Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- Our digestive system breaks proteins down into amino acids. We don’t just absorb the animal’s proteins directly; we use these building blocks to make human proteins. So no matter what we eat, our body remains human.
- Why are there only 20 natural amino acids?
- These 20 were selected by evolution because they give enough chemical diversity to build proteins. They are stable, easy to synthesise, and work well with the ribosome and genetic code. More could exist, but these 20 are just what life settled on.
- Can you make other non-natural amino acids? Design some new amino acids.
- Yes! Scientists can make synthetic amino acids to tweak protein properties. Examples I thought of: Seleno-cysteine variant → replacing sulfur with selenium to improve redox activity or make enzymes more reactive and, Fluoro-leucine → adds a fluorine atom to increase hydrophobic interactions and thermal stability.
- Where did amino acids come from before enzymes that make them, and before life started?
- Amino acids could form in the prebiotic world through reactions like Strecker synthesis. They might have also come from meteorites or formed from simple gases (CH₄, NH₃, H₂O) with energy sources like lightning or UV light. So amino acids existed before life could make them.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- α-helices made from L-amino acids are right-handed. D-amino acids are mirror images, so the helix would be left-handed.
- Can you discover additional helices in proteins?
- Yes! We can find new helices in proteins that weren’t noticed before. Experimental methods like X-ray crystallography, NMR, or cryo-EM can reveal hidden or flexible helices. Computational tools like AlphaFold or PSIPRED can predict helical regions just from the protein sequence. Some helices only form under certain conditions, like when a protein binds a ligand or embeds in a membrane. So combining experiments and predictions, we can discover or even design new helices to improve protein stability or function.
- Why are most molecular helices right-handed?
- Life uses L-amino acids, which naturally favour right-handed helices because of steric constraints and backbone angles. Left-handed helices are less stable with L-amino acids, so they are rare in nature.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
- β-sheets expose backbone hydrogen bonds and sometimes hydrophobic side chains. These can interact with other β-sheets, leading to aggregation. This stacking is a major reason some proteins clump together.
- The main forces are hydrogen bonding between β-strands, hydrophobic interactions among side chains, and the entropy gain from releasing water molecules when sheets pack together.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
- Misfolded proteins often expose β-strands that like to stack. These β-sheets form insoluble fibrils, which resist degradation and build up as plaques in diseases like Alzheimer’s.
- Yes! Their stability and ability to self-assemble make them useful in materials science. They can form hydrogels, nanofibers, or scaffolds for tissue engineering, which are biodegradable and strong.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
- Briefly describe the protein you selected and why you selected it.
- I selected the human ABO glycosyltransferase, the enzyme responsible for determining the ABO blood group system. This enzyme modifies the H antigen on red blood cells by transferring a sugar residue. The A variant transfers N-acetylgalactosamine, while the B variant transfers galactose. Very small amino acid changes in this protein determine whether someone has blood type A, B, AB, or O. I chose this protein because it is clinically important and demonstrates how small sequence changes can drastically alter enzyme specificity.
- Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Does your protein belong to any protein family?
- UniProt Entry: P16442 (ABO glycosyltransferase, Homo sapiens). The full-length protein contains 354 amino acids.
- From sequence analysis, common residues include:Leucine (L), Glycine (G) and Serine (S) Leucine( 41 times) is abundant because it stabilises the hydrophobic core. Glycine appears frequently in flexible loop regions, particularly near the catalytic site.
- The results showed many homologous proteins with sequence identities ranging from 54.3% to 100%. The alignment scores ranged from 205 to 1879, and the E-values were extremely low (from 4.4e-14 down to 0), indicating highly significant similarity. These results show that ABO glycosyltransferase is strongly conserved across species and belongs to a well-established glycosyltransferase enzyme family.
- Yes. It belongs to the glycosyltransferase family, specifically the GT6 family (according to CAZy classification). These enzymes transfer sugar moieties from activated nucleotide sugars (like UDP-sugars) to acceptor substrates.
- Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å).Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
- The structure of human ABO glycosyltransferase can be found in the RCSB Protein Data Bank under PDB ID 1LZ7. This structure was solved in 2002 using X-ray crystallography at a resolution of approximately 2.0 Å. Since good-quality structures are typically below 2.7 Å resolution, this is considered a high-quality structure. In addition to the protein, the structure contains a UDP-sugar substrate analog, a metal ion (Mn²⁺), and water molecules. Structurally, the enzyme belongs to the GT-B fold glycosyltransferase family, which consists of two Rossmann-like domains forming a catalytic cleft.
- Open the structure of your protein in any 3D molecule visualization software: Visualize the protein as “cartoon”, “ribbon” and “ball and stick”. Color the protein by secondary structure. Does it have more helices or sheets? Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
PDB ID: 1LZ7 (human ABO A-transferase) - Cartoon
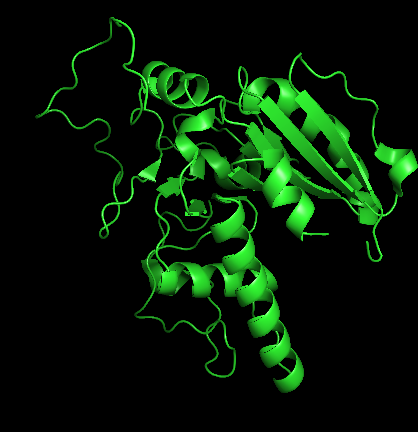
Ribbon

It has more helices coloured in red.
- Ball and stick (for protein + ligand)

- Color by Secondary Structure
Red = α-helices, Yellow = β-sheets and Green = loops
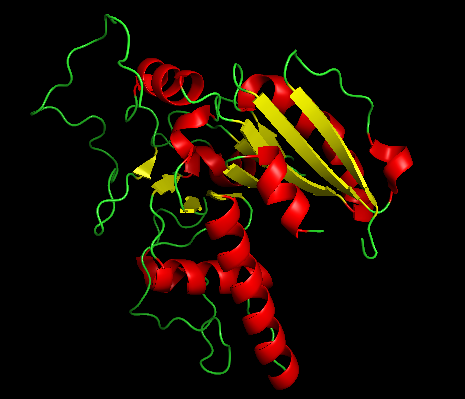
- Color by Residue Type (Hydrophobic vs Hydrophilic)
Hydrophobic residues:
orange, resn ALA+VAL+LEU+ILE+MET+PHE+TRP+PRO
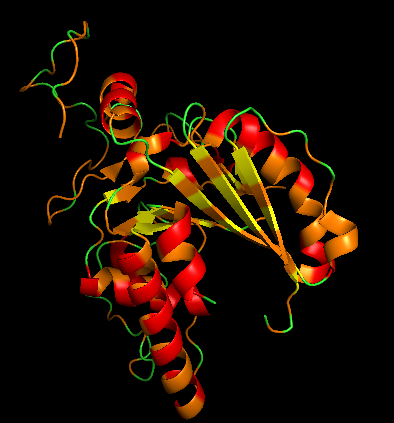
Polar/charged residues:
cyan, resn SER+THR+ASN+GLN+TYR+CYS
blue, resn LYS+ARG+HIS
red, resn ASP+GLU
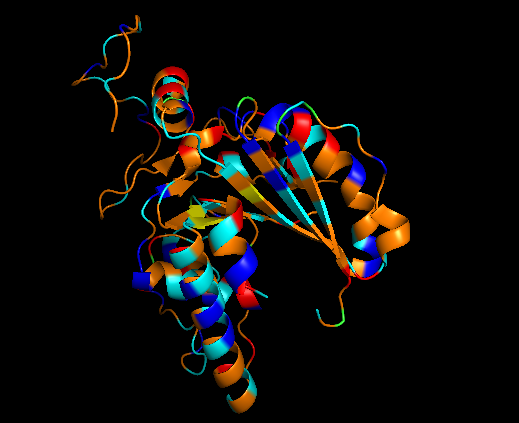
It has almost the same amount of residues but polar/charged residues are little higher than hydrophobic residues.
I observed one binding pocket, that’s it.
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
[x] Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
[x] Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
- Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
- I used the ESM2 protein language model to generate an unsupervised deep mutational scan of the human ABO glycosyltransferase sequence. The heatmap shows the predicted effect of every possible amino acid substitution across the protein sequence. Positions with yellow or light green colors correspond to mutations that are more tolerated, while dark blue or purple indicates mutations that are predicted to be unfavorable.
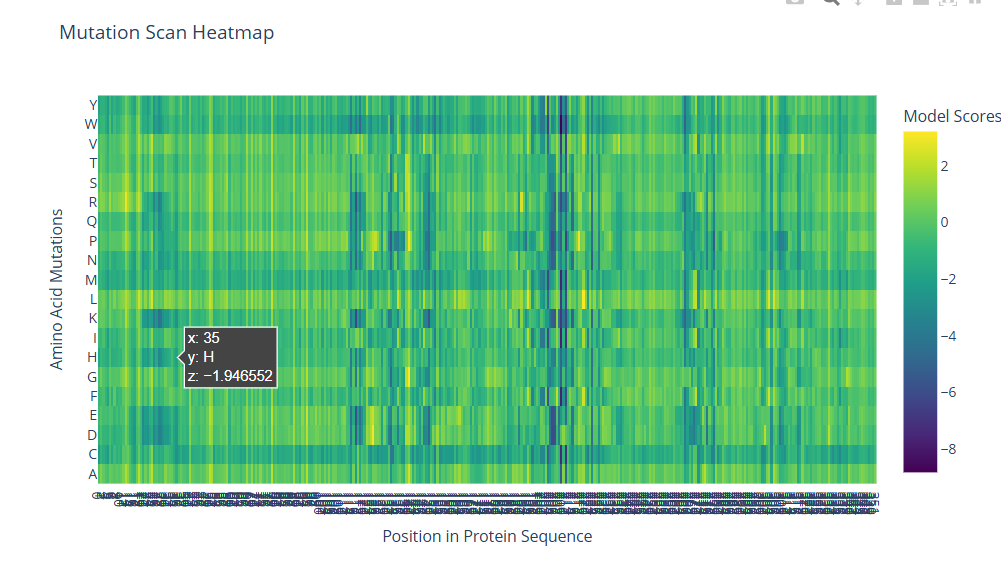
Several vertical bands of darker colors appear across the heatmap, indicating positions where most mutations are not tolerated. These likely correspond to structurally important or functionally critical residues in the protein. In contrast, many other regions show more neutral scores, suggesting they are more tolerant to mutation and may be located in flexible or surface regions.
For example, at position 35, mutating the residue to histidine produces a negative score (approximately −1.95), suggesting this substitution is unfavorable. This could be because histidine introduces a charged side chain that disrupts local packing or interactions.
As a bonus analysis, I attempted to find experimental deep mutational scan datasets for the human ABO glycosyltransferase. However, comprehensive experimental scans for this protein were not readily available in public databases. Therefore, a direct comparison between the ESM2 predictions and experimental mutation effects could not be performed. Nevertheless, previous studies on other proteins have shown that protein language models like ESM2 often correlate well with experimental mutational scans, particularly for identifying conserved and functionally important residues.
- Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
- Using the provided dataset, protein sequences were embedded into a reduced dimensional latent space using a protein language model and visualized using t-SNE. In this representation, each point corresponds to a protein sequence, and proteins with similar sequence features appear close together.
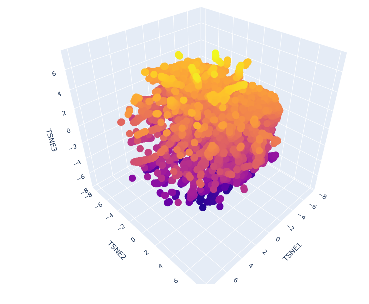
The resulting map shows a dense cluster of proteins with a gradual spread across the three t-SNE dimensions. This suggests that many of the proteins in the dataset are related and share common structural or functional features.
My selected protein, the human ABO glycosyltransferase, appears within this cluster and is positioned near other proteins with similar sequence characteristics. These neighboring proteins likely belong to related glycosyltransferase families and share similar catalytic functions involving sugar transfer reactions.
C2. Protein Folding
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
- The protein sequence was folded using ESMFold to predict its 3D structure. The predicted coordinates were compared to the original experimental structure using structural alignment in PyMol. The two structures showed strong agreement with a low RMSD value, indicating that ESMFold accurately reproduced the overall protein fold.
To test structural robustness, several mutations were introduced into the sequence. Single amino acid substitutions caused only minor local structural changes, while the overall fold of the protein remained largely unchanged. This suggests that the protein structure is relatively resilient to small mutations.
When larger sequence modifications were introduced, such as altering or replacing longer segments of the sequence, the predicted structure became more distorted. In these cases, secondary structure elements were disrupted and the overall fold changed significantly.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
- The backbone structure of the selected protein was used as input for ProteinMPNN to generate candidate sequences that could fold into the same structure. ProteinMPNN predicted several sequences along with probability distributions for each residue position.
Comparison of the predicted sequences with the original sequence showed moderate sequence identity, with many conserved residues in structurally important regions. Positions located in the protein core or active site showed strong preferences for specific amino acids, while surface residues displayed greater variability and allowed multiple amino acid substitutions.
The top predicted sequence was then folded using ESMFold. The resulting structure closely matched the original protein structure, maintaining the same overall fold and secondary structure elements. This demonstrates that multiple sequences can encode the same protein structure and highlights the robustness of protein folds.
Part D. Group Brainstorm on Bacteriophage Engineering
- Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”). Write a 1-page proposal (bullet points or short paragraphs) describing: Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”). Why do you think those tools might help solve your chosen sub-problem? Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”). Include a schematic of your pipeline.This resource may be useful: HTGAA Protein Engineering Tools . Include your group’s short plan for engineering a bacteriophage.
Project Proposal: Precision Engineering of the MS2 L Protein
Selected Goals:
- Increased Stability (Targeting the transmembrane domain)
- Higher Toxicity (Targeting the DnaJ regulatory interaction)
1. Computational Tools & Approaches
- Protein Language Models (ESM-2): I will use ESM-2 to perform in silico deep mutational scanning of the 75-amino acid L protein. By calculating the pseudo-perplexity or log-likelihood of variants, I can identify mutations in the C-terminal hydrophobic region likely to increase structural stability without disrupting the membrane-anchored fold.
- AlphaFold-Multimer: I will model the interaction between the MS2 L protein and the host chaperone DnaJ. Specifically, I will look at the interface involving the DnaJ C-terminal domain (near residue P330). I will then design mutations in the basic N-terminal half of the L protein to computationally “break” this interaction.
- Structure-Based Truncation Design: Based on the knowledge that N-terminal truncations (like the $L^{\triangle dj}$ alleles) can accelerate lysis by up to 20 minutes, I will use structural modeling to define the minimal functional lytic peptide that retains the conserved LS dipeptide motif.
2. Rationale: Why These Tools?
- Stability: Small membrane proteins are notoriously difficult to stabilize experimentally. Using ESM-2 allows for a high-throughput search of the sequence space to find “evolutionarily plausible” mutations that reinforce the protein’s thermal and chemical stability.
- Toxicity: The MS2 L protein is naturally “retarded” by its binding to DnaJ. By using AlphaFold-Multimer to identify and then mutate the specific residues that facilitate this binding, I can create a “hyper-toxic” variant that escapes host regulation, leading to faster and more aggressive lysis.
3. Potential Pitfalls
- Unknown Lytic Mechanism: Unlike other amurins that inhibit peptidoglycan synthesis, the exact cellular target of MS2 L is still unknown. Engineering for toxicity is risky when the “kill target” is not yet identified in the literature.
- Modeling Membrane Proteins: AlphaFold-Multimer and ESM-2 can sometimes struggle with the highly flexible, basic N-terminal tails of small proteins, which may lead to inaccurate interface predictions for the DnaJ complex.
4. Pipeline Schematic
- Sequence Input: Wild-type MS2 L (75 aa).
- Variant Generation: ESM-2 scoring to identify stabilizing mutations in the C-terminus.
- Interaction Modeling: AlphaFold-Multimer to map the L-DnaJ interface.
- Interface Disruption: Targeted mutagenesis of the L protein N-terminus to prevent DnaJ binding.
- Final Selection: Identification of 2–3 “L-hyper” candidates for synthesis, characterized by increased stability scores and predicted loss of DnaJ affinity.