Week 2 HW: DNA Read, Write, & Edit
Part 0: Basics of Gel Electrophoresis
- Attend or watch all lecture and recitation videos. Optionally watch bootcamp.
Part 1: Benchling & In-silico Gel Art
- Make a free account at benchling.com
- Import the Lambda DNA.
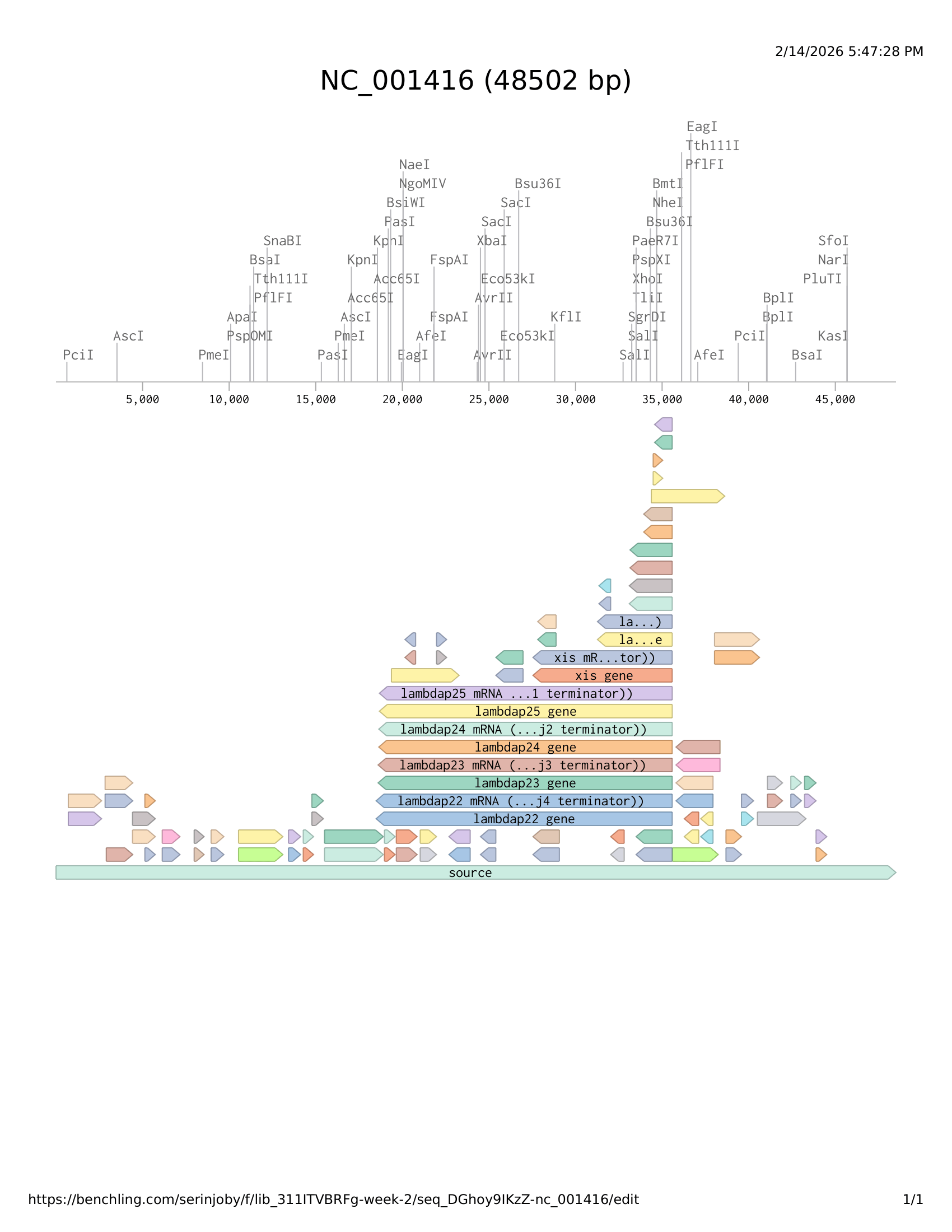
- Simulate Restriction Enzyme Digestion with the following Enzymes:
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- SalI
.png)
- Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
- You might find Ronan’s website a helpful tool for quickly iterating on designs!
Part 2: Gel Art – Restriction Digests and Gel Electrophoresis
Didnt have the lab access to perform the above experiment
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen, and why? Using one of the tools described in recitation (NCBI, UniProt, Google), obtain the protein sequence for the protein you chose.
(Example from our group homework, you may notice the particular format — The example below came from UniProt)
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
Answer
For this homework, I chose the ABO glycosyltransferase protein because it directly determines human blood group type by modifying cell-surface glycans. Since my broader project idea focuses on whether blood type A may influence gastrointestinal disease risk, this protein is central to that question. The ABO glycosyltransferase is responsible for adding specific sugar residues that create the A or B antigen. These glycan differences may influence host–microbe interactions, immune responses, or inflammation in the gut. I chose this protein because it represents the molecular basis of blood group identity, making it a logical starting point for exploring any mechanistic relationship between blood type and disease risk.
Here is the human ABO glycosyltransferase sequence (UniProt entry for human ABO):
sp|P16442|BGAT_HUMAN Histo-blood group ABO system transferase OS=Homo sapiens OX=9606 GN=ABO PE=1 SV=2 MAEVLRTLAGKPKCHALRPMILFLIMLVLVLFGYGVLSPRSLMPGSLERGFCMAVREPDH LQRVSLPRMVYPQPKVLTPCRKDVLVVTPWLAPIVWEGTFNIDILNEQFRLQNTTIGLTV FAIKKYVAFLKLFLETAEKHFMVGHRVHYYVFTDQPAAVPRVTLGTGRQLSVLEVRAYKR WQDVSMRRMEMISDFCERRFLSEVDYLVCVDVDMEFRDHVGVEILTPLFGTLHPGFYGSS REAFTYERRPQSQAYIPKDEGDFYYLGGFFGGSVQEVQRLTRACHQAMMVDQANGIEAVW HDESHLNKYLLRHKPTKVLSPEYLWDQQLLGWPAVLRKLRFTAVPKNHQAVRNP
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which a DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (Google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
(Example: Get to the original sequence of phage MS2 L-protein from its genome – phage MS2 genome - Nucleotide - NCBI)
Lysis protein DNA sequence
atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
Answer

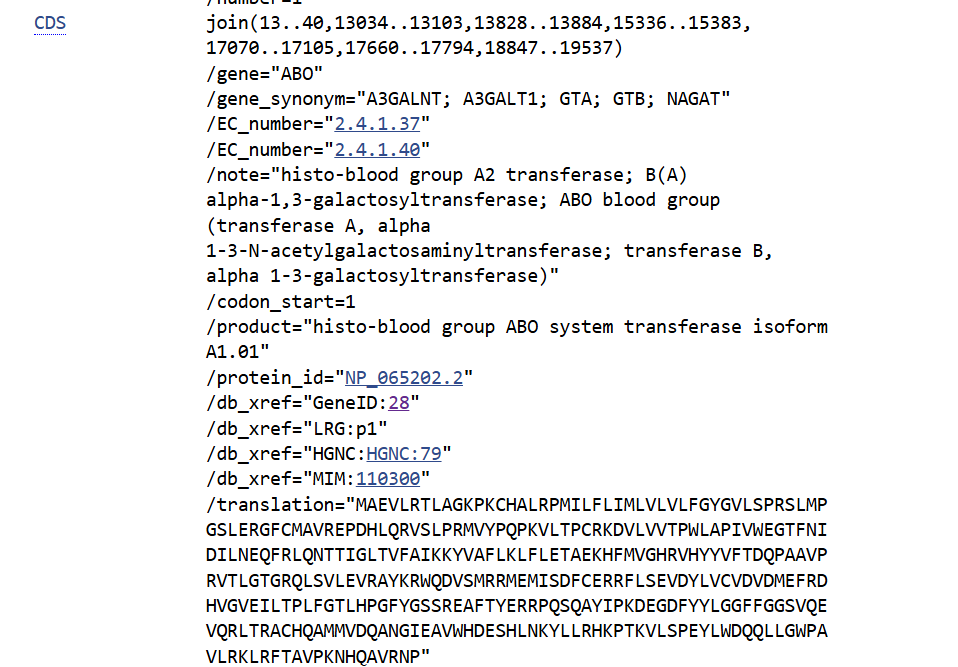
NG_006669.2:5026-5053,18047-18116,18841-18897,20349-20396,22083-22118,22673-22807,23860-24550 Homo sapiens ABO, alpha 1-3-N-acetylgalactosaminyltransferase and alpha 1-3-galactosyltransferase (ABO), RefSeqGene (LRG_792) on chromosome 9 ATGGCCGAGGTGTTGCGGACGCTGGCCGGAAAACCAAAATGCCACGCACTTCGACCTATGATCCTTTTCC TAATAATGCTTGTCTTGGTCTTGTTTGGTTACGGGGTCCTAAGCCCCAGAAGTCTAATGCCAGGAAGCCT GGAACGGGGGTTCTGCATGGCTGTTAGGGAACCTGACCATCTGCAGCGCGTCTCGTTGCCAAGGATGGTC TACCCCCAGCCAAAGGTGCTGACACCGTGTAGGAAGGATGTCCTCGTGGTGACCCCTTGGCTGGCTCCCA TTGTCTGGGAGGGCACATTCAACATCGACATCCTCAACGAGCAGTTCAGGCTCCAGAACACCACCATTGG GTTAACTGTGTTTGCCATCAAGAAATACGTGGCTTTCCTGAAGCTGTTCCTGGAGACGGCGGAGAAGCAC TTCATGGTGGGCCACCGTGTCCACTACTATGTCTTCACCGACCAGCCGGCCGCGGTGCCCCGCGTGACGC TGGGGACCGGTCGGCAGCTGTCAGTGCTGGAGGTGCGCGCCTACAAGCGCTGGCAGGACGTGTCCATGCG CCGCATGGAGATGATCAGTGACTTCTGCGAGCGGCGCTTCCTCAGCGAGGTGGATTACCTGGTGTGCGTG GACGTGGACATGGAGTTCCGCGACCACGTGGGCGTGGAGATCCTGACTCCGCTGTTCGGCACCCTGCACC CCGGCTTCTACGGAAGCAGCCGGGAGGCCTTCACCTACGAGCGCCGGCCCCAGTCCCAGGCCTACATCCC CAAGGACGAGGGCGATTTCTACTACCTGGGGGGGTTCTTCGGGGGGTCGGTGCAAGAGGTGCAGCGGCTC ACCAGGGCCTGCCACCAGGCCATGATGGTCGACCAGGCCAACGGCATCGAGGCCGTGTGGCACGACGAGA GCCACCTGAACAAGTACCTGCTGCGCCACAAACCCACCAAGGTGCTCTCCCCCGAGTACTTGTGGGACCA GCAGCTGCTGGGCTGGCCCGCCGTCCTGAGGAAGCTGAGGTTCACTGCGGTGCCCAAGAACCACCAGGCG GTCCGGAACCCGTGA
3.3. Codon optimisation.
Once a nucleotide sequence of your protein is determined, you need to codon optimise your sequence. You may, once again, utilise Google for a “codon optimisation tool”. In your own words, describe why you need to optimise codon usage. Which organism have you chosen to optimise the codon sequence for, and why?
(Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI)
Lysis protein DNA sequence with codon optimisation
ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
Answer
Once the nucleotide sequence of the protein is determined, codon optimisation is necessary because different organisms prefer different codons to encode the same amino acid. Although multiple codons can code for one amino acid, the frequency with which each codon is used varies between species. If a gene contains many codons that are rare in the host organism, translation can be inefficient, leading to low protein yield or incorrect folding. Codon optimisation adjusts the DNA sequence to better match the codon usage bias of the chosen expression host, without changing the amino acid sequence of the protein.
For this project, I chose to optimise the codon sequence for Escherichia coli, since it is one of the most commonly used organisms for recombinant protein expression. E. coli grows quickly, is inexpensive to culture, and has well-established cloning and expression systems. Optimising the ABO glycosyltransferase gene for E. coli would increase the likelihood of efficient transcription and translation, improving protein yield for experimental studies. Additionally, codon optimisation tools can help avoid problematic sequences such as strong secondary structures, rare codons, or unwanted restriction enzyme recognition sites.
Optimized codon:
ATGGCGGAAGTGCTGCGTACCCTGGCAGGTAAACCGAAGTGCCATGCCCTGCGTCCGATGATTCTGTTCCTGATTATGCTGGTGCTGGTGCTGTTCGGTTATGGCGTGCTGAGCCCGCGTAGCCTGATGCCGGGCTCTCTGGAACGTGGTTTCTGCATGGCGGTGCGCGAACCGGACCATCTGCAGCGTGTGAGCCTGCCGCGCATGGTGTATCCGCAGCCGAAAGTTCTGACCCCGTGCCGCAAAGATGTGCTGGTGGTGACGCCGTGGCTGGCGCCGATTGTGTGGGAAGGCACCTTTAATATTGATATTCTGAATGAACAGTTTCGCCTGCAGAATACCACCATTGGCCTGACCGTGTTTGCGATTAAAAAATACGTGGCGTTTCTGAAACTGTTTCTGGAAACGGCGGAAAAACATTTCATGGTGGGCCATCGCGTGCACTACTACGTCTTCACCGATCAGCCGGCGGCGGTGCCGCGCGTTACCCTGGGCACGGGCCGCCAGCTGAGCGTGCTGGAAGTGCGCGCGTATAAACGTTGGCAGGATGTTAGCATGCGCCGCATGGAAATGATTAGCGATTTTTGCGAACGTCGCTTTCTGAGCGAAGTGGATTATCTGGTGTGCGTGGATGTGGATATGGAATTTCGCGATCATGTGGGCGTGGAAATTCTGACCCCGCTGTTTGGCACCCTGCATCCGGGCTTCTATGGCAGCAGCCGCGAAGCATTCACCTACGAACGCCGCCCGCAGAGCCAGGCCTACATTCCGAAAGATGAAGGCGATTTCTATTATCTGGGCGGCTTCTTTGGCGGCTCAGTTCAGGAAGTGCAGCGTCTGACCCGCGCCTGCCATCAGGCGATGATGGTGGACCAGGCGAACGGCATTGAAGCCGTTTGGCATGATGAAAGCCATCTGAACAAATACCTGCTGCGTCATAAACCGACCAAAGTTCTGTCGCCGGAATATCTGTGGGATCAGCAGCTGCTGGGCTGGCCGGCGGTGCTGCGTAAACTGCGCTTTACCGCGGTGCCGAAAAACCATCAGGCGGTACGTAATCCGTAA
After codon optimisation using the VectorBuilder tool, the sequence showed a GC content of 56.53% and a Codon Adaptation Index (CAI) of 0.94. The GC content falls within the preferred range for E. coli expression (typically ~30–70%), suggesting the sequence should be stable and efficiently transcribed. The CAI value is close to 1.0, which indicates that the codons used in the optimised gene closely match the codon usage bias of the host organism. A high CAI generally correlates with improved translation efficiency because the host has abundant tRNAs for these codons.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words how the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Answer
To produce the protein from the DNA sequence, the optimised gene would first be cloned into an expression vector containing a promoter, ribosome binding site, and terminator. The plasmid would then be introduced into a host such as E. coli through transformation. Inside the cell, RNA polymerase binds to the promoter and transcribes the DNA into messenger RNA (mRNA). The ribosome then binds to the mRNA and reads the codons, while tRNAs deliver the corresponding amino acids to build the polypeptide chain. The growing chain folds into the functional ABO glycosyltransferase protein after translation.
An alternative method is a cell-free expression system, where purified transcription and translation machinery are mixed with the DNA template in vitro. In this system, RNA is synthesised from the DNA and immediately translated into protein without living cells. Cell-free expression is faster and easier to control, while cell-based expression generally produces larger quantities of protein.
In both approaches, the central dogma applies: DNA is transcribed into RNA, and RNA is translated into the protein.
3.5. How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level. Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated protein!!!
Answer
A single gene can produce multiple proteins at the transcriptional level, mainly through alternative splicing. During transcription, the DNA sequence is copied into a pre-mRNA that contains both exons (coding regions) and introns (non-coding regions). The cell’s splicing machinery can remove introns in different patterns and join different combinations of exons together. As a result, multiple mature mRNA transcripts can be produced from the same gene, and each mRNA can be translated into a slightly different protein with different structure or function. This allows one gene to increase protein diversity without changing the DNA sequence.
Below is a small illustrative alignment showing how DNA becomes RNA and then protein. Notice that T becomes U during transcription, and every 3 nucleotides (codon) form one amino acid during translation:
DNA: ATG AAA GCT TTT GGA TAA
RNA: AUG AAA GCU UUU GGA UAA
Protein: Met Lys Ala Phe Gly Stop
If an exon is skipped during splicing, the RNA sequence changes:
DNA: ATG AAA GGA TAA
RNA: AUG AAA GGA UAA
Protein: Met Lys Gly Stop
Even though the gene is the same, different mRNA transcripts lead to different proteins. This is one of the main ways cells generate protein diversity from a limited number of genes.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
4.2. Build Your DNA Insert Sequence
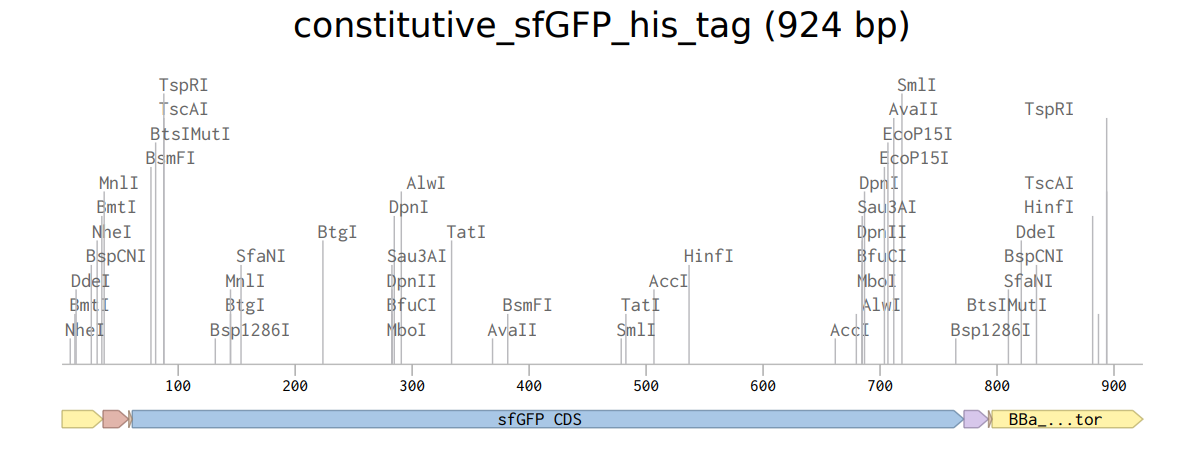
Click here to get the final sequence
FASTA file for the above sequence
constitutive_sfGFP_his_tag TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGCCATTAAAGAGGAGAAAGGTACCATGAGCAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCCGTGGAGAGGGTGAAGGTGATGCTACAAACGGAAAACTCACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCGTGGCCAACACTTGTCACTACTCTGACCTATGGTGTTCAATGCTTTTCCCGTTATCCGGATCACATGAAACGGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGCACTATATCTTTCAAAGATGACGGGACCTACAAGACGCGTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATCGTATCGAGTTAAAGGGTATTGATTTTAAAGAAGATGGAAACATTCTTGGACACAAACTCGAGTACAACTTTAACTCACACAATGTATACATCACGGCAGACAAACAAAAGAATGGAATCAAAGCTAACTTCAAAATTCGCCACAACGTTGAAGATGGTTCCGTTCAACTAGCAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCGACACAATCTGTCCTTTCGAAAGATCCCAACGAAAAGCGTGACCACATGGTCCTTCTTGAGTTTGTAACTGCTGCTGGGATTACACATGGCATGGATGAGCTCTACAAACATCACCATCACCATCATCACTAACCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
4.5. Import your sequence

4.6. Choose Your Vector
When I first uploaded the DNA sequence, it gave an error due to high GC content. I then used Twist’s built-in codon optimization tool for E. coli to optimize the sequence, and the new sequence is provided below:
ATGGCAGAAG TTCTTCGCAC TTTAGCAGGC AAGCCCAAAT GTCACGCATT ACGGCCAATG ATATTATTTC TCATCATGCT CGTTTTGGTA CTCTTTGGCT ACGGTGTACT CAGTCCTCGC TCTTTGATGC CTGGTAGTTT AGAGAGAGGG TTTTGTATGG CCGTCCGGGA GCCAGATCAC CTGCAAAGAG TATCATTGCC TCGGATGGTT TACCCCCAAC CTAAGGTGTT AACTCCTTGT CGAAAGGACG TTCTTGTAGT AACTCCTTGG CTTGCCCCTA TCGTATGGGA AGGTACATTC AACATCGACA TCCTTAACGA GCAATTCCGG TTGCAAAACA CGACTATAGG TCTTACAGTT TTCGCAATAA AGAAGTATGT TGCCTTCCTC AAGTTATTCC TCGAGACAGC TGAGAAGCAC TTTATGGTCG GTCACCGGGT TCATTATTAT GTGTTTACTG ACCAACCAGC AGCCGTTCCT CGTGTCACTT TAGGTACTGG TCGTCAATTA TCCGTTCTCG AGGTCCGGGC CTACAAGCGC TGGCAAGACG TATCTATGCG TCGAATGGAG ATGATCAGTG ACTTCTGTGA GCGGAGATTC CTTTCAGAGG TTGACTACTT GGTCTGTGTA GACGTTGACA TGGAGTTCCG GGACCACGTA GGTGTTGAGA TCTTAACGCC ATTATTCGGA ACTCTTCACC CCGGTTTCTA CGGGAGTTCG CGCGAGGCTT TTACATATGA GCGTAGACCT CAATCCCAAG CATATATACC TAAGGACGAG GGTGACTTTT ACTACTTAGG TGGATTCTTC GGTGGGTCCG TACAAGAGGT TCAACGCTTA ACTCGGGCAT GTCACCAAGC AATGATGGTC GATCAAGCAA ATGGGATCGA GGCAGTCTGG CACGACGAGT CTCACTTAAA TAAGTATTTG CTTCGGCACA AGCCAACAAA GGTGCTTAGT CCCGAGTACT TGTGGGACCA ACAATTACTC GGATGGCCTG CAGTCCTTAG AAAGCTCCGT TTCACGGCAG TTCCCAAGAA TCACCAAGCT GTTCGGAACC CATGA
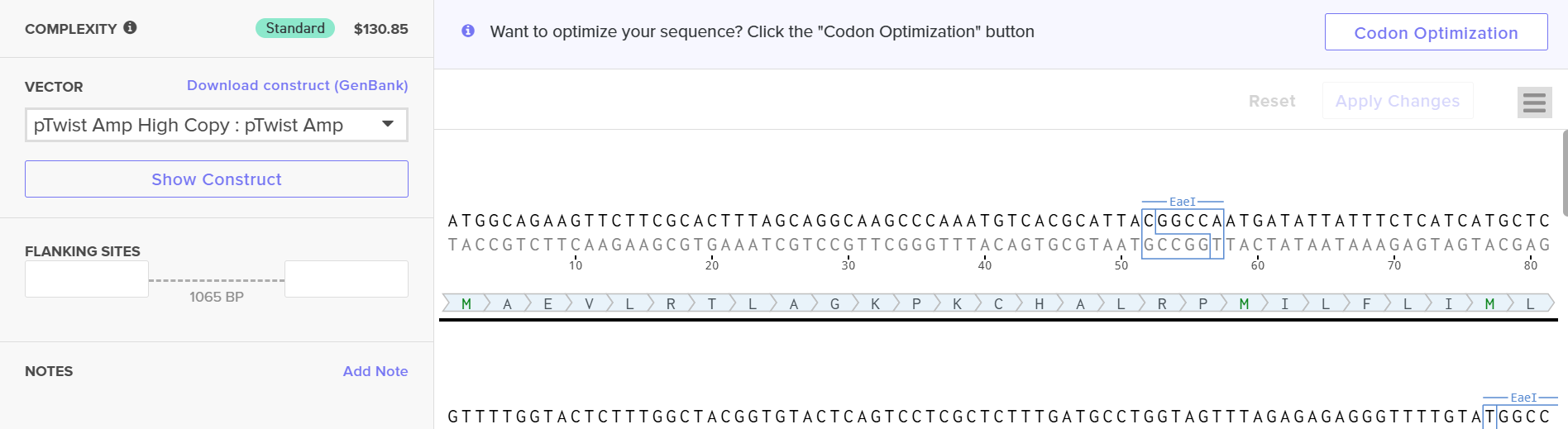
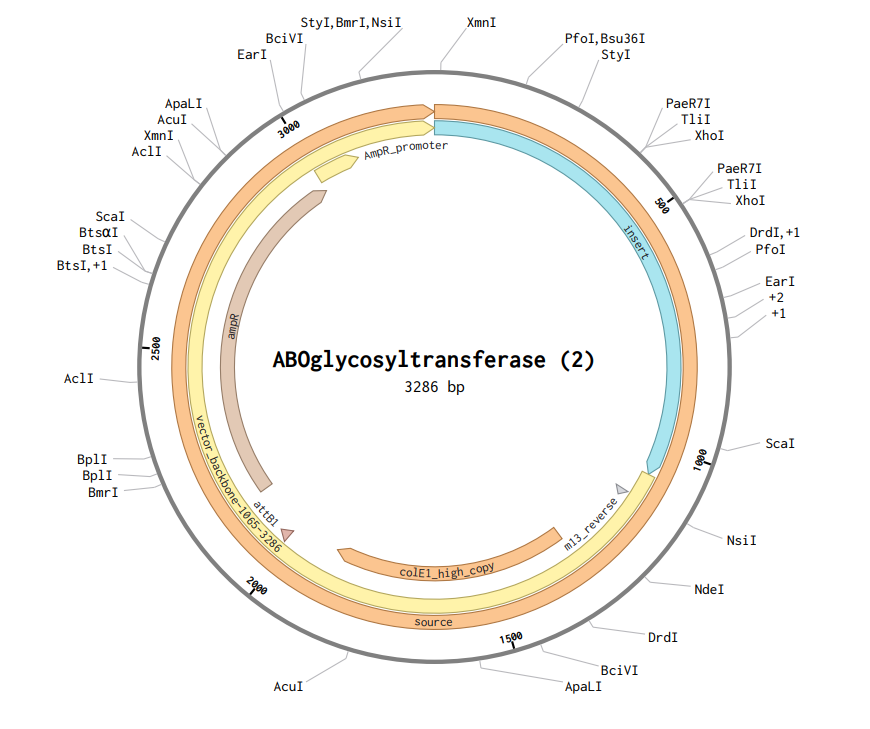
After downloading the construct from Twist, I uploaded it to Benchling, and the plasmid map obtained is shown below.
Part 5: DNA Read/Write/Edit
5.1. DNA Read
- What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
- For my project, I would want to sequence gut microbiome DNA from people with different ABO blood groups, especially comparing blood type A with non-A individuals. My research question is whether the reported association between blood type A and gastrointestinal disease risk has an actual biological mechanism rather than being only a population-level correlation. ABO blood groups are defined by differences in glycan structures, and these glycans are not only present on red blood cells but also on intestinal mucosal surfaces. Many gut microbes interact directly with host glycans by binding to them or metabolizing them as nutrients. Because of this, I suspect that different blood group glycans could shape the microbial community in the gut. Sequencing microbiome DNA would allow me to determine whether certain bacteria, especially glycan-binding or inflammation-associated species, are enriched in individuals with blood type A. In addition, metagenomic sequencing would reveal functional genes such as glycan-degrading enzymes or virulence factors that might trigger inflammatory responses. This information would help identify biological markers that could be used as inputs for a synthetic biology sensing system designed to test the mechanism experimentally.
- In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
(i) Is your method first-, second- or third-generation or other? How so?
(ii) What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
(iii) What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
(iv) What is the output of your chosen sequencing technology?
For my project, I would use Illumina sequencing (sequencing by synthesis) to sequence gut microbiome DNA from individuals with different ABO blood groups. Since my goal is to compare microbial communities and identify possible functional genes linked to host glycan interactions, I need a method that can accurately sequence many DNA fragments from a mixed sample at high depth. Illumina sequencing is widely used for metagenomics because it provides high accuracy, strong statistical power, and the ability to detect small differences in microbial composition between groups.
Illumina sequencing is a second-generation (next-generation sequencing, NGS) technology. It is considered second-generation because it performs massively parallel sequencing of millions of DNA fragments at the same time and requires clonal amplification before sequencing. The technology uses bridge amplification to generate clusters and reversible terminator nucleotides to read one base at a time. Unlike first-generation Sanger sequencing, which reads single fragments individually, Illumina reads many short fragments simultaneously, making it suitable for complex microbiome samples.
The input for this method would be total DNA extracted from stool samples containing gut microbiome material. First, DNA is isolated from the sample and then fragmented into short pieces of approximately 200–500 base pairs. The fragment ends are repaired and modified by adding an A-tail, followed by ligation of Illumina-specific adapters to both ends. The adapter-ligated fragments are PCR amplified to enrich correctly prepared molecules and create the sequencing library. After quality control and quantification, the library is loaded onto the flow cell for sequencing.
The sequencing process begins with cluster generation on the flow cell. DNA fragments bind to complementary oligonucleotides attached to the surface and undergo bridge amplification, forming clonal clusters of identical DNA molecules. During sequencing by synthesis, fluorescently labeled nucleotides with reversible terminators are added. Only one nucleotide can be incorporated in each cycle. After incorporation, a camera records the fluorescent signal, which corresponds to a specific base (A, T, C, or G). The fluorescent label and terminator are then chemically removed, allowing the next cycle to occur. By repeating this process, the machine determines the sequence base by base through detection of fluorescence signals, a process known as base calling.
The output of Illumina sequencing is a large collection of short DNA sequence reads stored in FASTQ files. Each read contains the nucleotide sequence along with a quality score indicating confidence in each base call. These reads can then be analyzed bioinformatically to identify microbial species, compare microbiome composition between blood groups, and detect functional genes such as glycan-degrading enzymes or inflammation-associated factors. This information helps evaluate whether differences in microbiome behavior could explain the observed association between blood type A and gastrointestinal disease risk.
5.2. DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize!
For my project, I would synthesize a bacterial genetic sensing circuit that detects blood-group-related glycans and activates a measurable reporter when inflammatory conditions are present. The goal is not to diagnose disease yet, but to experimentally test whether molecules associated with blood type A environments change microbial behavior in a biologically meaningful way.
ABO blood groups differ in terminal sugar structures on host glycans. Blood type A contains N-acetylgalactosamine (GalNAc) as the terminal sugar. Many gut bacteria recognize or metabolize host glycans, so my idea is to engineer a bacterium (for example a lab strain of E. coli) with a circuit that turns on a fluorescent signal only when two conditions occur: detection of A-associated glycans and detection of inflammation-related signals (such as nitrate or reactive oxygen stress). This would function as a controllable research platform to experimentally connect host glycans to microbial inflammatory responses.
The DNA I would synthesize is therefore a two-input AND-gate genetic circuit consisting of: A glycan-responsive promoter (activated by GalNAc metabolism regulator), an inflammation-responsive promoter (stress/nitrate inducible), a transcriptional logic gate (split activator system), and a GFP reporter gene
If fluorescence appears only when both signals are present, it would support the hypothesis that specific host glycan environments influence microbial inflammatory behavior.
Example construct design:
- Part 1 – Constitutive regulator expression: Promoter → regulator protein sensing GalNAc
- Part 2 – Inflammation promoter controlling activator half: Stress promoter → Activator fragment A
- Part 3 – Glycan promoter controlling activator half: GalNAc promoter → Activator fragment B
- Part 4 – Output reporter: AND gate → GFP expression
Below is a simplified example of a reporter cassette that could realistically be synthesized (promoter + RBS + GFP + terminator):
TTGACATGATAAGTAAGGAGGTTTAAACATGAGTAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCAGTGGAGAGGGTGAAGGTGATGCAACATACGGAAAACTTACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCATGGCCAACACTTGTCACTACTTTCTCTTATGGTGTTCAATGCTTTTCAAGATACCCAGATCATATGAAACAGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTACAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGTGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAGCACCCAGTCCAAGCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGCTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAATTTTTTGCTAGC
(This represents a GFP reporter module; regulatory promoters would be placed upstream depending on the sensing design.)
Synthesizing this circuit allows experimental testing of the hypothesis: Blood-type-specific glycans influence microbial inflammatory behavior.
Instead of relying on epidemiological correlations, the engineered system creates a controllable biological readout. If activation differs in A-glycan conditions compared to others, it would provide mechanistic evidence that host glycan composition can shape disease-related microbial responses.
(ii) What technology or technologies would you use to perform this DNA synthesis, and why? Also answer the following questions:
- What are the essential steps of your chosen sequencing methods?
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, and scalability?
- To synthesise my designed genetic circuit, I would use array-based phosphoramidite DNA synthesis followed by fragment assembly (such as Gibson Assembly). Because my construct is a designed sequence rather than naturally occurring DNA, it must be chemically built from short oligonucleotides and then assembled into a complete gene cassette. This approach allows precise control over regulatory elements such as promoters, ribosome binding sites, and reporter genes, which is necessary for constructing a synthetic sensing circuit. The process begins with chemical synthesis of short oligonucleotides (about 60–200 bp) using phosphoramidite chemistry, where nucleotides are added one base at a time to a growing DNA strand attached to a solid surface. After deprotection and cleavage, the oligos are PCR amplified and designed with overlapping regions. These fragments are then assembled into the full construct using Gibson Assembly, in which exonuclease creates complementary overhangs, polymerase fills gaps, and ligase seals the backbone. The assembled plasmid is transformed into bacteria, and colonies are collected for sequence verification.
- To read and verify the synthesised DNA, I would use Illumina sequencing (sequencing-by-synthesis). The plasmid DNA would first be extracted and fragmented, adapters would be ligated, and a sequencing library would be prepared. The fragments bind to a flow cell and undergo bridge amplification to form clusters. During sequencing, fluorescent reversible terminator nucleotides are incorporated one at a time, and each cycle is imaged to identify the added base. The fluorescent signal detected at each cycle is converted into nucleotide identity through base calling, generating short sequence reads that can be aligned to the designed construct to confirm its correctness.
- The main limitations of Illumina sequencing relate to read length and assembly rather than accuracy. Although it provides very high accuracy and throughput, it produces short reads, so reconstruction of long repetitive regions can be difficult. For my application this is manageable because plasmids are small and have a known reference sequence. In terms of speed, library preparation and sequencing runs take several hours to days, which is slower than simple PCR validation but provides much more reliable confirmation. Scalability is excellent since many constructs can be sequenced simultaneously, but costs increase when sequencing only a very small number of samples.
5.3. DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
- For my project, I would want to edit bacterial DNA rather than human DNA, specifically genes involved in glycan recognition and inflammatory sensing in a model gut bacterium such as E. coli. The goal of my work is to test whether ABO blood-group glycans , especially the type A terminal sugar N-acetylgalactosamine (GalNAc) , can influence microbial behavior linked to gastrointestinal disease. Instead of modifying patients, I would engineer a controllable microbial system that mimics how gut bacteria might respond inside the intestine. The main edits I would introduce are regulatory and sensing modifications. First, I would insert a glycan-responsive sensing module so the bacterium can detect A-type glycans. This could involve adding or modifying carbohydrate-binding proteins or transport/metabolism regulators that activate transcription when GalNAc is present. Second, I would add an inflammation-response module that detects gut stress signals such as nitrate or oxidative stress, which are commonly elevated during intestinal inflammation. Finally, I would connect both inputs to a reporter output (for example fluorescence), forming a logical AND gate so the cell responds only when both host glycan signals and inflammatory conditions occur together. These edits would allow the bacterium to act as a biological probe of the gut environment. If the engineered cells activate differently in A-type glycan conditions compared to others, it would suggest a mechanistic relationship between blood group chemistry and microbial inflammatory behavior. This approach avoids ethical concerns of editing human genomes and instead creates a reversible experimental model that helps transform epidemiological correlations into testable biological mechanisms.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions:
- How does your technology of choice edit DNA? What are the essential steps?
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
- To introduce the edits in my engineered gut bacterium, I would use CRISPR-Cas9 genome editing combined with homologous recombination. This approach is widely used in bacteria because it allows precise insertion of synthetic genetic circuits at defined genomic locations rather than relying only on plasmids. For my project, stable integration is useful so the sensing system behaves consistently across experiments and does not get lost during cell growth.
- CRISPR-Cas9 edits DNA by creating a targeted double-strand break at a specific sequence determined by a guide RNA. The cell then attempts to repair this break. If a repair template containing designed DNA is provided, the bacterium uses homologous recombination to copy that template into its genome. In my case, the repair template would contain the glycan-sensing promoter, inflammation-response module, and reporter gene arranged as a logic circuit. The essential steps are: designing a guide RNA targeting a safe insertion site, delivering Cas9 and the guide into the bacteria, introducing a donor DNA template with homologous flanking regions, cleavage of the genome at the target site, and repair using the donor DNA to integrate the synthetic construct.
- Preparation involves several design stages. First, I would computationally select a genomic locus that does not disrupt essential genes. Then I would design the single guide RNA (sgRNA) sequence that uniquely matches that region. Next, I would synthesize a donor DNA template containing my circuit flanked by homology arms (~500–1000 bp) matching the insertion site. The experimental inputs therefore include: a plasmid expressing Cas9, a plasmid or cassette encoding the sgRNA, the donor DNA template, competent bacterial cells, and standard transformation reagents. After transformation, edited cells would be selected and verified by sequencing.
- The main limitations of this editing method are efficiency and off-target activity. Not all cells successfully incorporate the donor DNA after cutting, so screening is required to isolate correct clones. Homologous recombination efficiency in bacteria can also vary depending on strain and insert size, making larger constructs harder to integrate. Although CRISPR is precise, imperfect guide design can cause unintended cuts at similar sequences, potentially damaging the genome. Finally, multiplex editing (editing many sites at once) becomes less reliable because each additional edit lowers overall success probability. Despite these limitations, CRISPR-Cas9 provides sufficient precision and flexibility for constructing a stable synthetic sensing platform.