Week 6 HW: Genetic Circuits Part I: Assembly Technologies
DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity (HF) PCR Master Mix is a ready-to-use 2× mix designed for accurate and efficient DNA amplification. Typical components include:
- Phusion DNA polymerase: A thermostable, proofreading enzyme with 3′→5′ exonuclease activity for extremely high fidelity.
- dNTPs (deoxynucleotide triphosphates): Building blocks for new DNA synthesis.
- Optimized reaction buffer: Provides Mg²⁺ and pH stabilization for optimal enzyme activity.
- Stabilizers and enhancers: Improve yield, especially for GC-rich or complex templates.
2. What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature ((T_a)) depends on several molecular properties:
- Primer length: Longer primers have higher melting temperatures ((T_m)).
- GC content: G–C pairs contribute more hydrogen bonds (3 vs. 2 for A–T), increasing (T_m).
- Salt concentration: Stabilizes DNA duplexes, raising (T_m).
- Mismatches: Intentional (e.g., for mutagenesis) or unintentional mismatches lower (T_m).
- Primer concentration and secondary structure: Hairpins or dimers reduce effective binding.
Rule of thumb: (T_a = T_m – 2\text{–}5°C), and the two primers’ (T_m) should be within 5°C of each other.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
| Aspect | PCR | Restriction Enzyme Digest |
|---|---|---|
| Mechanism | Primers + polymerase amplify specific DNA regions | Enzymes cut at specific recognition sequences |
| Protocol | Template + primers + dNTPs + polymerase → thermocycling | DNA + enzyme + buffer → incubation (37°C) |
| Ends produced | Customizable (blunt/sticky via polymerase or overhangs) | Defined sticky/blunt ends based on enzyme |
| Customization | Can introduce mutations/overlaps via primers | Limited to existing restriction sites |
| Yield | Exponential amplification | Linear cutting (yield depends on starting material) |
| Purity | Requires cleanup (DpnI, purification) | Requires cleanup (phenol/chloroform, columns) |
When to use PCR: No restriction sites available, need mutations (amilCP color variants), or custom overlaps for Gibson.
When to use restriction digest: Routine subcloning with existing sites, rapid verification.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To make our digested or PCR-generated DNA compatible with Gibson Assembly:
- Design primers so that each fragment has 20–40 bp of overlapping sequence identity.
- Check orientation and reading frame in software (like Benchling) to ensure correct assembly.
- Purify fragments to remove enzymes, primers, and salts that inhibit the Gibson reaction.
- DpnI-treat PCR products to eliminate methylated template plasmid DNA.
- Verify concentration and quality (≥30 ng/µL) via spectrophotometry (Nanodrop/Qubit) before assembly.
- Use 2:1 insert:vector molar ratio (calculate via NEB calculator).
5. How does the plasmid DNA enter the E. coli cells during transformation?
During transformation, chemically competent E. coli cells are made permeable:
- Heat shock (ice → 42°C for 45s → ice) temporarily opens pores in the cell membrane.
- Plasmid DNA diffuses through these transient pores into the cytoplasm.
- Cells recover in SOC medium (nutrient-rich) at 37°C for 60 minutes, repairing membranes.
- Antibiotic selection ensures only transformed cells (with plasmid resistance gene) survive on chloramphenicol plates.
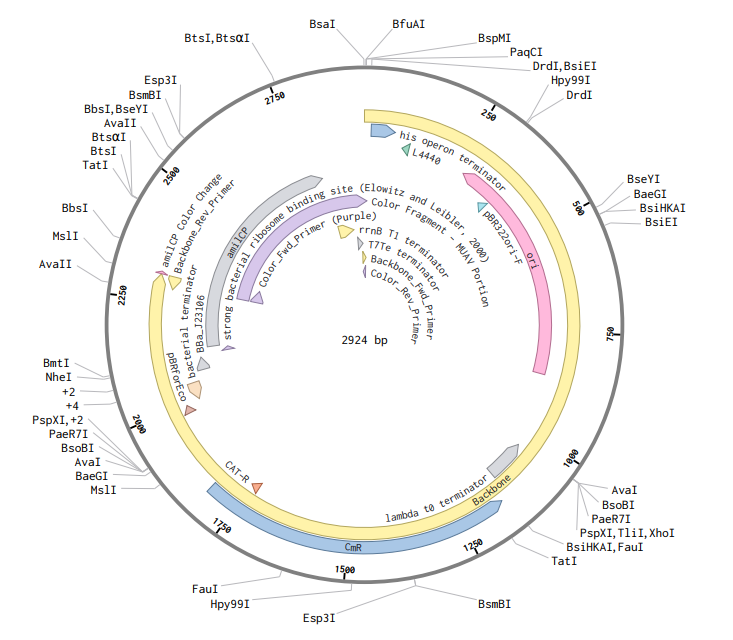
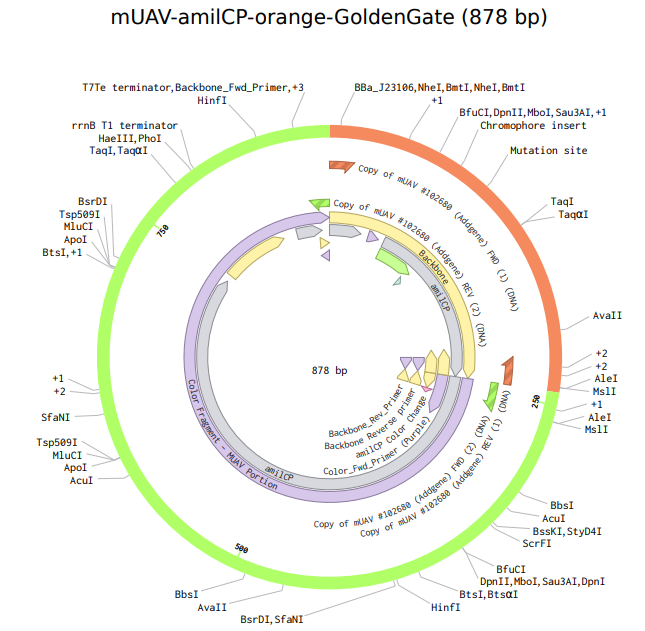
6. Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly (GGA) is a one-pot, scarless, multi-fragment cloning method using Type IIS restriction enzymes.
Key Components:
- Type IIS enzymes (BsaI, BpiI): Cut outside recognition sites, creating custom 4-bp overhangs.
- T4 DNA ligase: Joins compatible overhangs.
- Modular parts: Promoter, RBS, CDS, terminator with unique junction sequences.
Mechanism (5-step cycle):
Advantages over Gibson:
- Multi-fragment (up to 10+ parts simultaneously)
- No PCR needed for internal fragments
- Hierarchical (assemble subparts → final construct)
- Standardized (MoClo system)
Example: Promoter-RBS-amilCP-Terminator → single reaction yields complete expression cassette.
7. Explain the other method in 5-7 sentences plus diagrams (either handmade or online).
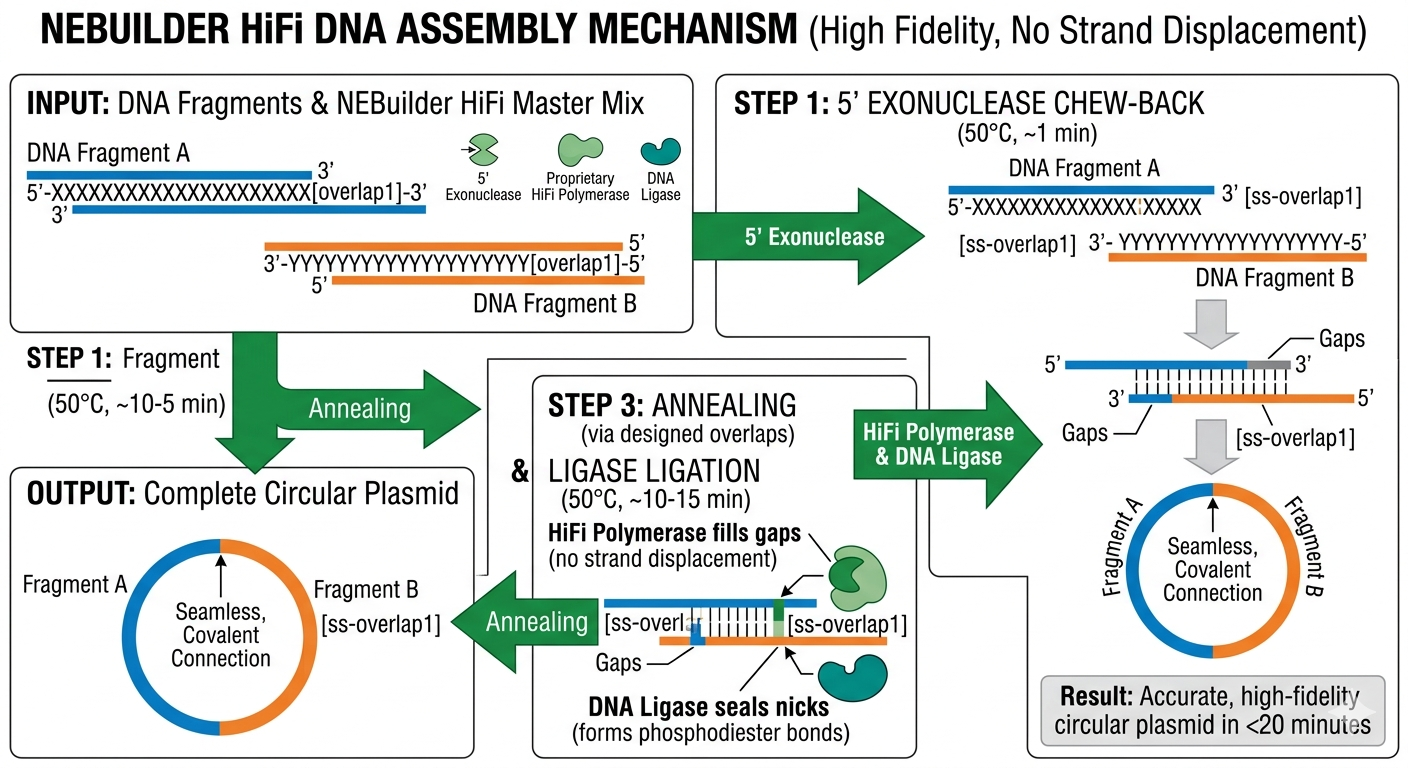
NEBuilder HiFi DNA Assembly (alternative to Gibson) uses a similar chew-back → anneal → fill-in → ligate mechanism but with a proprietary 5′ exonuclease-free HiFi polymerase for higher accuracy.
- Exonuclease chews back 5′ ends of overlapping fragments (~15-20 bp).
- Complementary single-stranded overhangs anneal via designed overlaps.
- HiFi polymerase fills in gaps without strand displacement.
- Ligase seals nicks, forming circular plasmids.
- Single 50°C, 15-min reaction (faster than Gibson’s 60 min).
- Works with 2-6 fragments, ideal for 1-2 inserts like this lab.
8. Model this assembly method with Benchling or Asimov Kernel!