Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
- Design short peptides that bind mutant SOD1.
- Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
- PepMLM: target sequence-conditioned peptide generation via masked language modeling
- PeptiVerse: therapeutic property prediction
- moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
- To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Superoxide dismutase 1
UniProt ID: P00441
Original:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Position 4: Alanine (A) → Valine (V)
Mutant:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
In the PepMLM notebook:
[x] Paste the mutant SOD1 sequence
[x] Set peptide length = 12
[x] Generate 4 peptides.
[x] Add known binder FLYRWLPSRRGG
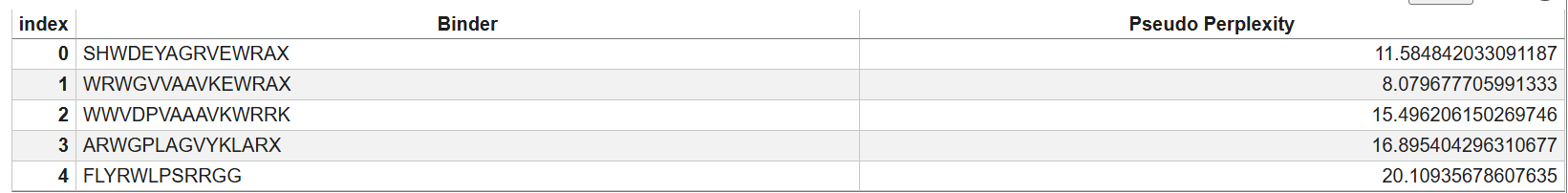
Lower perplexity = higher confidence binder.
Ranking:
1️⃣ WRWGVVAAVKEWRA → 8.08 (best)
2️⃣ SHWDEYAGRVEWRA → 11.58
3️⃣ WWVDPVAAAVKWRRK → 15.50
4️⃣ ARWGPLAGVYKLAR → 16.90
5️⃣ FLYRWLPSRRGG → 20.11 (known binder)
PepMLM assigned lower perplexity to several generated peptides than the known SOD1 binder FLYRWLPSRRGG, suggesting the model predicts these sequences may bind SOD1 with higher confidence.
Part 2: Evaluate Binders with AlphaFold3
- Navigate to the AlphaFold Server: alphafoldserver.com
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
| Peptide | ipTM | Interpretation |
|---|---|---|
| SHWDEYAGRVEWRA | 0.38 | weak interaction |
| WRWGVVAAVKEWRA | 0.25 | very weak |
| WWVDPVAAAVKWRRK | 0.39 | weak |
| ARWGPLAGVYKLAR | 0.42 | strongest among generated peptides |
| FLYRWLPSRRGG | 0.31 | weak |
AlphaFold3 predictions showed relatively low interface confidence scores for all tested peptides, with ipTM values ranging from 0.25 to 0.42. The peptide ARWGPLAGVYKLAR produced the highest ipTM score (0.42), suggesting a slightly stronger predicted interaction with mutant SOD1 compared to the other generated peptides. The known SOD1-binding peptide FLYRWLPSRRGG showed an ipTM score of 0.31, which is comparable to several PepMLM-generated peptides. Overall, the predictions suggest weak but possible surface interactions between the peptides and the protein. This indicates that some generated peptides may have comparable binding potential to the known binder, although the interaction confidence remains moderate.
Visualization of the AlphaFold3 predictions showed that several peptides did not appear to form stable contacts with the SOD1 surface. In many models, the peptide was positioned away from the protein, suggesting weak or uncertain binding. This observation is consistent with the low ipTM scores obtained for the predicted complexes. Small peptides are hard for AlphaFold to dock correctly, especially without experimental constraints. Therefore low ipTM values and weak interactions are expected.
The AlphaFold3 predictions produced relatively low ipTM scores for all peptide–SOD1 complexes, ranging from 0.25 to 0.42. Among the PepMLM-generated peptides, ARWGPLAGVYKLAR showed the highest ipTM value (0.42), suggesting a slightly stronger predicted interaction compared to the others. The known SOD1-binding peptide FLYRWLPSRRGG produced an ipTM score of 0.31. Interestingly, several PepMLM-generated peptides showed ipTM scores comparable to or higher than the known binder, indicating that the model may have generated sequences with similar or potentially improved binding potential to mutant SOD1.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes [x] Predicted binding affinity
[x] Solubility
[x] Hemolysis probability
[x] Net charge (pH 7)
[x] Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Outputs from Peptiverse:
| Peptide | Binding Affinity (pKd/pKi) | Solubility | Hemolysis Probability | Net Charge (pH 7) | Molecular Weight (Da) |
|---|---|---|---|---|---|
| SHWDEYAGRVEWRA | 5.86 (Weak) | Soluble | 0.035 | -1.45 | 1761.8 |
| WRWGVVAAVKEWRA | 6.92 (Weak) | Soluble | 0.167 | +1.76 | 1714.0 |
| WWVDPVAAAVKWRRK | 6.38 (Weak) | Soluble | 0.055 | +2.76 | 1868.2 |
| ARWGPLAGVYKLAR | 5.81 (Weak) | Soluble | 0.041 | +2.79 | 1557.8 |
| FLYRWLPSRRGG | 5.97 (Weak) | Soluble | 0.047 | +2.76 | 1507.7 |
Compared them with my AlphaFold ipTM results:
| Peptide | ipTM | Binding affinity (pKd/pKi) | Solubility | Hemolysis prob. | Net charge |
|---|---|---|---|---|---|
| SHWDEYAGRVEWRA | 0.38 | 5.86 | Soluble | 0.035 | -1.45 |
| WRWGVVAAVKEWRA | 0.25 | 6.92 (highest) | Soluble | 0.167 | +1.76 |
| WWVDPVAAAVKWRRK | 0.39 | 6.38 | Soluble | 0.055 | +2.76 |
| ARWGPLAGVYKLAR | 0.42 (highest) | 5.81 | Soluble | 0.041 | +2.79 |
| FLYRWLPSRRGG | 0.31 | 5.97 | Soluble | 0.047 | +2.76 |
Important observations:
- All peptides are predicted soluble → good for therapeutics.
- All are non-hemolytic (very low probability).
- Predicted binding affinities are weak but similar.
- The peptide with highest structural confidence (ARWGPLAGVYKLAR) does not have the strongest predicted affinity.
- WRWGVVAAVKEWRA has the strongest predicted affinity but very low ipTM, meaning structure prediction did not support strong binding.
Peptide to advance
Selected peptide: ARWGPLAGVYKLAR
ARWGPLAGVYKLAR was selected as the most promising candidate because it showed the highest ipTM score in the AlphaFold3 structural predictions, suggesting relatively stronger interaction with mutant SOD1. Additionally, PeptiVerse predicted good solubility, low hemolysis probability, and a reasonable net positive charge, which are favorable properties for therapeutic peptides. Therefore, this peptide provides the best balance between predicted binding potential and therapeutic safety.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
moPPIt Generated Peptides:
| Index | Peptide | Hemolysis | Solubility | Predicted Affinity | Motif Score |
|---|---|---|---|---|---|
| 0 | KECYDKTNDFNW | 0.887 | 0.83 | 6.11 | 0.56 |
| 1 | GQLKCYNKGTCR | 0.944 | 0.92 | 6.28 | 0.83 |
| 2 | SDKFRTCVQKRV | 0.936 | 0.75 | 7.61 | 0.90 |
- All peptides show good solubility scores (0.75–0.92).
- Predicted binding affinity values are comparable or slightly stronger than PepMLM peptides.
- SDKFRTCVQKRV has the highest predicted affinity and motif score, suggesting stronger targeted interaction with the binding site on Superoxide dismutase 1.
Peptides generated using moPPIt differ from those produced by PepMLM because the generation process is guided by specific design objectives. While PepMLM samples peptide sequences conditioned only on the target protein sequence, moPPIt allows the design process to be directed toward specific residues on the target protein and simultaneously optimizes multiple properties such as binding affinity, motif targeting, solubility, and hemolysis risk. As a result, the moPPIt-generated peptides display stronger motif scores and slightly improved predicted affinities compared to the earlier sampled peptides, suggesting more targeted binding to mutant Superoxide dismutase 1.
Before advancing these peptides toward clinical development, further computational and experimental validation would be required. Computationally, structural modeling using AlphaFold or molecular docking could be performed to confirm peptide binding to mutant SOD1. Molecular dynamics simulations could assess the stability of the peptide–protein complex. Experimentally, peptide binding could be validated using biochemical techniques such as surface plasmon resonance or isothermal titration calorimetry. Additionally, cellular assays would be required to evaluate toxicity, stability, and the ability of the peptides to inhibit SOD1 aggregation before progressing to in vivo studies.
Part C: Final Project: L-Protein Mutants
Lysis Protein Sequence (UniProtKB ID: https://www.uniprot.org/uniprotkb/P03609/entry)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
DnaJ sequence (UniProtKB ID: https://www.uniprot.org/uniprotkb/P03609/entry)
MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
Option 1: 5 Rational Mutants
| # | Exact Mutations (PDF format) | AA Positions | Region | Evidence |
|---|---|---|---|---|
| 1 | 38 C-T 13 P-L 1 + 43 T-G 15 S-A 1 | 13(P→L), 15(S→A) | Soluble | Lysis=1, Protein=1 |
| 2 | 52 A-G 18 R-G 1 + 55 C-A 19 R-S 1 | 18(R→G), 19(R→S) | Soluble | Lysis=1 |
| 3 | 131 T-C 44 L-P 1 + 133 G-C 45 A-P 1 | 44(L→P), 45(A→P) | TM | Lysis=1, Protein=1 |
| 4 | 136 A-T 46 I-F 1 | 46(I→F) | TM | Lysis=1, Protein=1 |
| 5 | 38 C-T 13 P-L 1 + 131 T-C 44 L-P 1 | 13(P→L), 44(L→P) | Combo | Best soluble+TM |
Option 2: DnaJ Interface
Triple mutant: P13L + S15A + R18G
Evidence: 38 C-T 13 P-L 1, 43 T-G 15 S-A 1, 52 A-G 18 R-G 1
Option 3: Random Mutagenesis
Output:
Mutant1: P->L(pos13)+R->W(pos20)
Mutant2: R->S(pos19)+R->G(pos18)
Mutant3: I->F(pos46)+L->P(pos44)
Mutant4: A->P(pos45)+E->V(pos25)
Mutant5: P->L(pos13)+S->A(pos15)
Good mutant = Lysis=1 mutations only, ≥2 changes, soluble+TM balance.