select residues at the interface. We will then use the spreadsheet to check the conservation of those residues and manually design mutations (e.g., swapping a large hydrophobic residue for a charged one) predicted to break the interaction.
Protein Language Models (PLMs)
Protein language models such as ESM or ProtBERT will be used to perform in silico mutagenesis on the MS2 L protein sequence. These models can suggest mutations that preserve structural and functional constraints learned from large protein datasets.
This approach allows us to generate multiple candidate mutations across the L protein, avoid mutations likely to disrupt folding, and explore sequence space beyond naturally occurring variants
AlphaFold Structure Prediction
Each candidate L variant will be analyzed using AlphaFold to predict protein structure and membrane topology. Since the C-terminal transmembrane region is essential for lytic activity, structural prediction will help identify mutations that preserve this functional domain.
Structural predictions will also help identify:
- misfolded variants
- mutations that destabilize the transmembrane region
- variants that may alter oligomerization or membrane insertion
Interaction Modeling with Host Proteins
Because MS2 L interacts with the DnaJ chaperone, which affects lysis timing, candidate variants can be evaluated using AlphaFold-Multimer to predict changes in the L–DnaJ interaction.
This could help identify variants that:
- maintain necessary folding assistance
- reduce excessive dependency on host chaperones
- improve robustness of lysis across physiological conditions
Proposed Computational Strategy
First, protein language models (e.g., ESM-2, ProtT5) will be used to perform directed in silico mutagenesis. These models capture evolutionary constraints and residue interactions, enabling the generation of structurally plausible variants while identifying mutation-tolerant and functionally critical positions. This step efficiently reduces the combinatorial search space.
Second, predicted variants will be structurally evaluated using AlphaFold2 for monomer folding and AlphaFold - Multimer to assess oligomerization and interaction with host factors such as DnaJ.
Third, membrane compatibility will be analyzed using membrane-aware modeling (RosettaMP) and selected molecular dynamics simulations.
Fourth, ΔΔG prediction tools (e.g., FoldX, Rosetta energy functions) will filter out destabilizing mutations.
In parallel, codon optimization algorithms will redesign selected variants for improved expression in E. coli, as toxicity depends on both structure and intracellular concentration.
Potential Pitfalls
Pitfall 1: Dynamic Regions and Model Quality
The L protein is small and likely has flexible/disordered regions, especially in its N-terminal domain.
Pitfall 2: Stability vs. Function Trade-off
A mutation that makes the protein more stable in its monomeric state might prevent it from undergoing the necessary conformational changes to oligomerize and form a pore in the membrane.
Pitfall 3: Lack of Membrane Context
Our stability predictions (Rosetta) are performed in a virtual “aqueous” environment and do not account for the energetic complexity of the lipid bilayer.
Limited biological data: There is still limited structural and mechanistic knowledge about MS2 L.
Cellular context not captured computationally Protein modeling tools may not fully capture membrane environment.
One limitation is the scarcity of quantitative datasets linking specific mutations to measured lysis kinetics.
L-Protein Mutants
To generate the first two mutations in the L protein of bacteriophage MS2 within the transmembrane region, I selected the top candidates predicted by the Python models and the spreadsheet analysis for that region. I applied the same approach to the soluble region, ensuring that all mutations were introduced at amino acid positions with less constrained mutability.
METRFPQQSQQTPASTNRRRPFKHEDYPCRRNQRSSTLlVLIFLAIFLSlFTlQLLLSLLEAVIRTVTTLQQLLT METRFPQQSQQTPASTNRRRPFKHEDYPCRRNQRSSTLheLnlvpnFLleFTNQLhLSLLEAeIRTVTTLQQLLT METRqPQQqQQTPASTNRRRPFKHEDYPrRRNQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT lEiRqPQQqQQTPASTNRRRPFKHEDYPrRRNQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT For the final mutation, which was the most aggressive, I introduced mutations in both regions across all possible amino acid positions.
lEiRqPQQqQQTPASTNRRRPFKHEDYPrRRNQRSSTLleLnlvpnFLleFTlQLhLSLLEAeIRTVTTLQQLLT