https://www.gla.ac.uk/colleges/mvls/graduateschool/clinicalacademictraining/
1.Describe a biological engineering application or tool you want to develop and why.
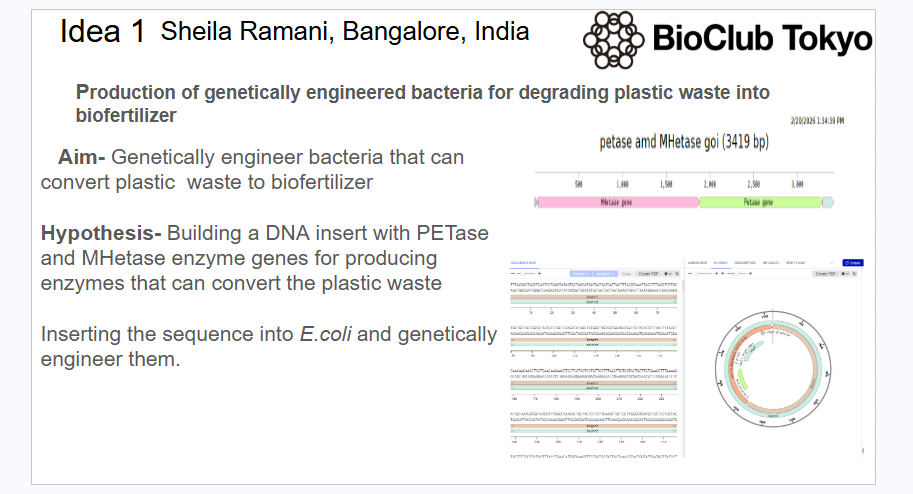
I want to engineer a bacteria to produce enzymes to convert plastic wastes to a fertilizer. Microbes secrete extracellular enzymes—such as PETase, MHETase, cutinases, lipases, and esterases—to hydrolyze (break) the chemical bonds of plastics, releasing monomers (e.g., ethylene glycol, terephthalic acid) and oligomers.
I want to create a bacteria that can function by engineering it to produce enzymes for break down of plastics and convert to useful biofertilizers for plants. I want to develop such an organism because plastic seems to pose serious threats as being non biodegradable for ages.
https://www.synbiobeta.com/read/opentrons-new-interface-for-automated-pipetting-robot-streamlines-workflow-programming
Create a Python file to run on an Opentrons liquid handling robot.
1.Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
2.Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons. You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept. If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
https://bio.libretexts.org/Workbench/Bio_11A_-_Introduction_to_Biology_I
Homework: Protein Design I Part A. Conceptual Questions 1.Why are there only 20 natural amino acids?
The 20 natural amino acids evolved as optimal sets very early, during the RNA world (4 billion years ago). The format was not changed and became frozen because it would disrupt all proteins and also due to tRNA recognition limitations further expansion was prohibited.
https://www.scinexx.de/news/medizin/designer-zellen-gegen-schuppenflechte/
Assignment: DNA Assembly Answer these questions about the protocol
1.What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The components of Phusion High-Fidelity PCR Master Mix contains Phusion DNA Polymerase (high-accuracy, 50x higher than Taq), dNTPs (nucleotide building blocks), and optimized reaction buffer with MgCl2(essential for enzyme activity).
https://bigthink.com/neuropsych/human-neuron-signals/https://www.designboom.com/art/jill-bliss-mushroom-nature-medleys-08-12-2017/
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1.What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs allow biological systems to act as analog processors, mimicking neural network learning and offering higher versatility, precision, and robustness in synthetic biology applications. Context-dependent responses rather than simple “on/off” outputs. Mimic artificial neural networks using transcriptional regulators, allow for complex, Nonlinear processing of multiple inputs, Higher fault tolerance, and the ability to perform regression analysis, which is not possible with traditional digital genetic circuits.
https://www.tudelft.nl/en/2024/tu-delft/creating-life-from-lifeless-biomolecules-with-ai-and-lab-evolution
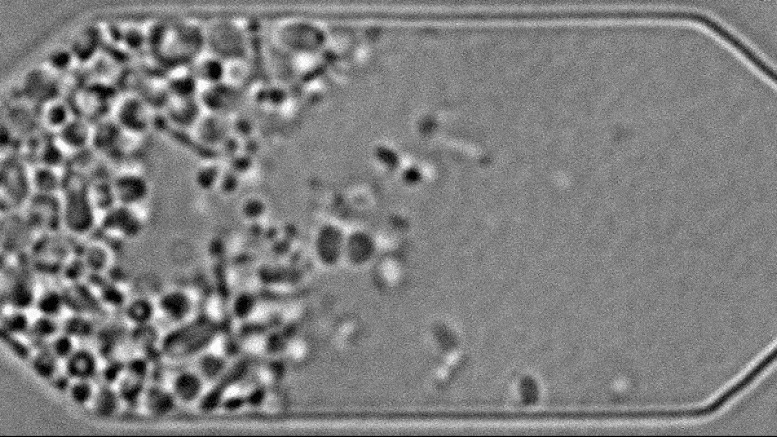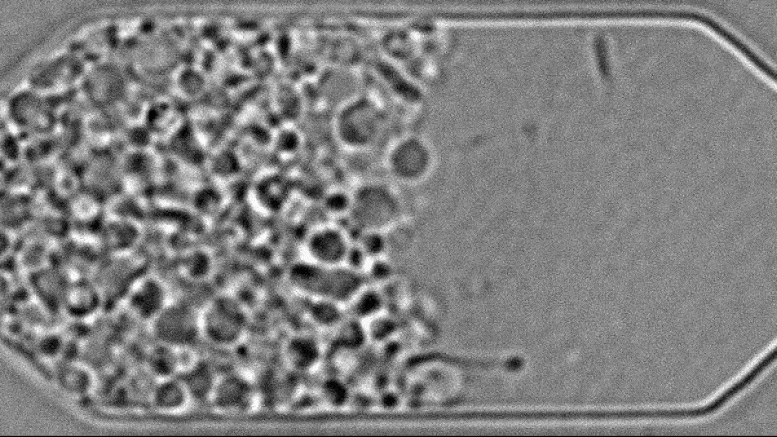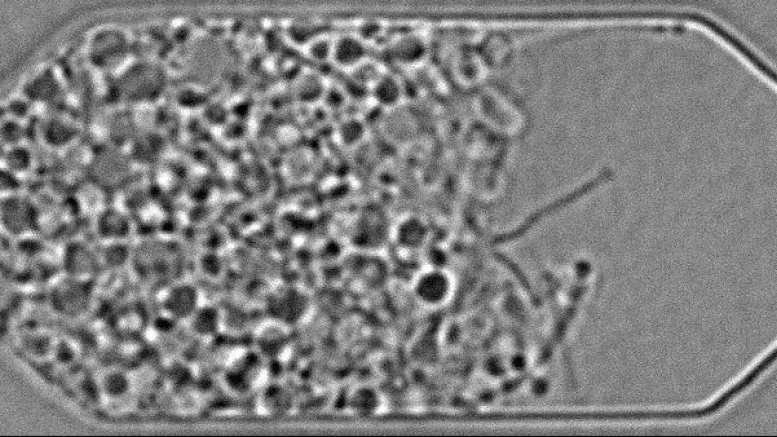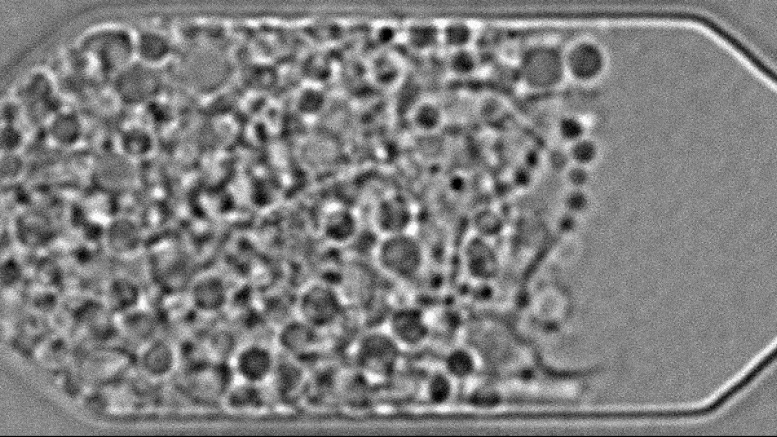
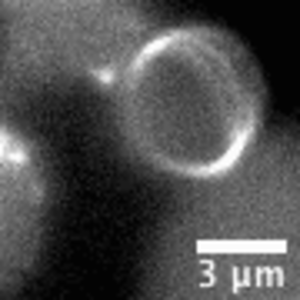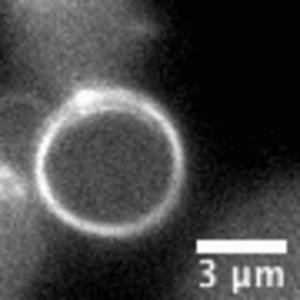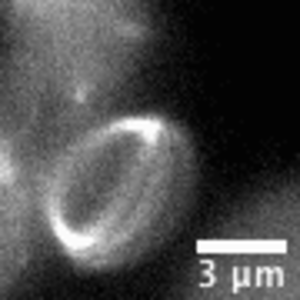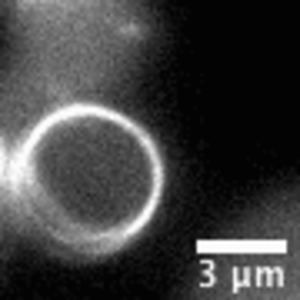
“Living material assembly of bacteriogenic protocells” by Can Xu, Nicolas Martin, Mei Li, and Stephen Mann, 14 September 2022, Nature. DOI: 10.1038/s41586-022-05223-w
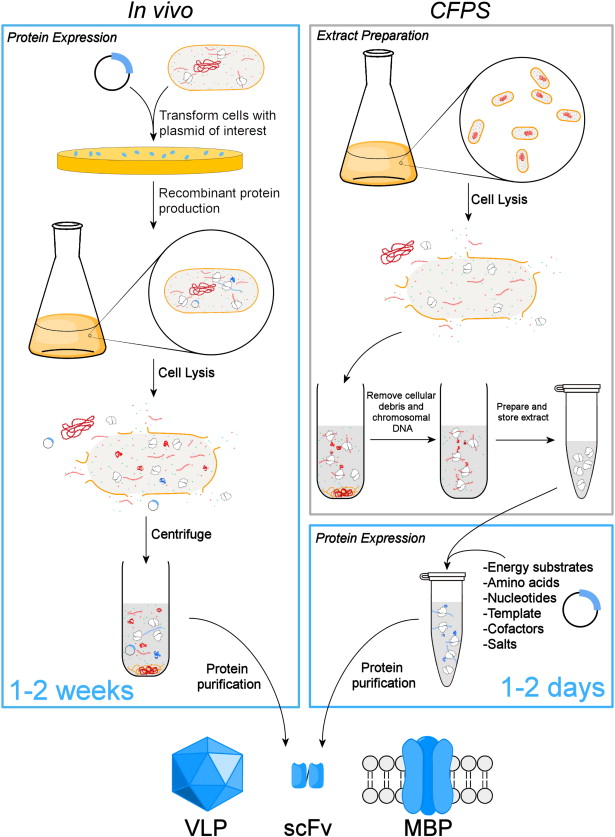
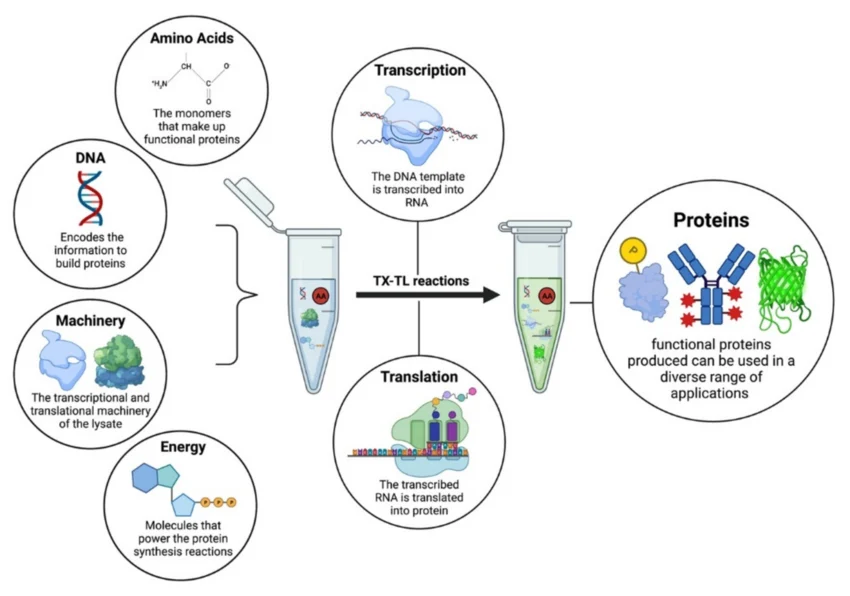
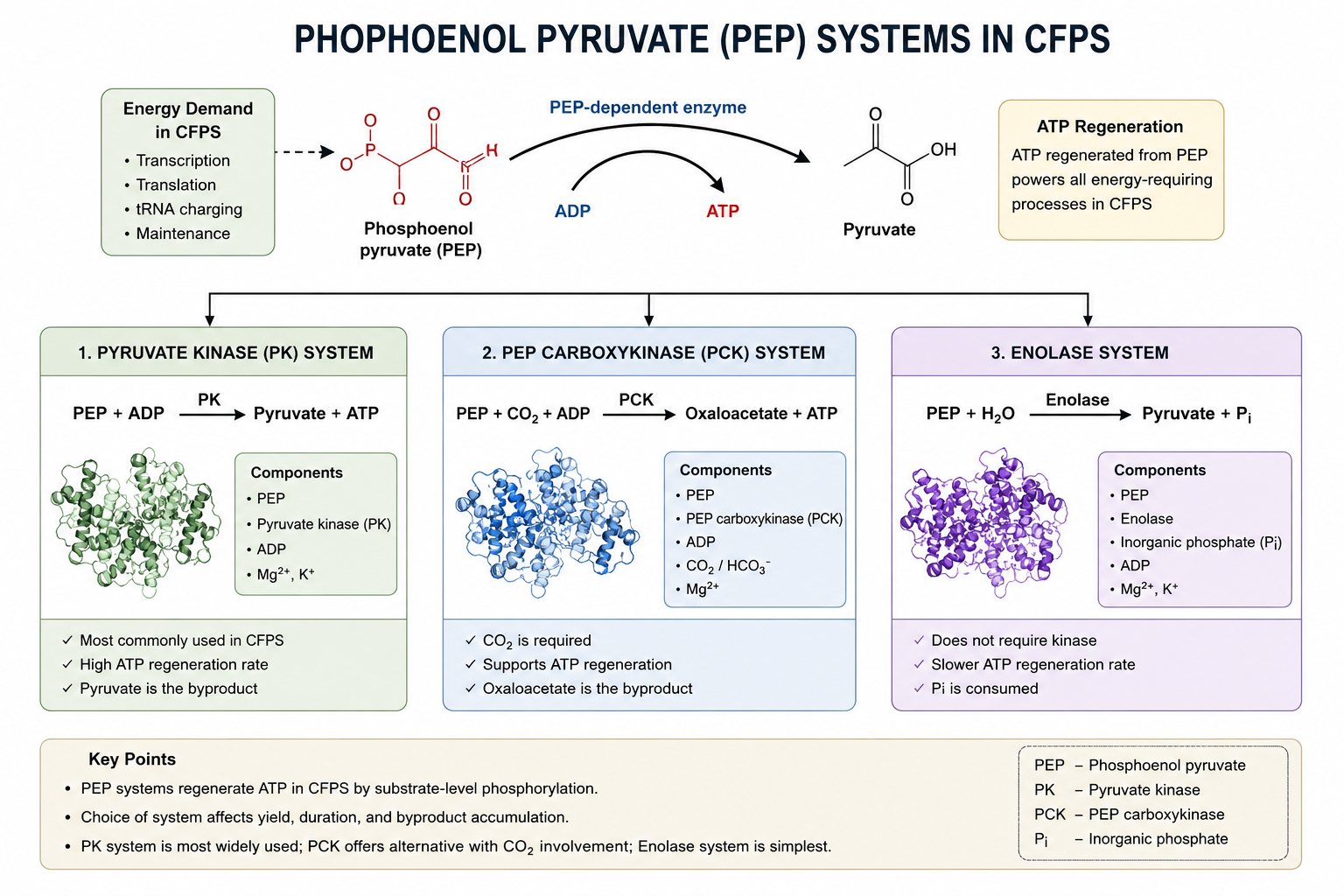
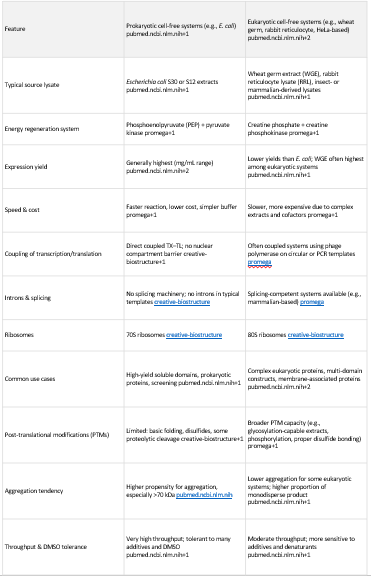
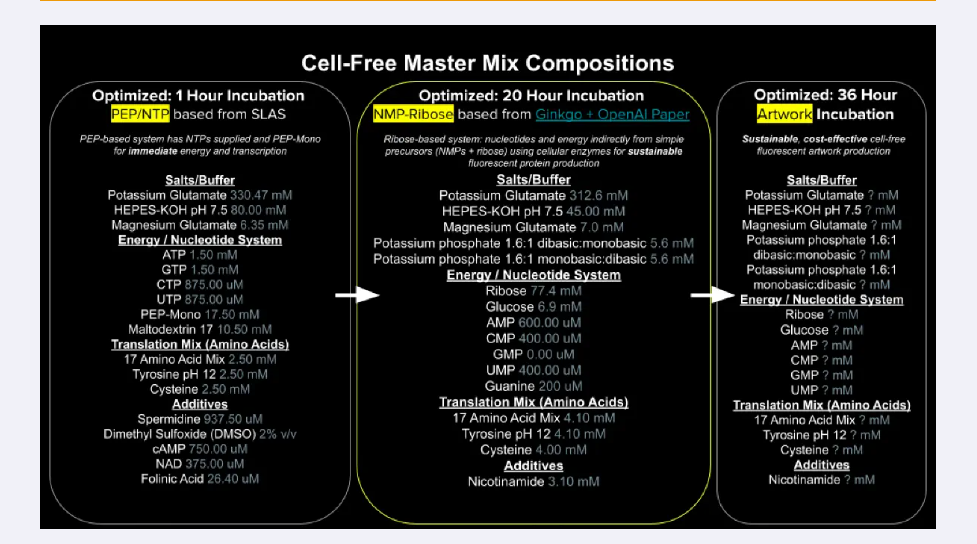
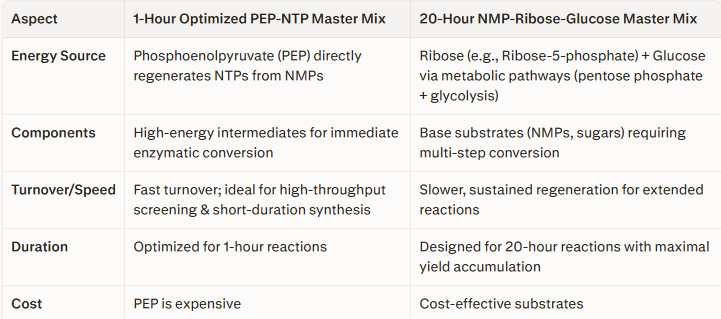
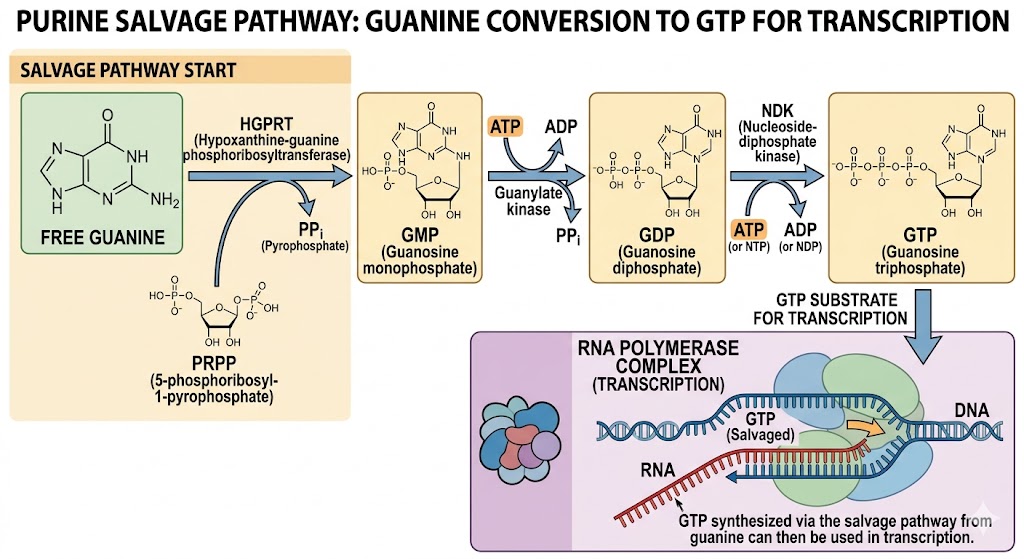
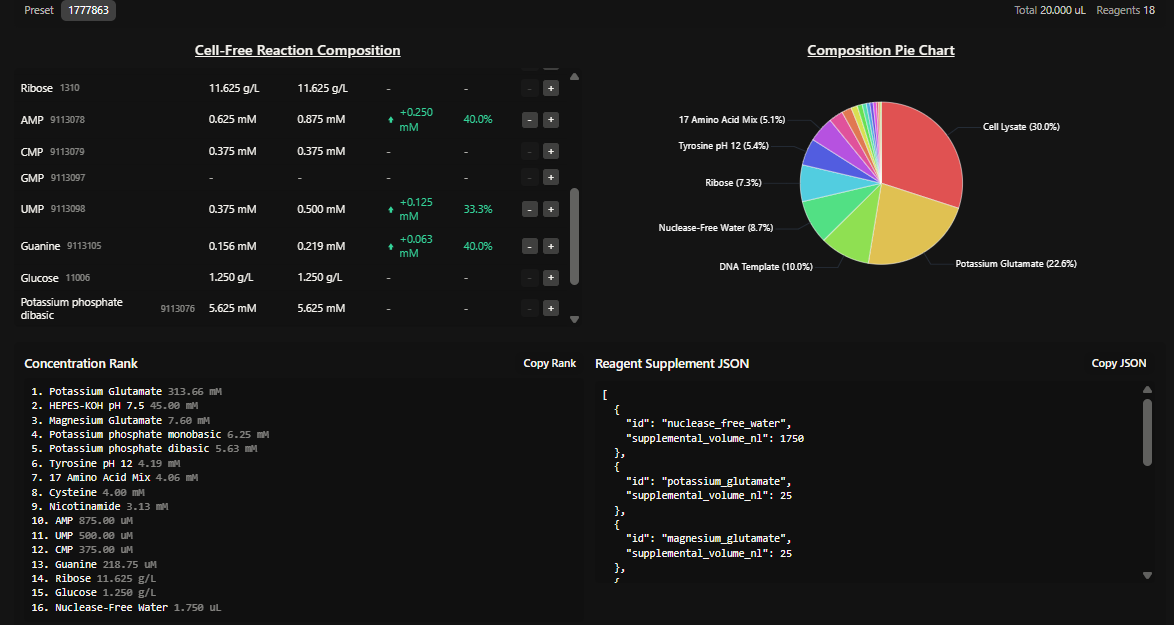
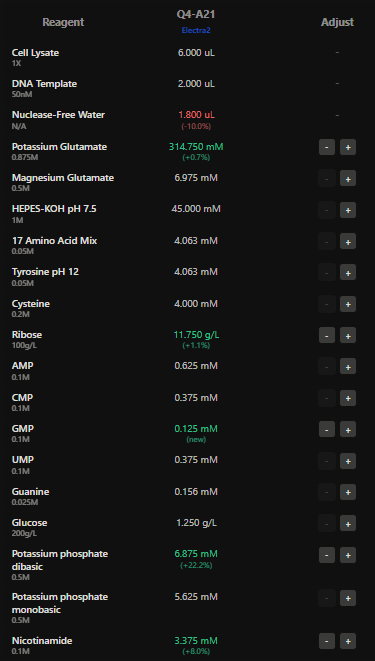
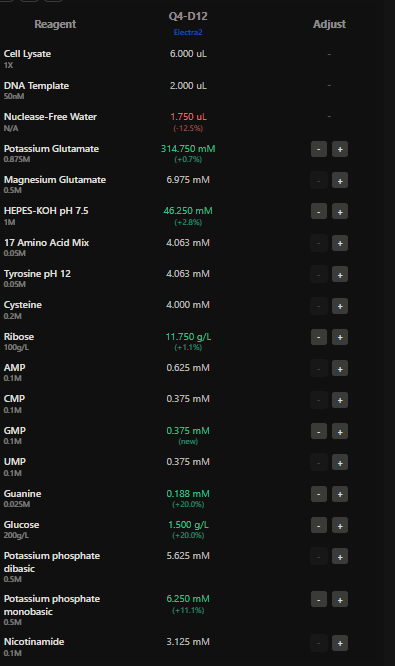
Homework Part A: General and Lecturer-Specific Questions General homework questions 1.Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
https://www.cygnustechnologies.com/hcp-identification-quantification
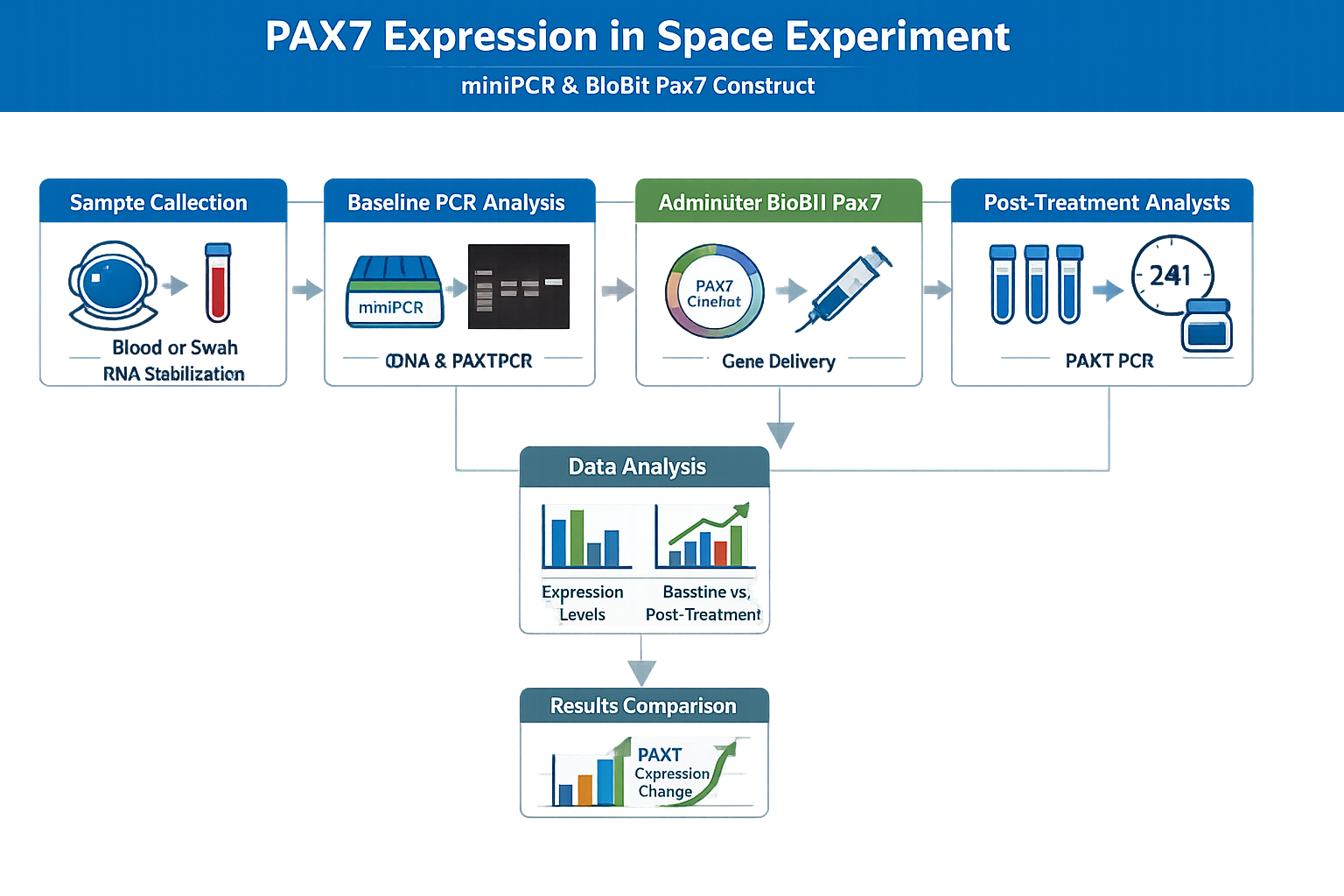
Homework: Final Project For your final project:
1.Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
In the final project the presence of specific mec A mRNA is detected using aptamer fluorescence.The fluorescence intensity is directly proportional to the amount of the mRNA present at the site.
1.Describe a biological engineering application or tool you want to develop and why.
I want to engineer a bacteria to produce enzymes to convert plastic wastes to a fertilizer. Microbes secrete extracellular enzymes—such as PETase, MHETase, cutinases, lipases, and esterases—to hydrolyze (break) the chemical bonds of plastics, releasing monomers (e.g., ethylene glycol, terephthalic acid) and oligomers.
I want to create a bacteria that can function by engineering it to produce enzymes for break down of plastics and convert to useful biofertilizers for plants. I want to develop such an organism because plastic seems to pose serious threats as being non biodegradable for ages.
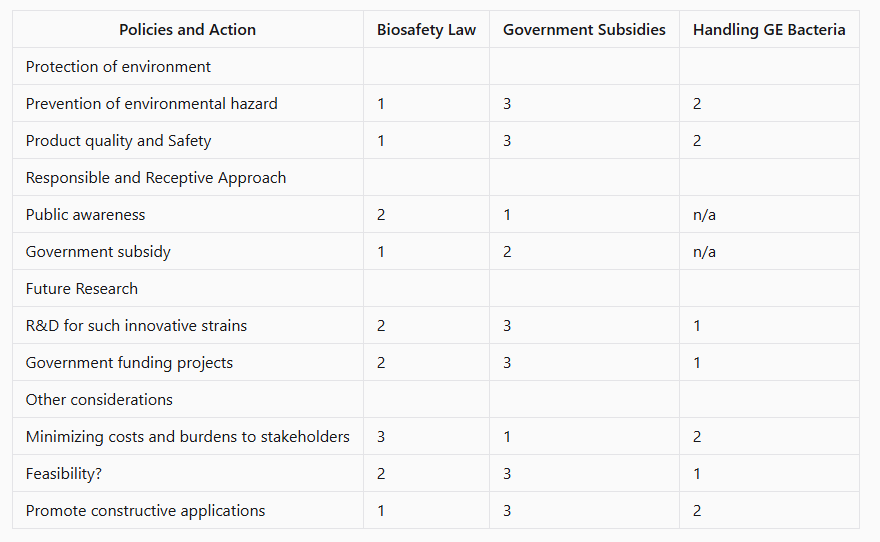
2.Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
The main goal to make this enzyme produced by engineered bacteria an ethical and safely managed product with minimal risks to environment,responsible use of it and a proper containment of the engineered organism.
This main goal can be broken down to sub goals:
Protect the environment:
As the product is being created to reduce environmental pollution of plastics , release of these organisms in the soil should not harm the natural ecosysytem of soil bacteria - prevent horizontal transfer of genes.
Auxotrophs of the engineered bacteria dependent on unnatural amino acids should be created so that in their absence the bacteria die.
Farmers should be well informed about the fertilizer created by the bacteria, concentration to be used to prevent any harm to natural ph of the soil while maintaining its fertility.
Responsible and Receptive Approach
Companies or industries producing the product should meake the information about the product available to the public
Awareness about the product to the end user (farmer)and government should provide susbsidy to promote the product.
Future Research
Encourage researchers in the area to innovate better strains to produce an optimised enzyme for production of the fertilizer from such waste.
Government funding to such projects which aims at decreasing the environmental pollution and giving back useful products to nature.
3.Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
A new law on the manufacturing of “Fertilizers from plastic waste " should be created
Purpose:
The main aim of this law would be to involve a regulatory body to check the safety standards of the fertilizers before application to soil- check the amount of contaminants present in the end product, concentration application limits of the fertilizer, safety levels for the end user.
Design:
The regulatory body will decide on the threshold levels of the fertilizer used, also the soil ecosystem on which it is applied should not be affected. The government agricultural department should ensure the end user be fully informed about the product. The product should have a quality check inspection before release into the market.
Assumptions:
The law is accepted at the same level in all countries.The run off from these fertilizers are assumed to be low in heavy metals.
Risks of Failure and Success:
Risks of failures: The end users may not be receptive to the idea of using such fertilizers produced out of waste.
Risks of success: The law decreases the burden of plastic waste in landfills and provide a sustainable alternative to chemical fertilizers.
Government subsidies to farmers using the fertilizer and also incentives to companies
Purpose:
These strategies will promote the product and also make people aware and be receptive to such sustainable approaches.
Design:
The government can conduct awareness camps and demonstrate its application. Tax benefits can be provided to companies that sell these products.
Assumptions:
While initial setup requires high capital investment, the government assumes long-term savings in waste management and lower fertilizer costs for farmers.
Risks of Failure & Success:
Risks of Failures: Subsidies may be subject to change, and if environmental regulations on microplastics in fertilizer tighten, current products might become non-compliant.
Risk of Success: Many startups might adopt these fertilizers quickly without thinking for the benefit of obtaining incentives, without thinking about the long term impacts.
Handling of the engineered bacteria
Purpose:
Proper protocol should be followed to handle these “superbugs”(genetically engineered) to prevent them from mixing with natural biota in the ecosystem.This can lead to creating a pathogen by horizontal gene transfer.
Design:
The organisms created should be created as auxotrophs so that in the absence of the desired nutrient, kills the microbe. Incorporate genes in bacteria that kill it in specific environmental conditions- create a suicidal circuit.
Assumption:
It is assumed that the genetic modification does not significantly impair the growth rate or metabolic function of the bacteria, allowing for sufficient yield.
Risks of Failure & Success:
Risks of Failures: The engineered genes may be lost or mutated over successive generations, causing the bacteria to lose their intended function or gain unintended traits.
Risk of Success:The survival and effectiveness of Genetically Engineered (GE)Bacteria in the field depend on factors like temperature, pH, and nutrient availability.
3.Score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Courtesy: perplexity
3.Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
I think a combination of biosafety law of fertilizers made from plastic waste and governemnt subsidies I would prioritize because that would help in the promotion of the new innovative product as well as use it efficiently. The awareness about the product would also make the public receptive towards new technology and adopt sustainable practices.
Here many assumptions are made about the engineered bacteria beinga safe organism which will not be a hazard to the ecosystem and would prevent nutrient leaching and will not undergo mutation in the process to become pathogenic.These uncertainties can be mitigated by some of the methods mentioned above.The handling of the GE bacteria becomes important when you consider the soil ecosystem with its natural organisms. There are also uncertainties regarding process of conversion of fertilizer from plastic waste.
Professor Jacobson
1.Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of DNA polymerase typically ranges from 10-4 to 10-6 errors per base pair during initial nucleotide insertion. While proofreading the accuracy improves to 10-7 to 10-8 errors per base pair. The human haploid genome is 3.5 billion base pairs long so roughly 6.3 billion pairs long, so the DNA polymerase has an estimated error rate of 1 error per 109to 1010 nucleotides. Biology has a very efficient way of solving this discrepancy by using proofreading method in (3’- 5’ Exonuclease Activity) and mismatch repair mechanisms.
2.How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The standard genetic code uses 64 codons for 20 amino acids and 3 stop signals.
Calculation for Average Protein For a 375-amino-acid protein, the total number of coding DNA sequences is the product of codon choices per position: 1×1×29×31×45×63 =3.2 × 10195 So roughly 3374 =10^179.
In reality the codon usage varies from species to species meaning the organism prefers a certain codon over the otehr to produce the same amino acid which is used in protein engineering as codon optimization.
Synonymous codon variants often fail to produce functional, equivalent proteins in practice due to cellular biases and kinetic effects during gene expression.
Codon usage bias matches tRNA availability in human cells, so rare codons slow ribosome speed, reducing protein yield. Optimal codons boost expression up to 15-fold, while mismatched ones drop levels dramatically. Codon swaps can change protein structure, solubility, or stability, even if the amino acid sequence stays identical.
Dr.LeProust:
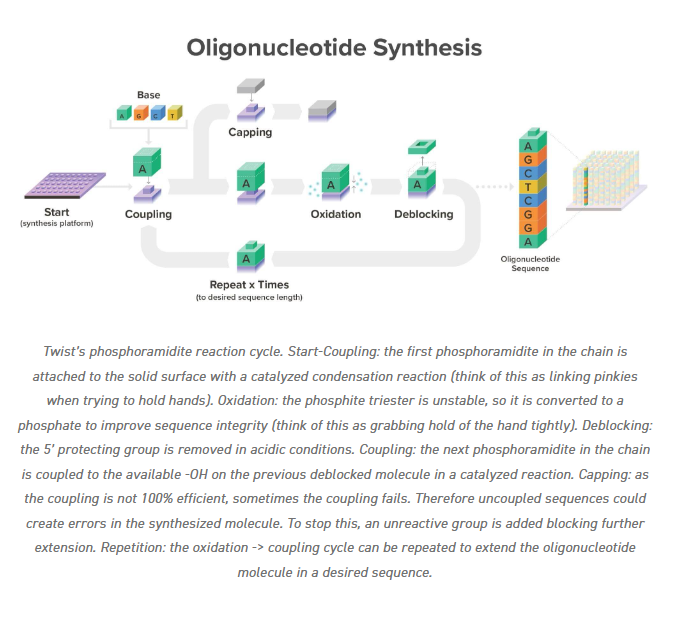
1.What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite solid-phase synthesis is the most commonly used method for oligonucleotide (oligo) synthesis today. This technique builds oligos stepwise on a solid support like controlled pore glass (CPG), adding protected nucleoside phosphoramidite monomers one at a time. Key steps include detritylation (removing the 5’-protecting group), coupling (adding the next nucleotide), oxidation (stabilizing the phosphite linkage), and capping (blocking failed sequences).
Courtesy: twistbioscience.com
2.Why is it difficult to make oligos longer than 200nt via direct synthesis?
Each nucleotide addition has a coupling yield of about 99%, but errors compound exponentially; a 200-mer requires roughly 200 cycles, dropping full-length product yield below practical levels (e.g., ~36% theoretical at 99% efficiency, far lower in practice). Longer sequences amplify deletions, truncations, and depurination from repetitive harsh cycles (oxidation, capping, deprotection)
Failure sequences (n-1 mers, mutations) dominate output, and no standard method like HPLC or gel electrophoresis resolves the tiny full-length fraction from closely related byproducts.
Longer strands form secondary structures that sterically hinder reagent diffusion and coupling, especially on porous solid supports like CPG, where diffusion slows dramatically.
3.Why can’t you make a 2000bp gene via direct oligo synthesis?
Deletions, insertions, and substitutions build up rapidly beyond 100-200 nt, as there’s no proofreading like in enzymatic replication. Long sequences form stable hairpins or folds that sterically hinder reagent access and coupling.
Professor Church
1.What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 amino acids often referenced for many animals (e.g., swine, dogs, rats) are those animals cannot synthesize sufficiently: lysine, methionine, tryptophan, threonine, valine, isoleucine, leucine, arginine, histidine, and phenylalanine.
The lysine contingency refers to a fictional genetic failsafe from the Jurassic Park franchise. In Jurassic Park, geneticist Henry Wu engineered dinosaurs unable to synthesize the essential amino acid lysine, making them dependent on external supplements provided by park staff. Without lysine, the dinosaurs would enter a coma and die, preventing their survival if they escaped Isla Nublar and disrupted ecosystems. Lysine is abundant in nature—found in plants like soy, bacteria, and prey animals—allowing dinosaurs (or any organism) to obtain it through diet. Humans and animals can’t synthesize lysine either but thrive without supplements by eating lysine-rich foods, undermining the contingency’s viability. Lysine is an essential amino acid critical for protein synthesis, collagen formation, and carnitine production, with deficiencies linked to anemia or impaired metabolism.
Information coutesy: perplexity lysine as essential amino acid and lysine contingency
Courtesy:NCBI - O’Leary NA, Cox E, Holmes JB, Anderson WR, Falk R, Hem V, Tsuchiya MTN, Schuler GD, Zhang X, Torcivia J, Ketter A, Breen L, Cothran J, Bajwa H, Tinne J, Meric PA, Hlavina W, Schneider VA. Exploring and retrieving sequence and metadata for species across the tree of life with NCBI Datasets. Sci Data. 2024 Jul 5;11(1):732. doi: 10.1038/s41597-024-03571-y. PMID: 38969627; PMCID: PMC11226681.
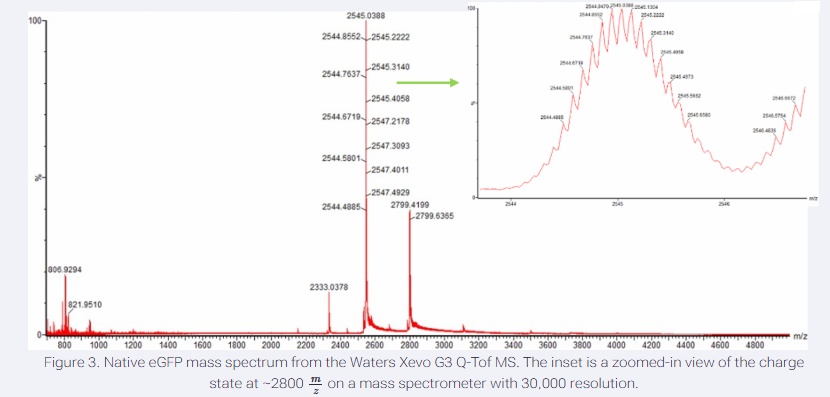
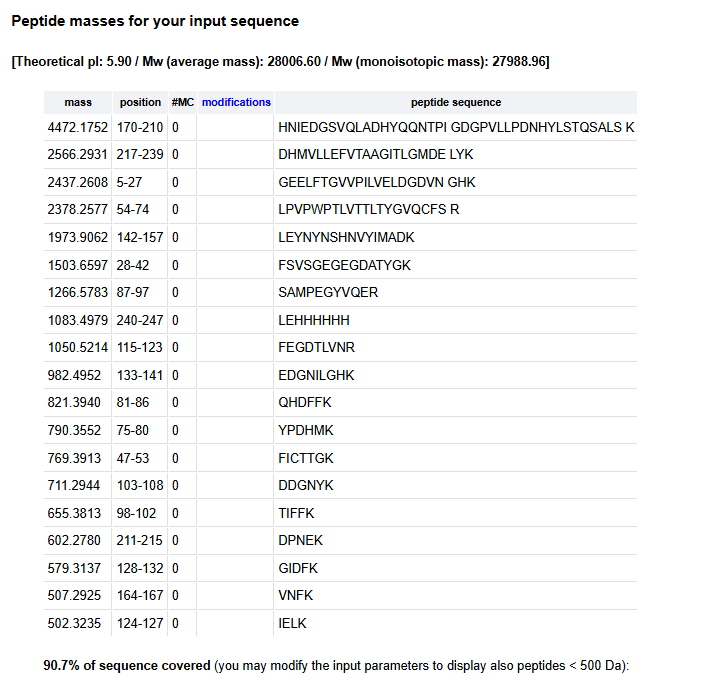
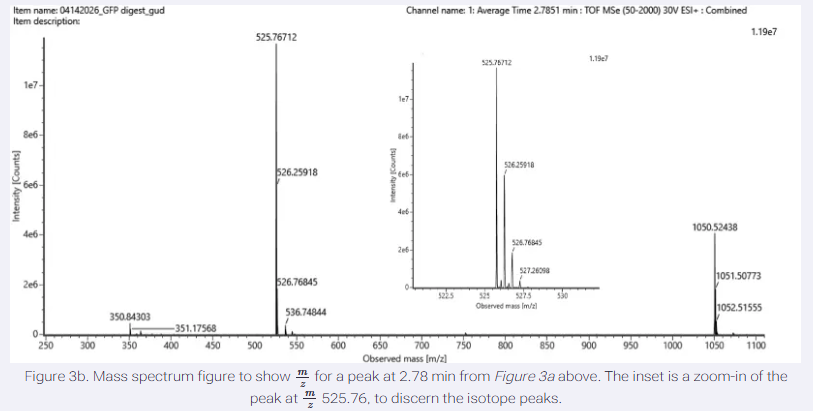
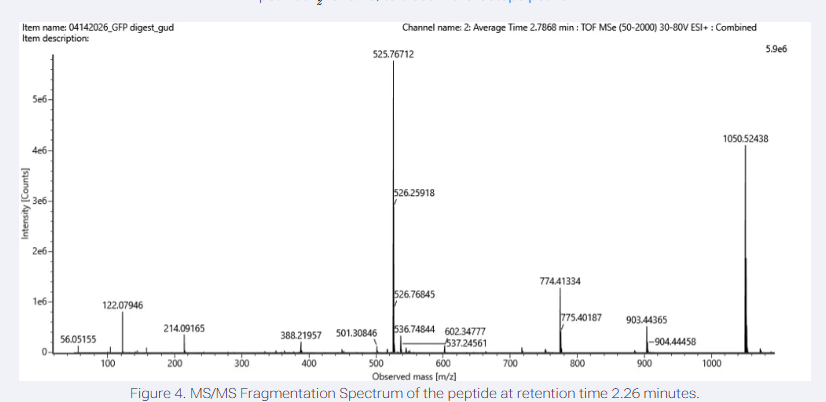
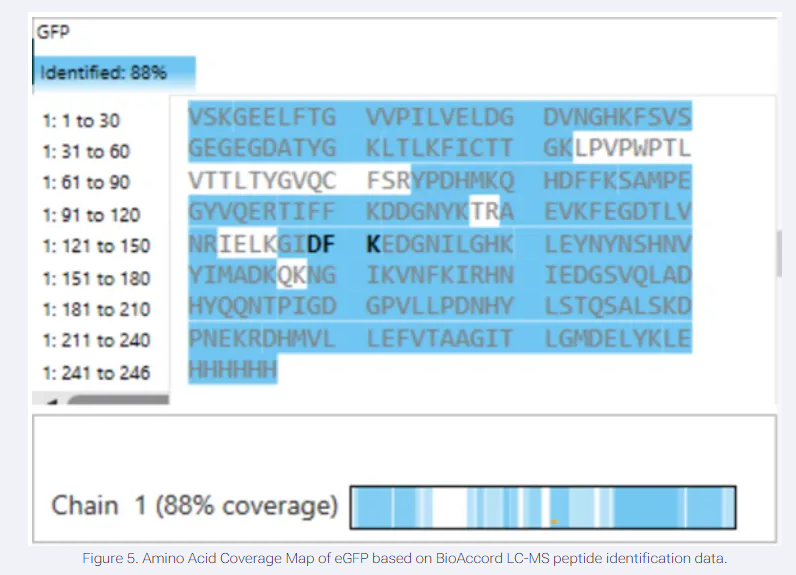
I have chosen wild type green fluorescent protein. This protein is used as a reporter gene in plasmids to study expression of genes as well as in biosensing, protein localization and also in live cell imaging.
I would like to research on GFP variants and understand for better use in biosensor field.
Sequence
Green fluorescent protein
Gene
GFP
Status
UniProtKB reviewed (Swiss-Prot)
Organism
Aequorea victoria (Water jellyfish) (Mesonema victoria)
FASTA sequence
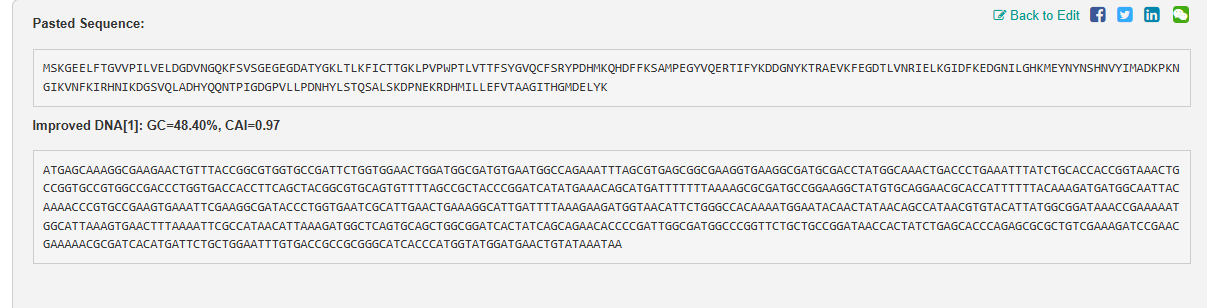
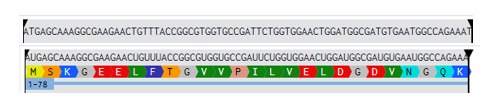
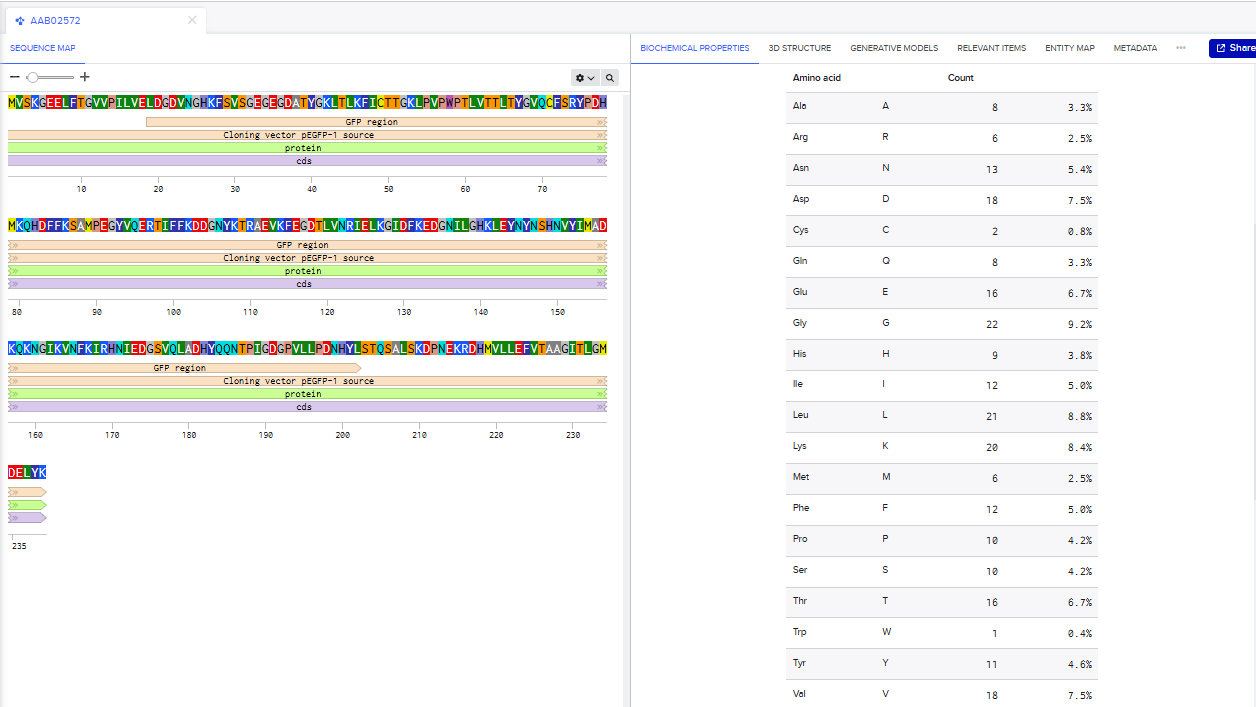
AAA58246.1 green-fluorescent protein [Aequorea victoria]
MSKGEELFTGVVPILVELDGDVNGQKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQC
FSRYPDHMKQHDFFKSAMPEGYVQERTIFYKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHK
MEYNYNSHNVYIMADKPKNGIKVNFKIRHNIKDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKD
PNEKRDHMILLEFVTAAGITHGMDELYK
Courtesy: The UniProt Consortium, “UniProt: the Universal Protein Knowledgebase in 2025,” Nucleic Acids Research, 2025.
3.2 Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Courtesy:NCBI - O’Leary NA, Cox E, Holmes JB, Anderson WR, Falk R, Hem V, Tsuchiya MTN, Schuler GD, Zhang X, Torcivia J, Ketter A, Breen L, Cothran J, Bajwa H, Tinne J, Meric PA, Hlavina W, Schneider VA. Exploring and retrieving sequence and metadata for species across the tree of life with NCBI Datasets. Sci Data. 2024 Jul 5;11(1):732. doi: 10.1038/s41597-024-03571-y. PMID: 38969627; PMCID: PMC11226681.
3.3 Codon optimization
Codon optimization is done as each organism has a set of codon preferences for the same amino acids. Due to codon redundancy the same amino acid acn be coded for by multiple sets of codon. This method is used to maximise protein expression based on tailoring the DNA sequence based on tRNa abundance in the host organism.
It increases translation efficiency, improves protein yield, and eliminates negative regulatory elements (repressors), which is crucial for producing recombinant proteins, vaccines, and gene therapies.
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
There are many technologies that can be used to produce protein.
1.Recombinant DNA technology
The GFP gene is inserted into plasmid that can replicate inside host cell. They have strong promoters to ensure high levels GFP expression.Based on the expression of the selective marker plasmids are selected and transfected into host cells.
Heterologous Expression Systems (Host Cells)
Bacterial protein expression, primarily using E. coli, is a fast, cost-effective, and scalable method for producing recombinant proteins. It involves :
Cloning: Inserting the GFP gene into an expression vector.(E.coli plasmid)
Transformation: Introducing the plasmid into competent bacterial cells.
Expression: Culturing the cells and inducing GFP production.
Harvesting & Purification: Lysis of cells and purification of the GFP.
Cell-Dependent Method (In Vivo)
This occurs within the host cell
Transcription (DNA to mRNA):
Initiation: The enzyme RNA polymerase binds to a GFP DNA sequence called the promoter, signaling the start of the gene.
Elongation: RNA polymerase unwinds the DNA helix and reads one strand (the template strand) in the 3′ to 5′ direction, synthesizing a complementary RNA molecule in the 5′ to 3′ direction.
Termination: Upon reaching a “terminator” sequence, the RNA polymerase releases the newly formed pre-mRNA (GFP) strand.
Translation (mRNA to Protein):
The mature GFP mRNA moves to the cytoplasm, where it binds to a ribosome.
The ribosome reads the codons.
Transfer RNA (tRNA) molecules, carrying specific amino acids, match their anticodons to the mRNA codons.
The ribosome catalyzes a peptide bond between amino acids, building a polypeptide chain until a stop codon is reached.
Highly regulated, capable of complex post-translational modifications (folding, glycosylation) in eukaryotes, but slow and limited by cell viability.
Cell-Free Method (In Vitro)
Cell-free protein synthesis (CFPS) harnesses the machinery (ribosomes, tRNAs, enzymes) extracted from cells to produce proteins in a test tube, allowing direct control over the environment.
Preparation: Cells (e.g., E. coli,) are grown, lysed, and centrifuged to remove DNA, cell walls, and debris, leaving only the translational machinery.
Method: The extracted, active machinery is mixed with the GFP DNA template (plasmid or PCR-amplified), amino acids, energy sources (like ATP/GTP), and cofactors.
Process: The system can be coupled (transcription and translation occur together) or uncoupled (using mRNA directly). It bypasses the need for cell viability making it ideal for toxic, membrane, or complex proteins.
Fast (hours instead of days), open system allowing direct manipulation, and capable of producing toxic or high-yield proteins, but can be expensive for large-scale production.
3.5 [Optional] How does it work in nature/biological systems?
The protein works as a fluorescence producing mechanism in the jelly fish producing green light.
1. Describe how a single gene codes for multiple proteins at the transcriptional level.
A single gene codes for multiple proteins primarily through alternative splicing of pre-mRNA, where different combinations of exons are joined together after introns are removed.
Pre-mRNA is spliced in multiple ways to include or exclude specific exons. This produces different mRNA transcripts, which are then translated into different protein isoforms.
Alternative Promoters: A gene may have multiple promoters, allowing transcription to start at different points, resulting in mRNA molecules with different 5’ends.
Alternative Polyadenylation: This process alters the end of the mRNA, which can affect mRNA stability and localization, leading to different protein products.
2.Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!!
(i) What DNA would you want to sequence (e.g., read) and why?
I would like to explore DNA of Antibiotic resistant bacteria against penicillin. I want to study their sequence and understand how they become resistant to the antibiotics, study their interaction with antibiotics and compare their genes with their susceptible counterparts to understand which sequence produces which type of resistance to the organism towards antibiotics.
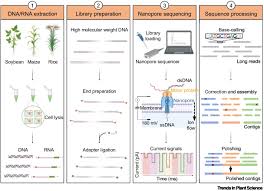
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Why: Produces very long reads (10 kb to >1 Mb) that can bridge repetitive regions, allowing for the easy reconstruction of plasmids and the identification of the genetic context of resistance genes (e.g., whether they are on a plasmid or chromosome).
Courtesy:Xu Y, Luo H, Wang Z, Lam HM, Huang C. Oxford Nanopore Technology: revolutionizing genomics research in plants. Trends Plant Sci. 2022 May;27(5):510-511. doi: 10.1016/j.tplants.2021.11.004. Epub 2021 Nov 23. PMID: 34836785.
1. Is your method first-, second- or third-generation or other? How so?
Oxford Nanopore Technologies (ONT) is considered a third-generation (or sometimes referred to as long-read) sequencing technology.
The first-generation (Sanger) or second-generation (Illumina/NGS) methods rely on DNA synthesis and detection of light signals, while ONT measures changes in electrical current as single molecules of DNA/RNA pass through a protein nanopore.
It is categorized as third-generation sequencing due to its ability to sequence long, single molecules of nucleic acids in real-time. It generates very long to ultra-long reads, superior assembly of complex genomes and structural variant detection.
2. What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input for the ONT method would be the genomic DNA (gDNA) or plasmid DNA, extracted from pure bacterial cultures or environmental samples.
Steps for Sequencing AMR Genes
Sample Preparation and DNA Extraction:
AMR Bacteria are cultured in LB broth and collected.
DNA is extracted using kits for high molecular weight DNA using Qiagen or MagAttract kits.
It is quantified and purified using Qubit and Nanodrop respectively.
Library Preparation:
Rapid Kits (e.g., Rapid Barcoding Kit SQK-RBK114.24): Transposases fragment and tag DNA with adapters in one step, ideal for quick turnaround.
Ligation Kits (e.g., Ligation Sequencing Kit SQK-LSK114): Provides higher output and longer reads for more comprehensive genome coverage.
3.What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
The prepared library is loaded onto an ONT flow cell (e.g., R10.4 or R9.4.1) on a device like the MinION or GridION.
As DNA passes through the nanopore, ionic current changes are measured and recorded.
1. Raw Signal Capturing and Preprocessing
Signal Acquisition: As DNA/RNA strands pass through a nanopore, they disrupt an electrical current. MinKNOW (ONT’s controlling software) records these changes.
File Format Conversion: Raw signals are traditionally stored in .fast5 files but are increasingly saved in the more efficient POD5 file format, which is designed for faster data handling and processing.
Data Preparation: Before basecalling, the raw signals are often organized and prepared, potentially involving filtering to remove uninformative data.
2. The Basecalling Process (Neural Network Decoding)
Deep Learning Models: Modern ONT basecallers (like Dorado, Guppy, or Bonito) use neural networks (such as Recurrent Neural Networks - RNNs, or Transformer models) to analyze the raw signal data.
“Squiggle” to Base Translation: The neural network maps the electrical signal changes to the corresponding nucleotide sequences, usually in real time while the sequencing is still running.
Move Tables: The process identifies when a new base enters the pore, producing a “move table” that indicates which part of the signal corresponds to which base.
Quality Scoring: Alongside the base sequence, the basecaller assigns a probability score to each base, often represented as a Phred score to indicate confidence in the call.
4.What is the output of your chosen sequencing technology?
Post-Processing and Output
FASTQ File Generation: The primary output is a FASTQ file, containing the sequences and their associated quality scores.
BAM/CRAM Output: Alternatively, basecallers can output files in SAM, BAM, or CRAM formats, which can include both the sequence data and the signal-level information.
Demultiplexing (Optional): If multiple samples were mixed in a single run (barcoding), the software identifies which reads belong to which barcode and separates them into individual files.
Polishing (Optional): Additional steps like Medaka or Nanopolish may be used to refine the sequence data further, especially for improving consensus accuracy.
5.2 DNA Write
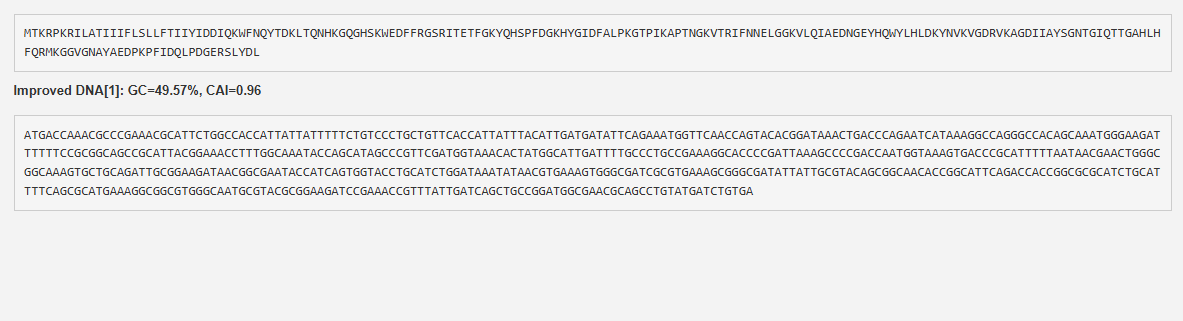
(i) What DNA would you want to synthesize (e.g., write) and why?
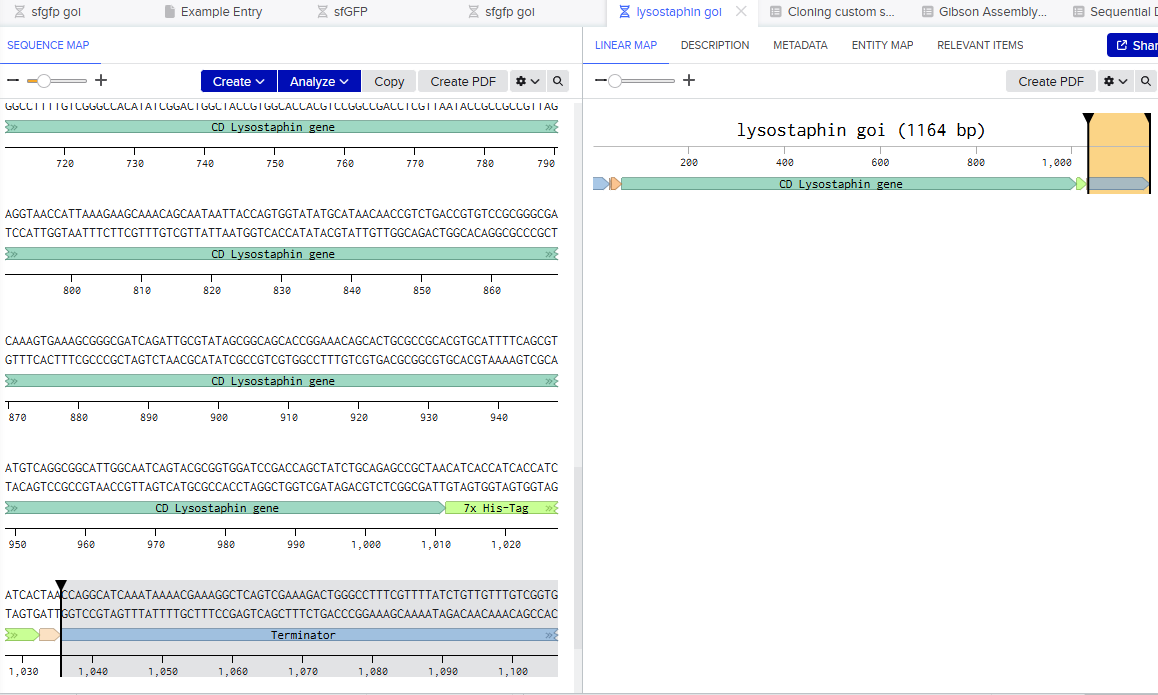
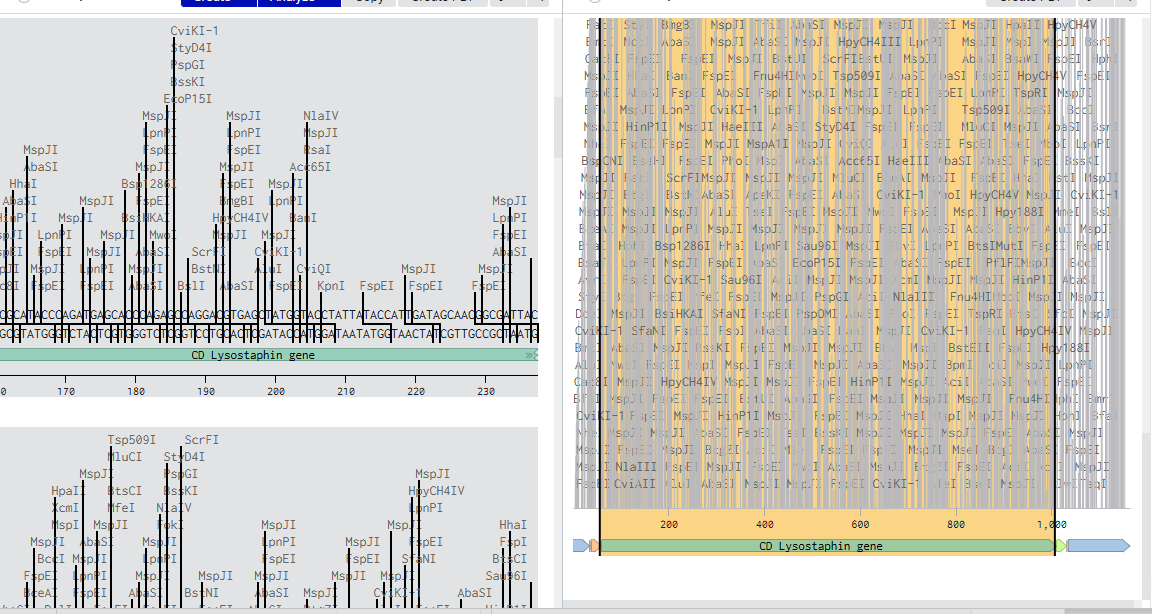
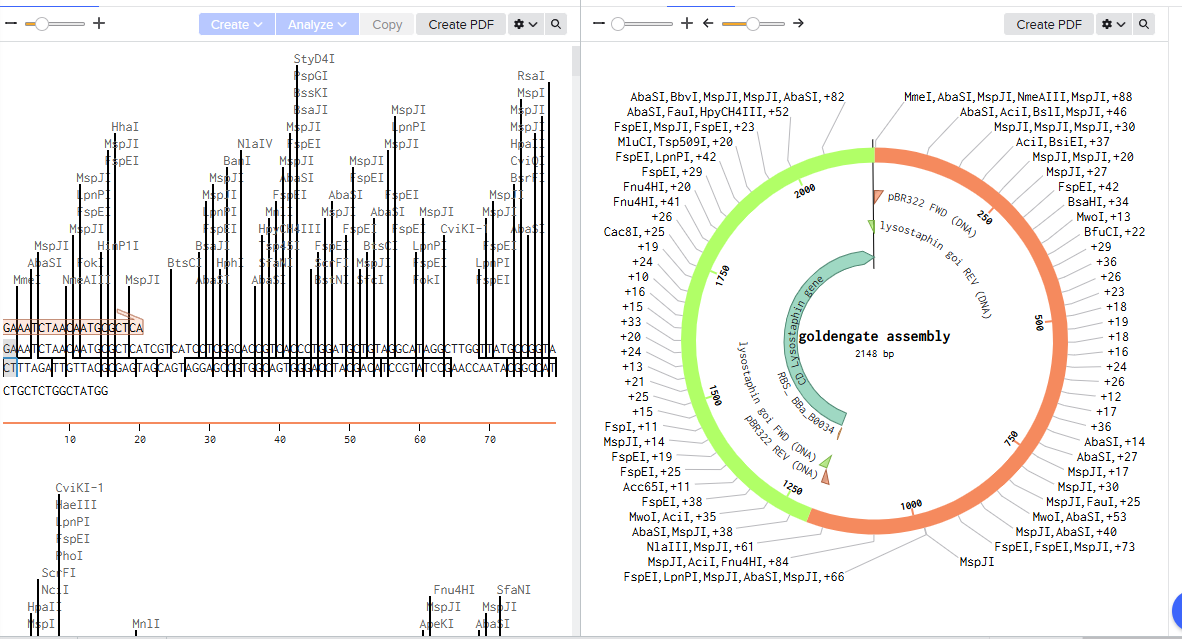
I want to synthesize the Staphylococcus staphylolyticus lysostaphin gene
Lysostaphin is highly active against antibiotic-resistant S. aureus (MRSA) and intermediately vancomycin-susceptible S. aureus (VISA), as its mechanism of action differs from traditional antibiotic resistance mechanisms.
Lysostaphin is a potent zinc-dependent metalloendopeptidase (specifically a glycylglycine endopeptidase) produced by Staphylococcus simulans. It acts as an antibacterial agent (bacteriocin) with high efficiency against Staphylococcus aureus (including MRSA).
ii) What technology or technologies would you use to perform this DNA synthesis and why?
The most widely used DNA synthesis technology for the lysostaphin gene is PCR-based gene assembly (or gene synthesis) from overlapping oligonucleotides. Because the wild-type lysostaphin gene natively contains high AT-rich regions and rare codons that make it difficult to express, researchers typically synthesize it “from scratch” using custom oligonucleotide assembly.
1.What are the essential steps of your chosen sequencing methods?
Codon Optimization: To increase expression efficiency, the native lss gene is often codon-optimized to match the preferences of the host microorganism, such as E. coli.
Vector Construction and Expression Systems: The gene is cloned into various expression vectors (e.g., pET-22b(+), pWB980, pET32a) and transformed into hosts like E. coli BL21(DE3) or Bacillus subtilis WB600, a strain engineered to be deficient in six extracellular proteases, reducing protein degradation.
Constitutive and Inducible Promoters: While many systems use inducible promoters (e.g., IPTG) to control production, recent advances include using constitutive, non-inducible promoters (e.g., pemIK-Sa1 from staphylococcal toxin-antitoxin systems) to reduce costs for large-scale production.
Restriction Enzyme Cloning: The synthetic lysostaphin gene is digested with restriction enzymes (e.g., EcoRI, XhoI, NdeI) and ligated into expression vectors like pPIC9 or pET22b(+) using T4 DNA ligase.
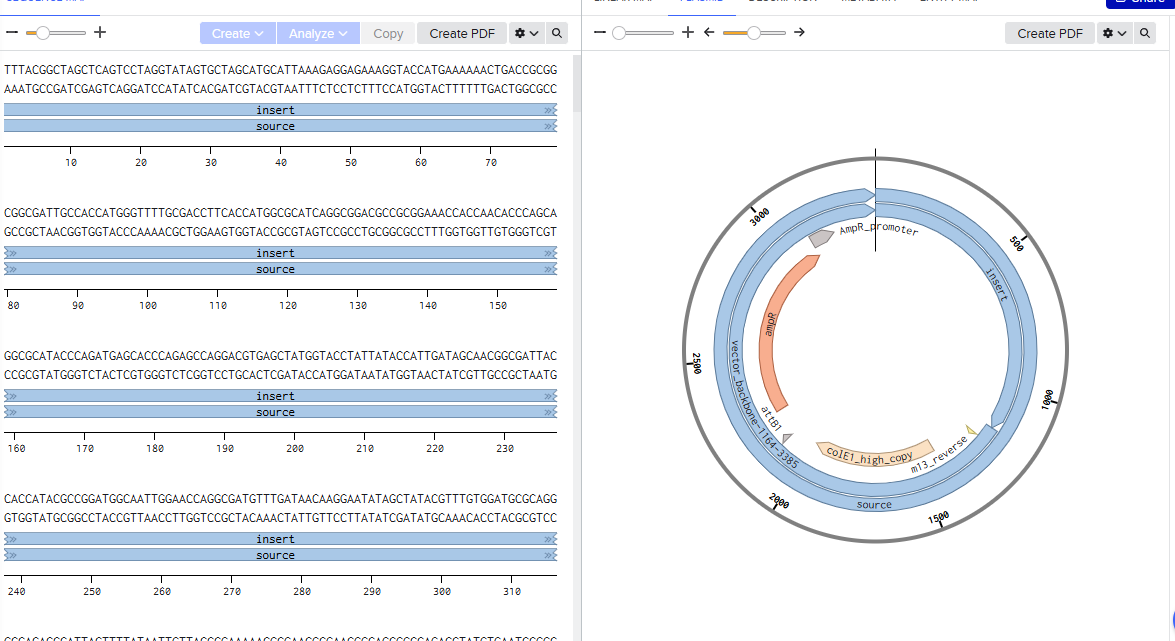
Gibson Assembly: The NEBuilder Assembly Tool is used to design primers for amplifying target regions, which are then assembled into plasmids (e.g., pMAD).
Homologous Recombination: Homologous recombinase is used to ligate optimized lysostaphin fragments into vectors.
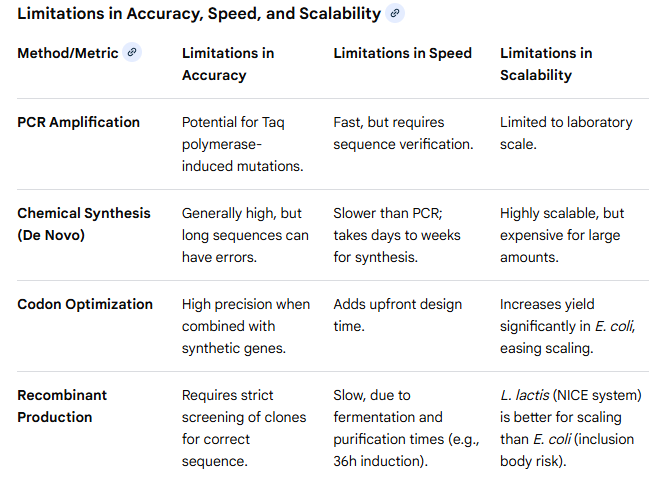
2. What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Courtesy: Perplexity
5.3 DNA Edit
i) What DNA would you want to edit and why?
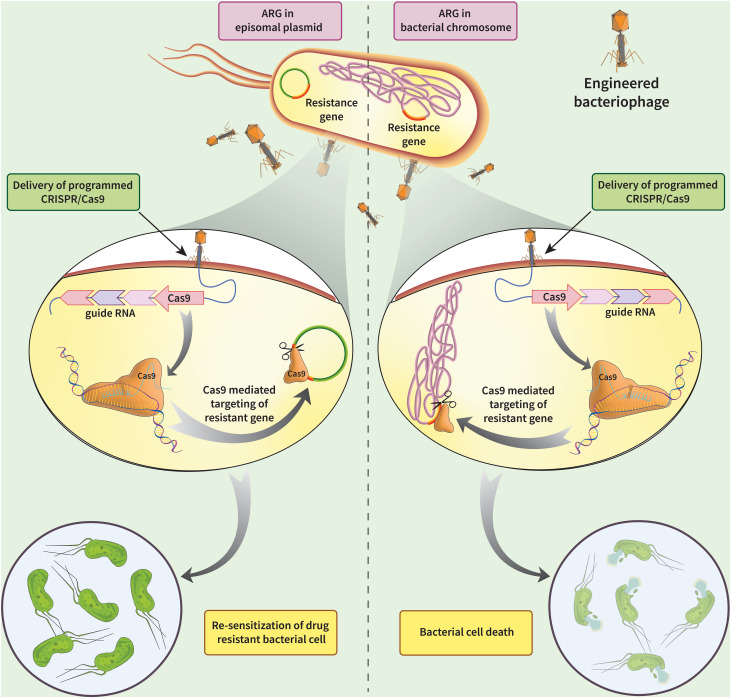
I would want to edit, silence, or delete antimicrobial resistance (AMR) genes in bacteria, specifically targeting resistance plasmids or chromosomal genes to restore antibiotic susceptibility.
Courtesy:Kadkhoda H, Gholizadeh P, Samadi Kafil H, Ghotaslou R, Pirzadeh T, Ahangarzadeh Rezaee M, Nabizadeh E, Feizi H, Aghazadeh M. Role of CRISPR-Cas systems and anti-CRISPR proteins in bacterial antibiotic resistance. Heliyon. 2024 Jul 16;10(14):e34692. doi: 10.1016/j.heliyon.2024.e34692. PMID: 39149034; PMCID: PMC11325803.
1.What technology or technologies would you use to perform these DNA edits and why?
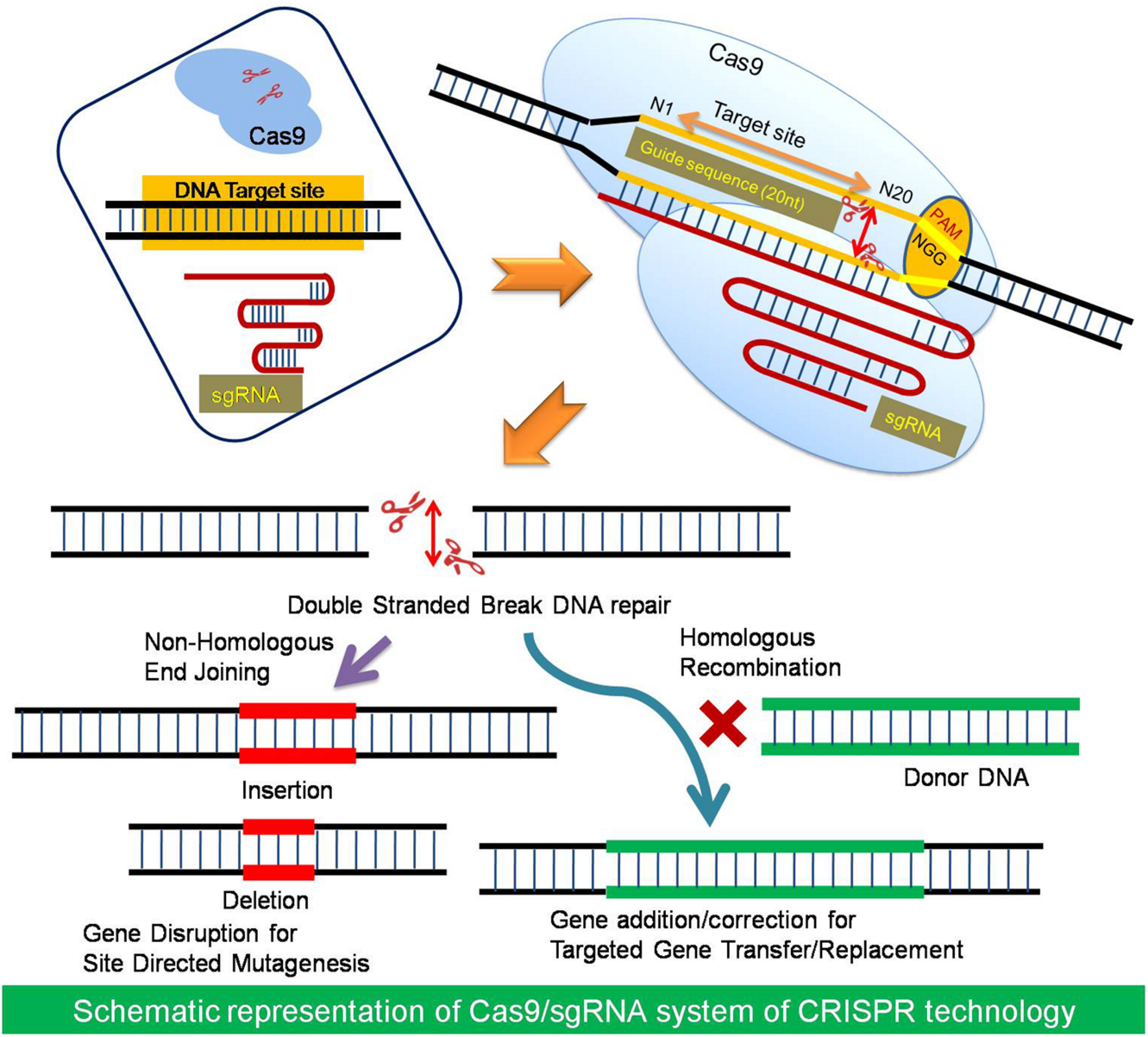
I would want to use CRISPR technology.CRISPR-Cas9 is a genome-editing technology that uses a guide RNA (gRNA) to direct the Cas9 enzyme to a specific DNA sequence, where it acts as molecular scissors to create a double-strand break. The cell then repairs this cut using either NHEJ (resulting in gene knockouts) or HDR (enabling precise gene insertion/correction).
It is faster, cheaper, and more accurate alternative to previous methods.
The other editing technologies are TALENs & ZFNs: Older, customizable nuclease technologies that bind to specific DNA sequences to induce breaks, though they are generally less flexible than CRISPR.
TALENs generally exhibit significantly lower off-target effects compared to both ZFNs and CRISPR, making them safer for certain applications.They are widely used for precise, large-scale, and stable genome engineering in plants and animals.
2. How does your technology of choice edit DNA? What are the essential steps?
CRISPR-Cas9 is a programmable gene-editing technology that uses a guide RNA (sgRNA) to direct the Cas9 enzyme to a specific DNA sequence, creating a targeted double-strand break (DSB). The system relies on a PAM sequence for binding, after which the cell repairs the cut using error-prone NHEJ (for gene disruption) or precise HDR (for gene correction).
Guide RNA (sgRNA): A synthetic RNA sequence designed to be complementary to the target DNA, directing the Cas9 enzyme to the precise location in the genome.
Cas9 Nuclease: An enzyme acting as molecular scissors that creates a double-strand break (DSB) in the DNA, specifically three bases upstream of a required
Protospacer Adjacent Motif (PAM) sequence.
Target Recognition: The CRISPR-Cas9 complex scans the genome for a PAM sequence (commonly 5’-NGG-3’). Once found, it checks if the sgRNA matches the adjacent DNA sequence.
Once the DNA is cut, the cell attempts to repair it, which allows for gene editing:
Non-Homologous End Joining (NHEJ): A fast,, error-prone repair mechanism that often introduces small deletions or insertions (indels), disrupting or “knocking out” the target gene.
Homology-Directed Repair (HDR): A precise repair mechanism used if a repair template is provided, allowing for the insertion of new, desired genetic information or correction of mutations.
Khatodia S, Bhatotia K, Passricha N, Khurana SMP and Tuteja N (2016) The CRISPR/Cas Genome-Editing Tool: Application in Improvement of Crops. Front. Plant Sci. 7:506. doi: 10.3389/fpls.2016.00506
3. What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
The preparation step involves the identification of the AMR gene to be targeted, then analyse the sequence for protospacer adjacent motif(PAM).
Design gRNAs complementary to AMR genes.Benchling tool can be used to reduce off-target effects and maximise specificity.
Construct plasmids for cloning gRNA and Cas9 gene inserts using restriction enzyme and ligase.
Transformation of E.coli cells with the plasmids, purication and verification of sequences using Sanger sequencing.
Preparation of delivery systems: chemical transformation, electroporation,bacteriophage particle for phage mediated delivery.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Inputs for AMR editing
Cas Nuclease: Cas9 (commonly S. pyogenes Cas9) is the primary protein, or Cas12a/Cpf1.
Guide RNA (gRNA/sgRNA): Specifically designed 20-nt guide sequence with a scaffold.
Plasmids: Expression vectors containing both the Cas9 gene and the gRNA sequence (e.g., pX330).
Primers: For PCR verification of the edit.
Enzymes: Restriction enzymes (e.g., BpiI, BsmBI) and T4 DNA ligase for cloning.
Delivery Vehicle: Phages (e.g., temperate/lytic phage), nanoparticles, or conjugated plasmids.
Limitations of CRISPR
Off-Target Effects: The CRISPR-Cas9 complex may bind to and modify genomic sites that are not the intended target, leading to potential, unintended, and sometimes harmful mutations.
Delivery Challenges: Delivering the large CRISPR-Cas9 components into specific cells or tissues is difficult, which limits its application in many clinical contexts.
Low Efficiency: The process is not 100% efficient, particularly with homology-directed repair (HDR), leading to cells that may not have the desired edit.
Mosaicism: In animal models, not all cells may be edited equally, resulting in mosaicism where only some cells carry the desired modification, making it difficult to identify, study, or rely on the desired edit.
PAM Sequence Requirements: The Cas9 protein must bind to a specific protospacer adjacent motif (PAM) sequence located next to the target DNA, which may not be present at the desired location.
Persistent Binding: In some instances, the Cas9 protein binds to the cut site persistently, preventing the DNA repair machinery from functioning, leading to editing failure.
Khatodia S, Bhatotia K, Passricha N, Khurana SMP and Tuteja N (2016) The CRISPR/Cas Genome-Editing Tool: Application in Improvement of Crops. Front. Plant Sci. 7:506. doi: 10.3389/fpls.2016.00506
Kadkhoda H, Gholizadeh P, Samadi Kafil H, Ghotaslou R, Pirzadeh T, Ahangarzadeh Rezaee M, Nabizadeh E, Feizi H, Aghazadeh M. Role of CRISPR-Cas systems and anti-CRISPR proteins in bacterial antibiotic resistance. Heliyon. 2024 Jul 16;10(14):e34692. doi: 10.1016/j.heliyon.2024.e34692. PMID: 39149034; PMCID: PMC11325803.
NCBI - O’Leary NA, Cox E, Holmes JB, Anderson WR, Falk R, Hem V, Tsuchiya MTN, Schuler GD, Zhang X, Torcivia J, Ketter A, Breen L, Cothran J, Bajwa H, Tinne J, Meric PA, Hlavina W, Schneider VA. Exploring and retrieving sequence and metadata for species across the tree of life with NCBI Datasets. Sci Data. 2024 Jul 5;11(1):732. doi: 10.1038/s41597-024-03571-y. PMID: 38969627; PMCID: PMC11226681.
Xu Y, Luo H, Wang Z, Lam HM, Huang C. Oxford Nanopore Technology: revolutionizing genomics research in plants. Trends Plant Sci. 2022 May;27(5):510-511. doi: 10.1016/j.tplants.2021.11.004. Epub 2021 Nov 23. PMID: 34836785.
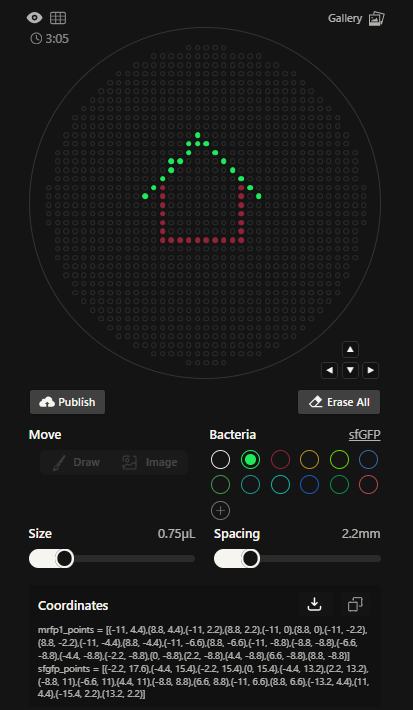
Create a Python file to run on an Opentrons liquid handling robot.
1.Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
2.Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
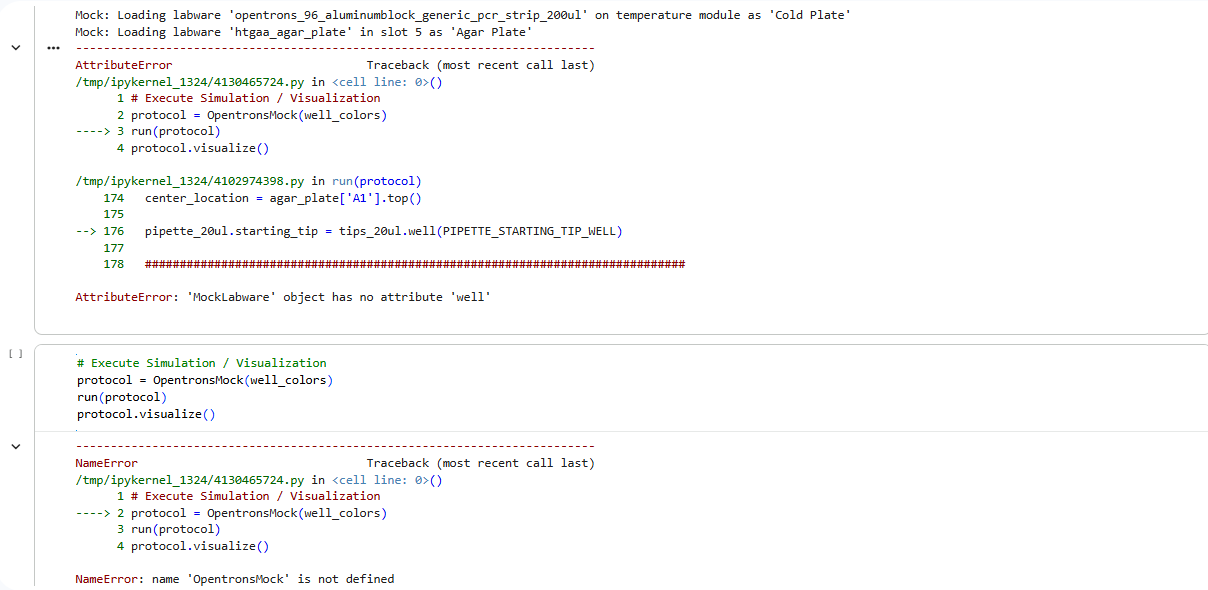
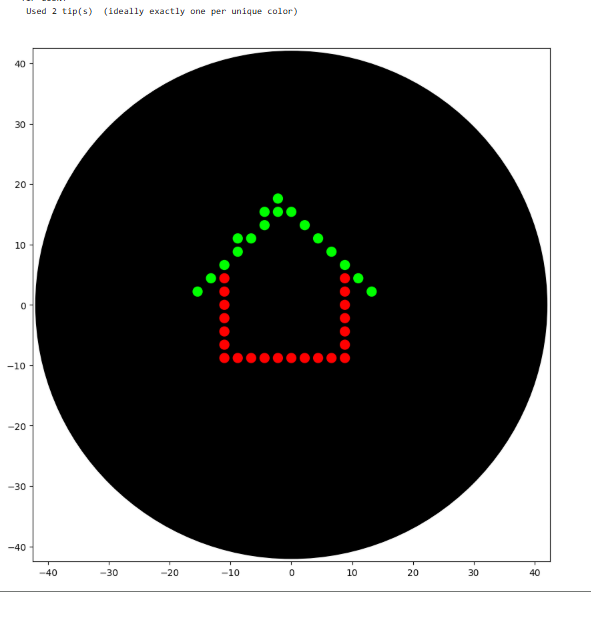
Errors
Script
Output
4.If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
I used AI Gemini to help me with the codes. Any corrections in the code were made using Gemini.
Post-Lab Questions
1.Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Journal Article
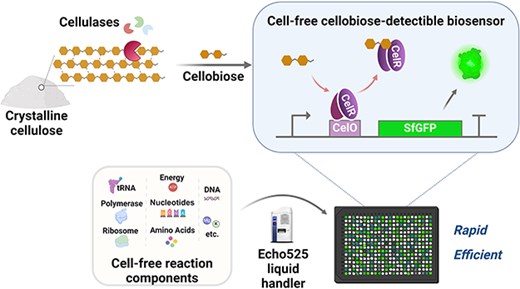
Cell-free biosensor with automated acoustic liquid handling for rapid and scalable characterization of cellobiohydrolases on microcrystalline cellulose
Taeok Kim, Eun Jung Jeon, Kil Koang Kwon, Minji Ko, Ha-Neul Kim, Seong Keun Kim, Eugene Rha, Jonghyeok Shin, Haseong Kim, Dae-Hee Lee, Bong Hyun Sung, Soo-Jung Kim, Hyewon Lee, Seung-Goo Lee, Cell-free biosensor with automated acoustic liquid handling for rapid and scalable characterization of cellobiohydrolases on microcrystalline cellulose, Synthetic Biology, Volume 10, Issue 1, 2025, ysaf005, https://doi.org/10.1093/synbio/ysaf005
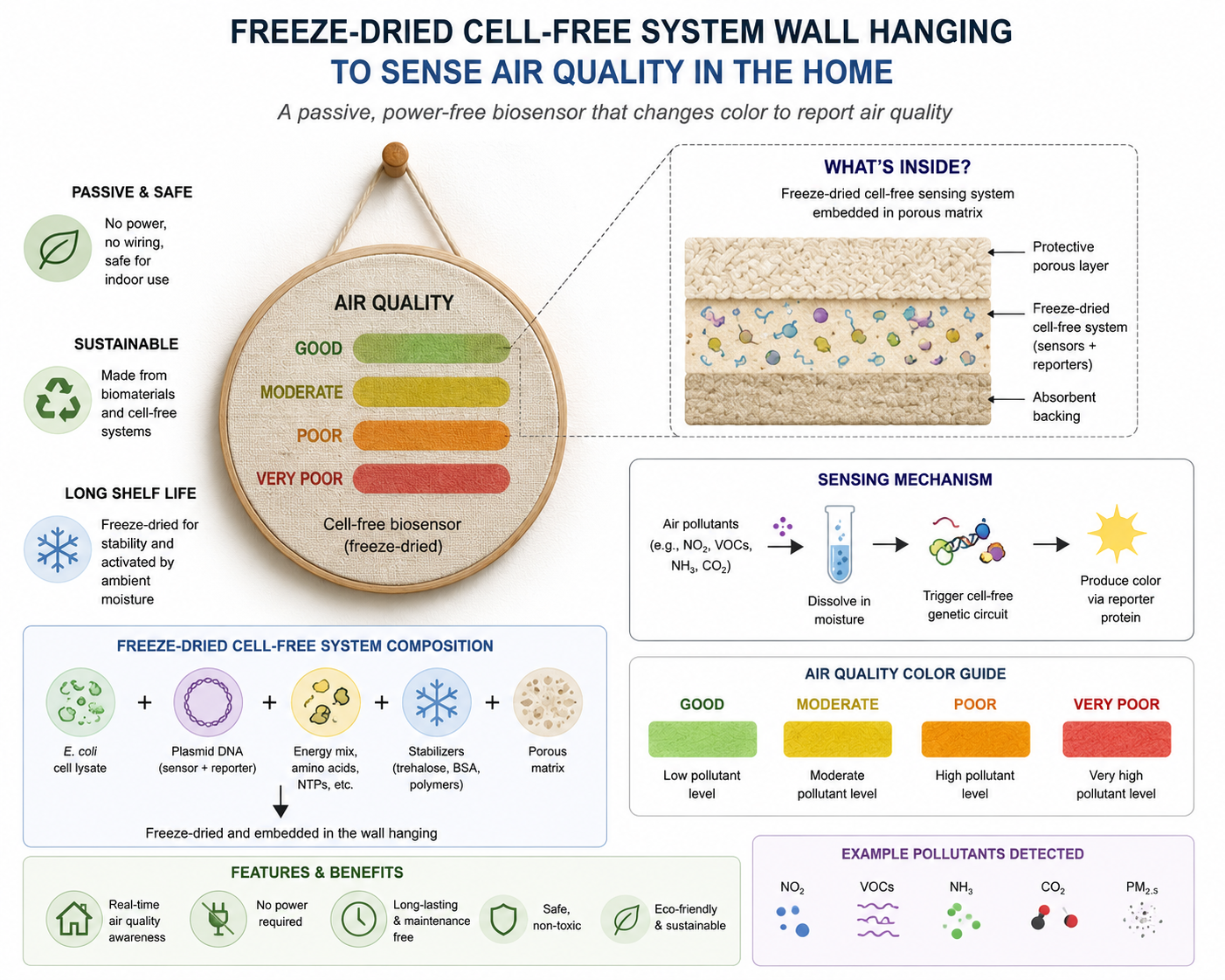
This paper talks about the high throughput screening challenges involving engineering enzymes that help in degrading cellulose in paper sludge or microplastics in sewage sludge as solid substrates are not readily accessible in cell-based biosensor systems. In the paper, a cell free cellobiose-detectable biosensor (CB-biosensor) for rapid characterization of cellobiohydrolase (CBH) activity, enabling direct detection of hydrolysis products without cellular constraints was adopted.The biosensor distinguishes between CBH subtypes (CBHI and CBHII) based on their modes of action. Echo 525 liquid handler enables precise and reproducible sample processing, with fluorescence signals from automated preparations comparable to manual experiments.Assay volumes can be reduced to just a few microlitres—impractical with manual methods. Echo 525 minimizes reagent consumption, accelerates testing, and facilitates reliable large-scale screening, advancing enzyme screening and accelerating the Design-Build-Test-Learn cycle for sustainable biomanufacturing.
2.Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
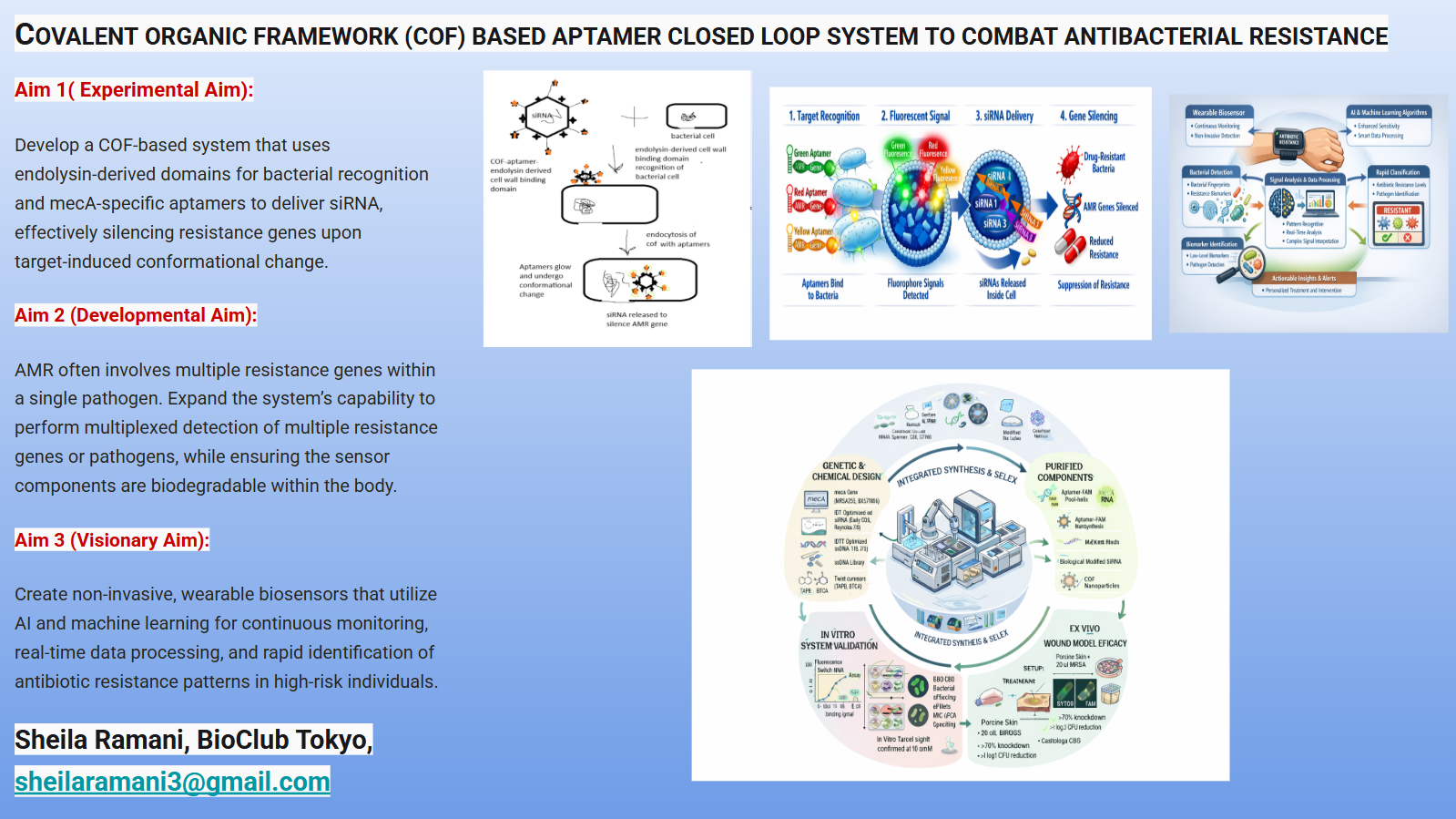
The final project idea which involves design of COF based biosensor with aptamer for detection of AMR genes and drug delivery, Opentron can be used to automate time consuming SELEX(Systematic Evolution of Ligands by Exponential Enrichment)process for aptamer selection. This increases binding affinity,improve specificity, removing sequences that bind to non-target components. PCR/RT-PCR amplification of the bound sequences is carried out automatically to generate the pool for the next round.
Robots control the functionalization of the COF surface with various reagents(e.g., amine-functionalization).
Immobilization: Automated, high-throughput liquid handling ensures consistent covalent attachment of the aptamer to the COF, ensuring uniform batch production.
Robots precisely load the COF-aptamer complex with siRNA for gene silencing of AMR, ensuring consistent dosages for therapeutic applications.
3.Taeok Kim, Eun Jung Jeon, Kil Koang Kwon, Minji Ko, Ha-Neul Kim, Seong Keun Kim, Eugene Rha, Jonghyeok Shin, Haseong Kim, Dae-Hee Lee, Bong Hyun Sung, Soo-Jung Kim, Hyewon Lee, Seung-Goo Lee, Cell-free biosensor with automated acoustic liquid handling for rapid and scalable characterization of cellobiohydrolases on microcrystalline cellulose, Synthetic Biology, Volume 10, Issue 1, 2025, ysaf005, https://doi.org/10.1093/synbio/ysaf005.
4.Google. (2026,February,20th). AI generated codes error rectification.Gemini version 2.0
“Design Biosensor for pcod with toe hold switch for mir-155” Microsoft Copilot, Microsoft, [23-2-2026], https://copilot.microsoft.com/
Abe, Takumi & Takashima, Rikito & Kamiya, Takehiro & Foong, Choon Pin & Numata, Keiji & Aoki, Daisuke & Otsuka, Hideyuki. (2021). Plastics to fertilizers: chemical recycling of a bio-based polycarbonate as a fertilizer source. Green Chemistry. 23. 10.1039/D1GC02327F.
Ogunwole, Olufemi & Okpe, Aboje. (2025). Conversion of Waste PET Plastics into Activated Carbon for Fertilizer Delivery Applications.
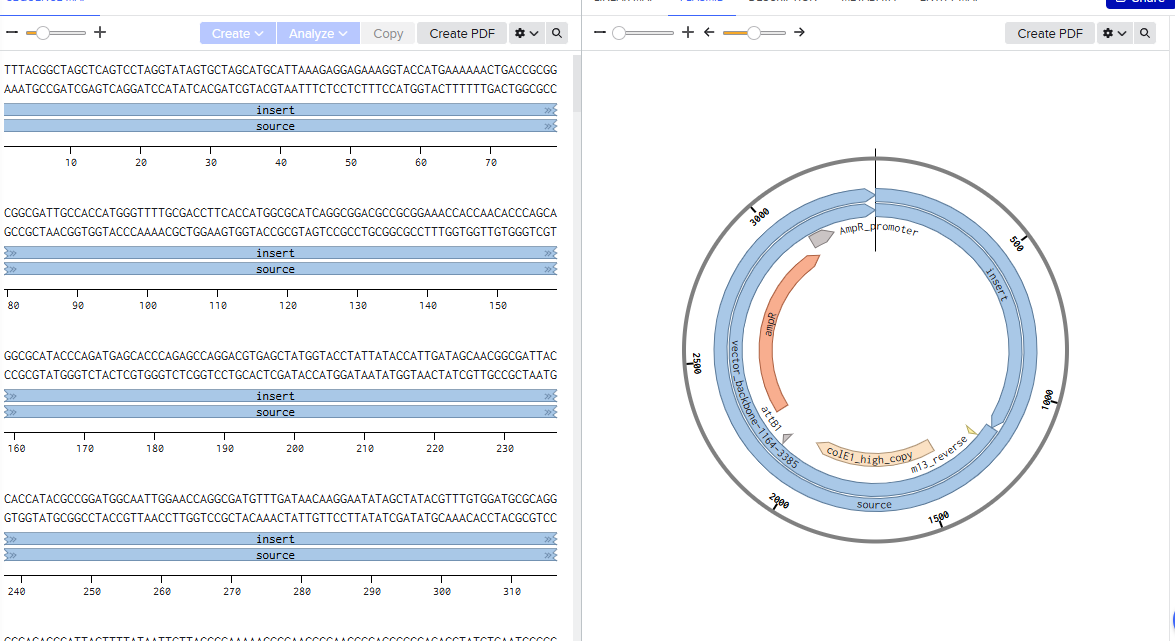
Benchling [Biology Software]. (2026).- Constructing the petase and MHetase plasmid.
Adila Nazli, David L. He, Dandan Liao, Muhammad Zafar Irshad Khan, Chao Huang, Yun He,Strategies and progresses for enhancing targeted antibiotic delivery, Advanced Drug Delivery Reviews,Volume 189,2022,114502,ISSN 0169-409X,https://doi.org/10.1016/j.addr.2022.114502.
Wang S, Emery NJ, Liu AP. A Novel Synthetic Toehold Switch for MicroRNA Detection in Mammalian Cells. ACS Synth Biol. 2019 May 17;8(5):1079-1088. doi: 10.1021/acssynbio.8b00530. Epub 2019 May 6. PMID: 31039307.
Sørensen AE, Wissing ML, Salö S, Englund AL, Dalgaard LT. MicroRNAs Related to Polycystic Ovary Syndrome (PCOS). Genes (Basel). 2014 Aug 25;5(3):684-708. doi: 10.3390/genes5030684. PMID: 25158044; PMCID: PMC4198925.
The 20 natural amino acids evolved as optimal sets very early, during the RNA world (4 billion years ago). The format was not changed and became frozen because it would disrupt all proteins and also due to tRNA recognition limitations further expansion was prohibited.
2.Where did amino acids come from before enzymes that make them, and before life started?
The amino acids were formed by abiotic processes on early Earth(4.5 billion years ago) using gases, minerals and energy sources present at that time.
Miller-Urey experiment simulated the similar environment in their experiment and created glycine,alanine and 33 other amino acids by condensation and reduction.
The paper cited gives a good insight into this chicken and egg theory
Singh, J., Thoma, B., Whitaker, D. et al. Thioester-mediated RNA aminoacylation and peptidyl-RNA synthesis in water. Nature 644, 933–944 (2025). https://doi.org/10.1038/s41586-025-09388-y
3.If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
L-amino acids form righthanded α-helices because their chirality favours such formation to prevent steric clashes in the side chains. In contrast the D-amino acids should prefer left handed helices to prevent steric clashes in the side chains.
4.Can you discover additional helices in proteins?
There are other helix types like the 3₁₀-helices, π-helices, and polyproline II (PPII) helices. They are formed by specific hydrogen bonding patterns and amino acid sequences.
Coutesy:Perplexity Pro prompt alpha helices
5.Why most molecular helices are right-handed?
The molecular helices in biology are right handed because of the L-chirality of amino acids and D-sugars. These molecular conformations sterically favour the right handed twist for stability and folding efficiency.
6.Why do β-sheets tend to aggregate?What is the driving force for β-sheet aggregation?
β-sheet aggregate due to hydrogen bond donors/acceptors at their edges, promote edge to edge interactions with other sheets or unfolded chains.Hydrophobic side chains on edges prefer being buried by intermolecular contacts, leading to intermolecular associations that extend sheets into fibrils or amyloids.
The primary driving force for β-sheet aggregation is thermodynamics.
The hydrogen bonds and Van der Waal’s forces lower free energy, further by cooperativity by dimerization.
Aggregation occurs when the hydrophobic residues bury themselves in a compact core-this “collapse” reduces solvent-exposed area and drives entropy gain from released water molecules.
7.Why do many amyloid diseases form β-sheets?Can you use amyloid β-sheets as materials?
Many amyloid diseases occur because of misfolding of proteins and adoption of β-sheet conformation and then self assembly into insoluble fibrils. Destabilization of native protein structure occurs first, then partial unfolding leading to exposure of β-strand regions that stack via hydrogen bonding into cross-β-sheet architectures. The fibrils formed are highly ordered parallel or antiparallel β-sheets,aggregate in a prion like manner, leading to plaque formation that disrupt tissue function in conditions like Alzheimer’s and type II diabetes.
Amyloid β-sheets as materials can be used as biomaterials because of their exceptional mechanical strength,biocompatibility, and nanoscale self-assembly. Non-pathogenic or engineered amyloid fibrils form robust scaffolds for tissue engineering, drug delivery, and biosensors. They mimic extracellular matrices to support cell adhesion and growth. They allow fabrications with bioplastics, hydrogels, and functional coatings for tunable properties via genetic modification or hybridization with nanoparticles.
Courtesy:Fallot LB, Natarajan C, Anderson CA, Nagelli EA, Burpo FJ, Limbocker R. From Pathology to Materials Science and Engineering: Harnessing the Amyloid State for Biotechnological Applications. ACS Appl Mater Interfaces. 2025 Nov 19;17(46):62839-62859. doi: 10.1021/acsami.5c11847. Epub 2025 Nov 10. PMID: 41211864; PMCID: PMC12635977.
8.Can you make other non-natural amino acids? Design some new amino acids.
Yes we can make.
The side chain of the amino acid has to be modified by methylation or some other functional group, or with another side chain that is bulky.
Advantages: Green, selective; challenges: Low yield, stability issues.
9.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We eat beef or fish whuch is later broken down by the enzymes in our body into the building blocks of the biomolecules present in each. The whole genome of the the fish or cow is not integerated within our DNA so that we become cow or fish.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
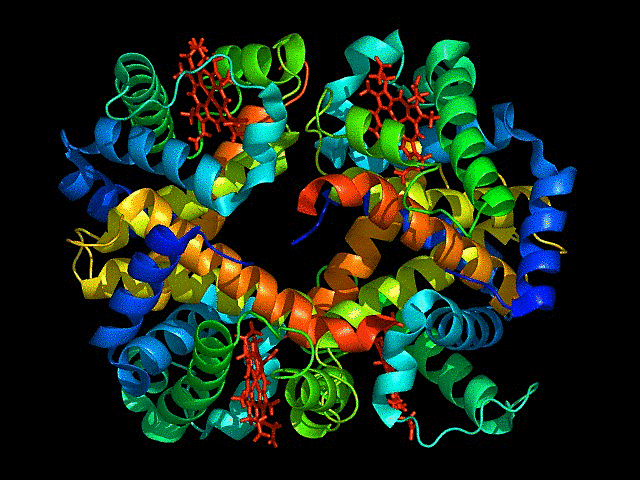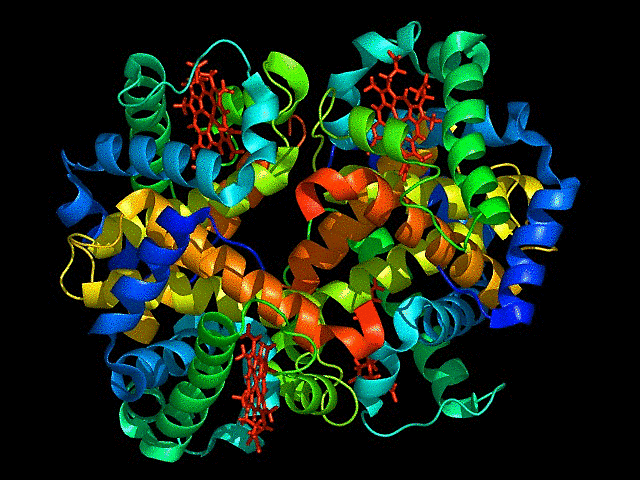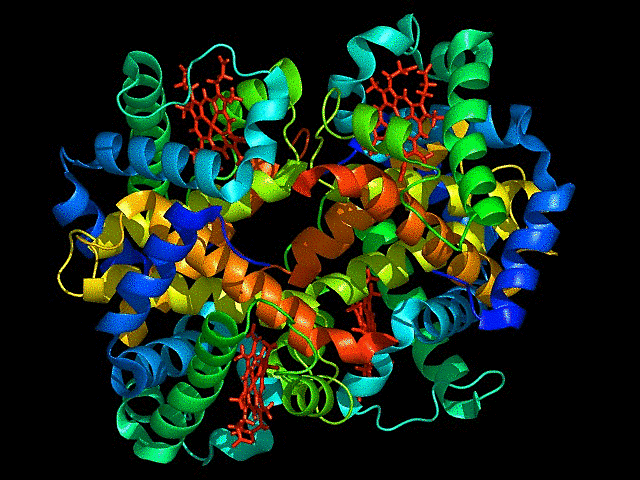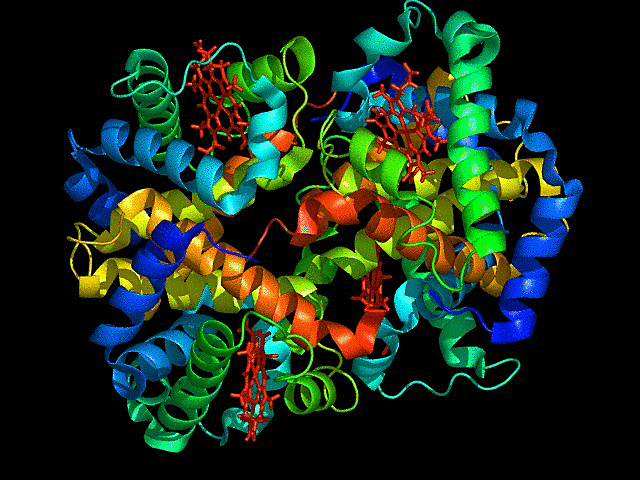
mCardinal is the far red fluorescent protein I have chosen. It is a bright, monomeric,derived from Entacmaea quadricolor, with an emission peak around 656 nm.
I chose this because its excitation at 604 nm and emission at 659 nm, is the optimal far-red range for deep-tissue penetration. It is far brighter than mKate2 and other early-generation far-red variants.The monomeric form of the fluorescent protein, minimizes toxicity and can be used as fusion tags with target proteins without causing aggregation. Highly photostable so can be used for long term imaging.
2.Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The protein is 268 amino acids long.
The most common amino acid is G, it occurs 25 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
courtesy:The UniProt Consortium
UniProt: the Universal Protein Knowledgebase in 2025
Nucleic Acids Res. 53:D609–D617 (2025)
It has many homologs and some of them are uncharacterised proteins too. Mostly the homologs belong to the red fluorescent protein family.
Does your protein belong to any protein family?
mCardinal belongs to the GFP-like protein family (specifically the Green Fluorescent Protein superfamily)
3.Identify the structure page of your protein in RCSB
Reference: H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne, The Protein Data Bank (2000) Nucleic Acids Research 28: 235-242 https://doi.org/10.1093/nar/28.1.235.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure was solved in 2014.it a good quality structure. its resolution is 2.21Å.
Are there any other molecules in the solved structure apart from protein?
No.
Does your protein belong to any structure classification family?
It belongs to family of Fluorescent proteins.
4.Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands) citation: Schrödinger, LLC. (2026). The PyMOL Molecular Graphics System, Version X.X. https://www.pymol.org/support.html.
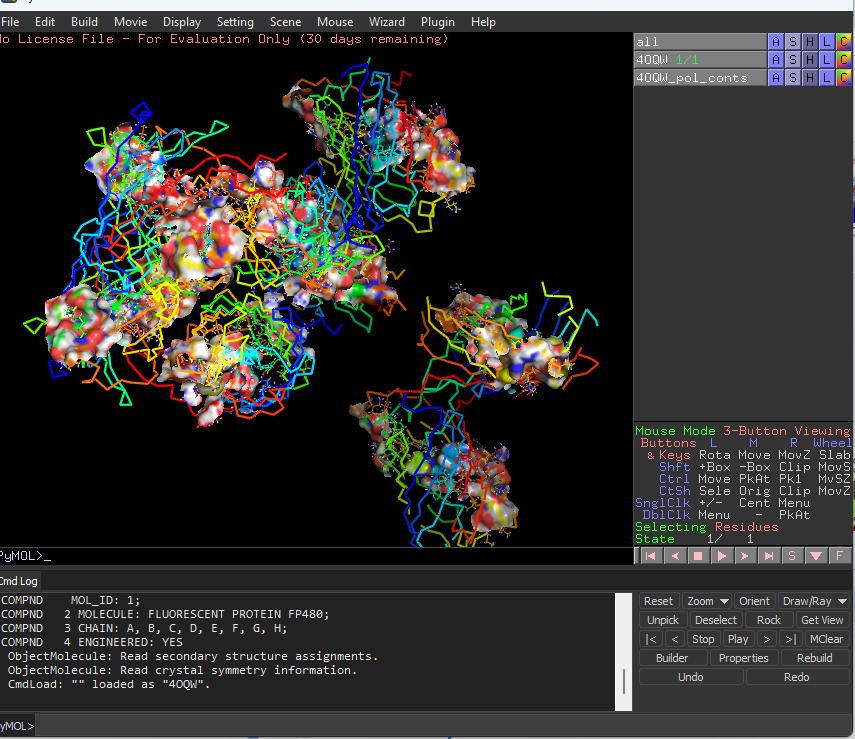
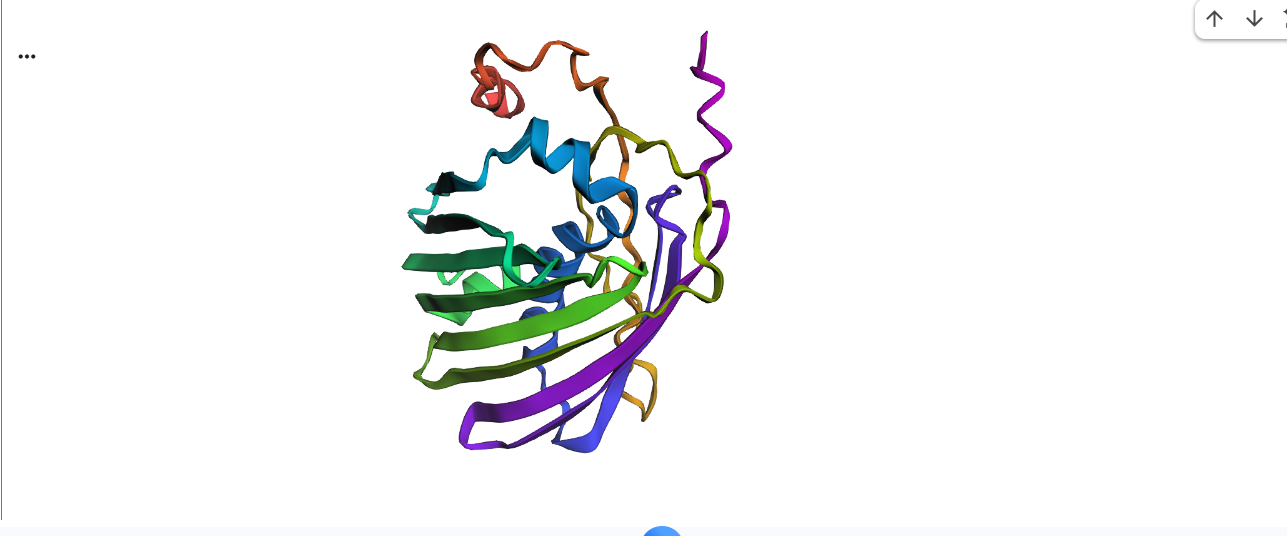
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
This represents ball and stick model of protein
This is cartoon represenattion of protein
This is ribbon representation of protein
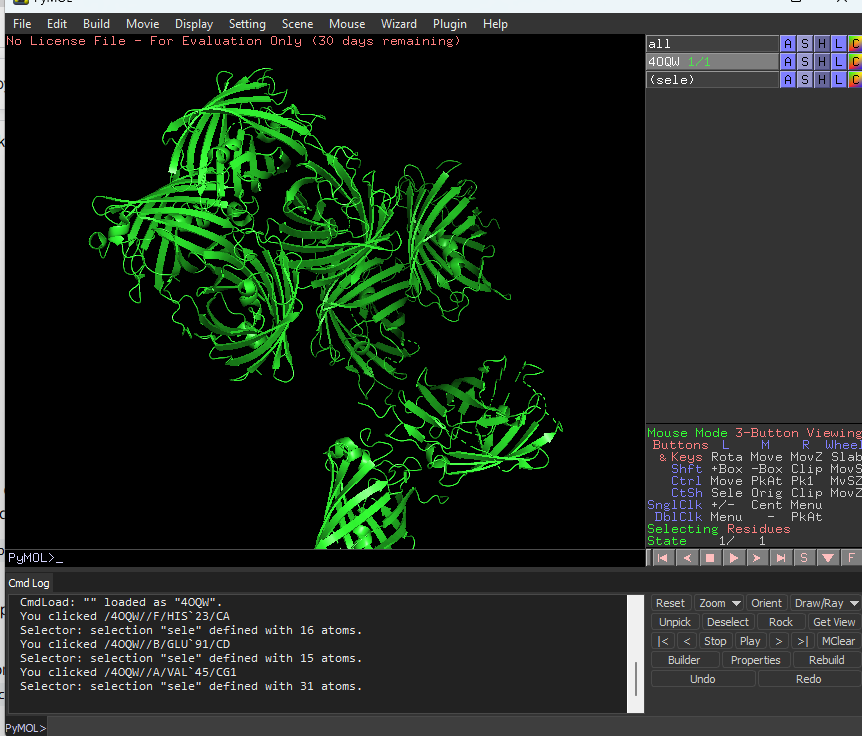
Color the protein by secondary structure. Does it have more helices or sheets?
It has more sheets.Helices are red, sheets are yellow and loops are green.
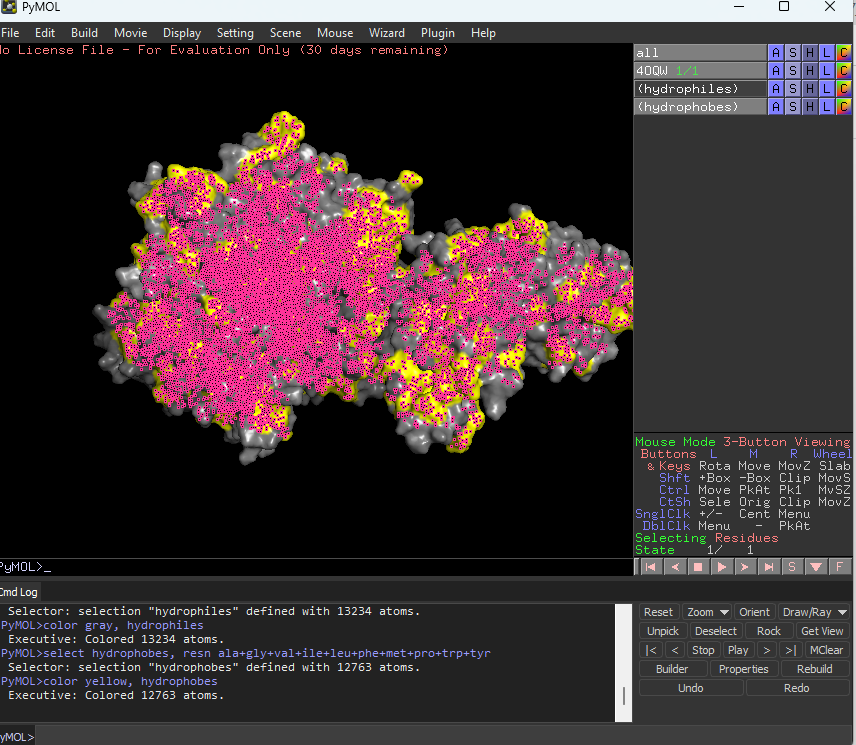
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The hydrophobic residues are yellow in colour and hydrophilic are gray in colour. This colour combination tells us that hydrophilic residues are more towards the outer side of protein and hydrophobic residues lie within the molecule buried inside.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes the protein has binding pockets for chromophore binding.The darker regions near the rainbow region is the cavity or pocket.
Part C. Using ML-Based Protein Design Tools
1.Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
2.Choose your favorite protein from the PDB
I am choosing the mCardinal far red fluorescent protein.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1 Protein Language Modeling
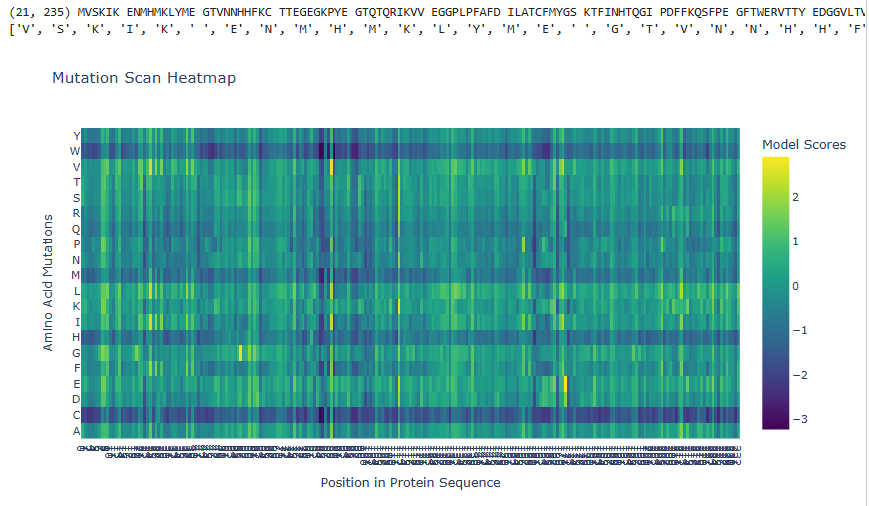
1.Deep Mutational Scans
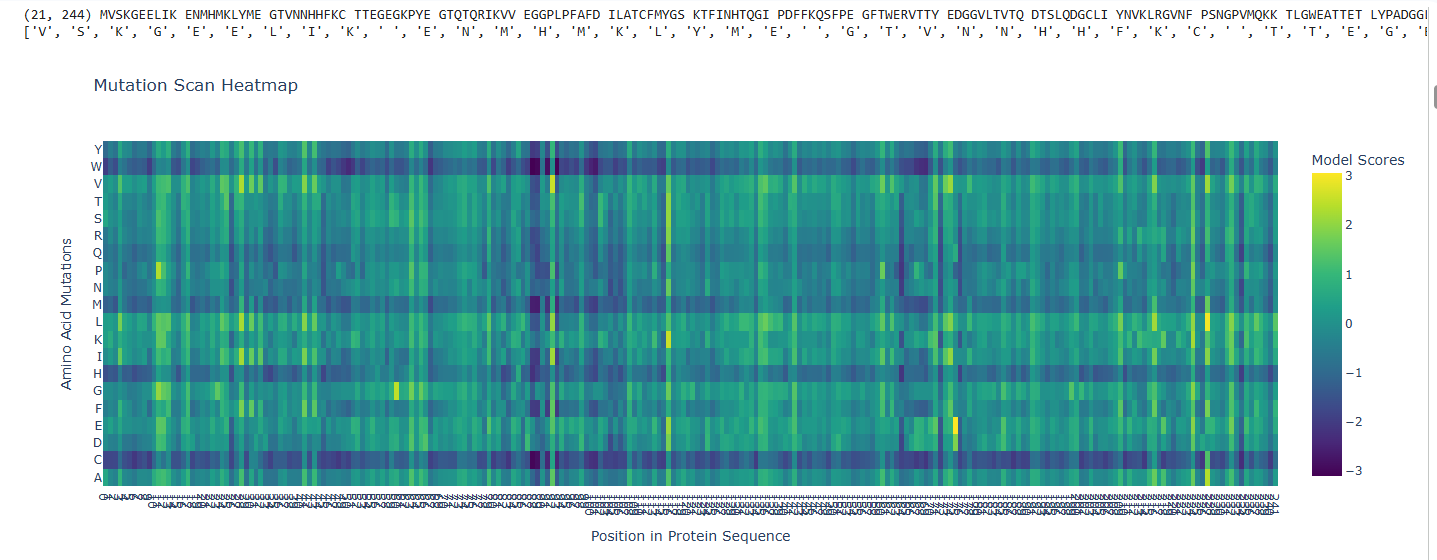
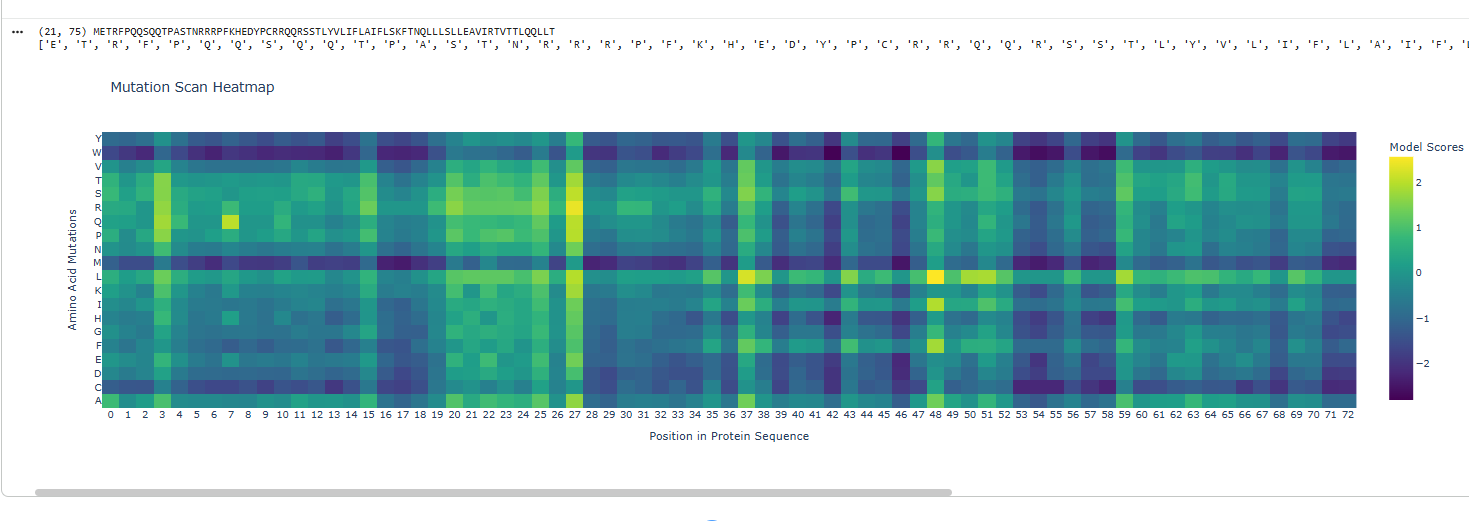
a.Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
b.Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The dark blue region corresponding to w on y- axis and m- axis. thes lead to probably disruptive mutations. This region is highly conserved. The darkest puple seen here has a negative value of -3.11.
c.(Bonus)Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
The UniProt Consortium
UniProt: the Universal Protein Knowledgebase in 2025
Nucleic Acids Res. 53:D609–D617 (2025)
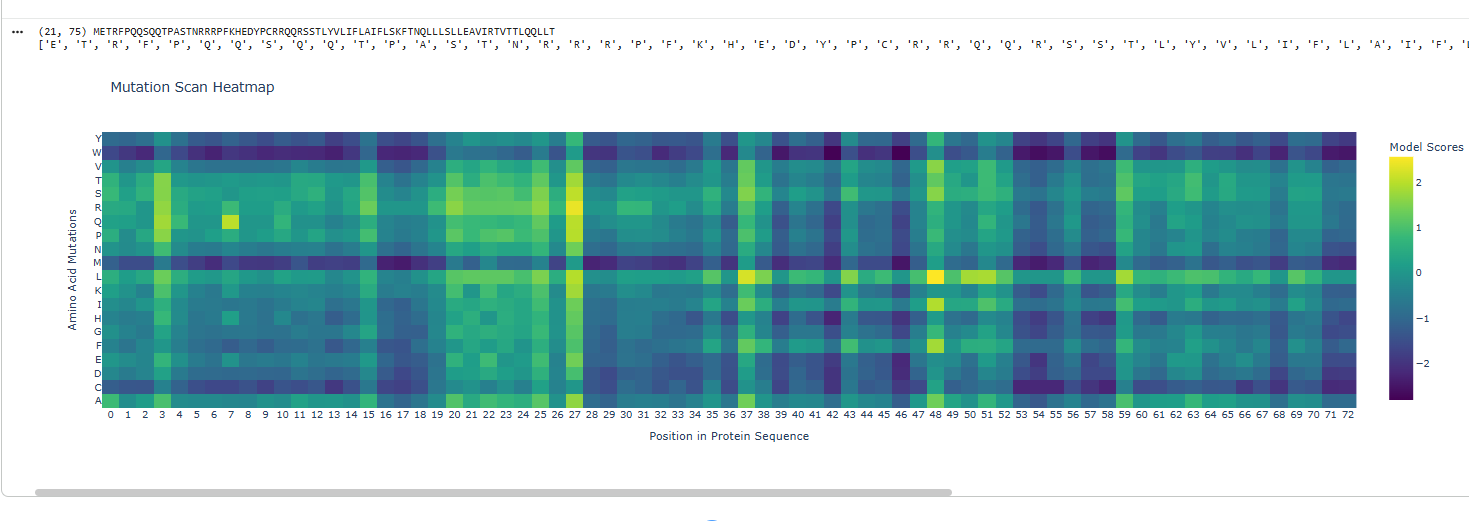
The protein sequence provided corresponds to the Circadian clock oscillator protein KaiB in cyanobacteria. It is frequently used as a test sequence for analyzing protein structure, stability, and mutations in AI-driven protein language models.
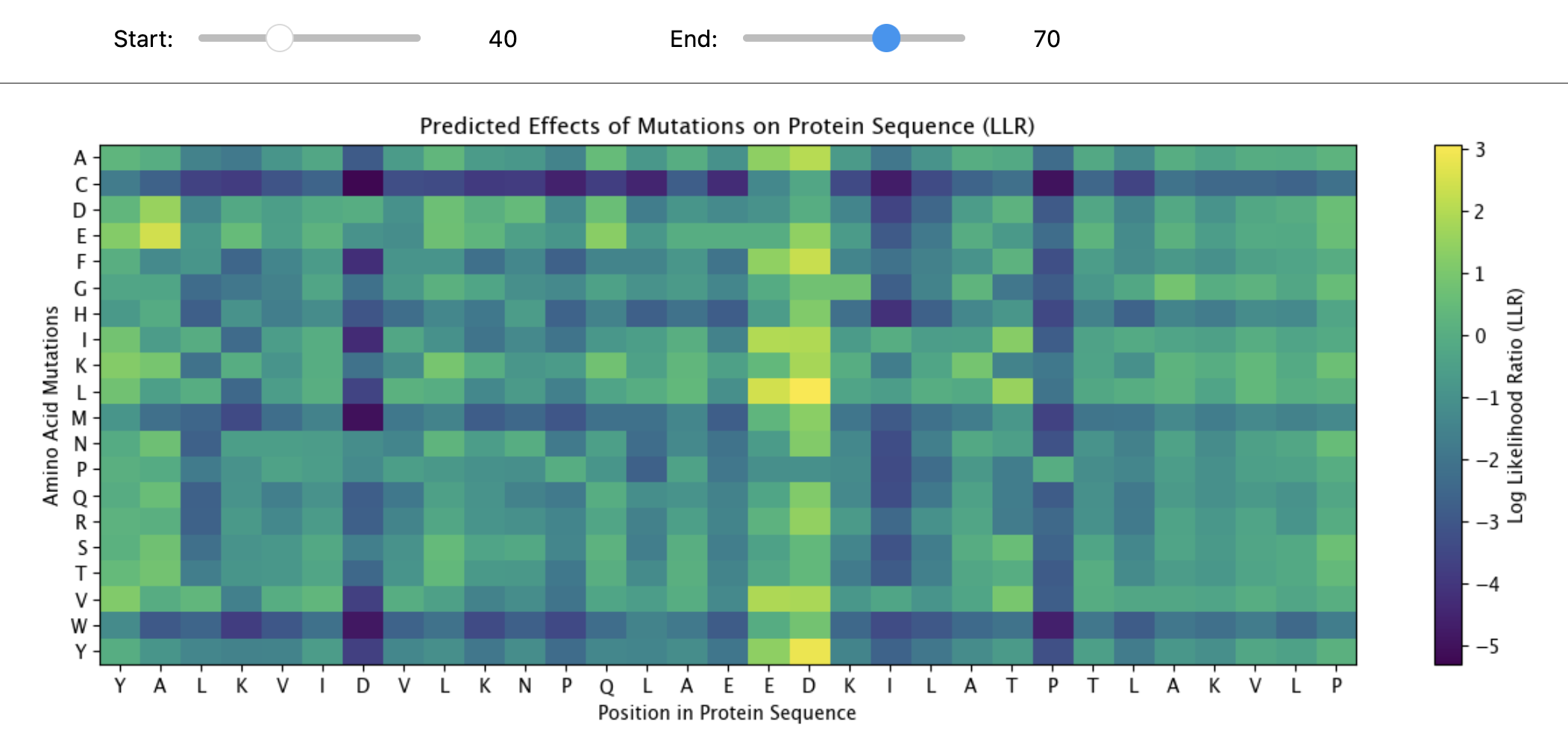
Yes, the two images are structurally similar in that they are both heatmaps designed to visualize the effects of amino acid mutations on a protein sequence.
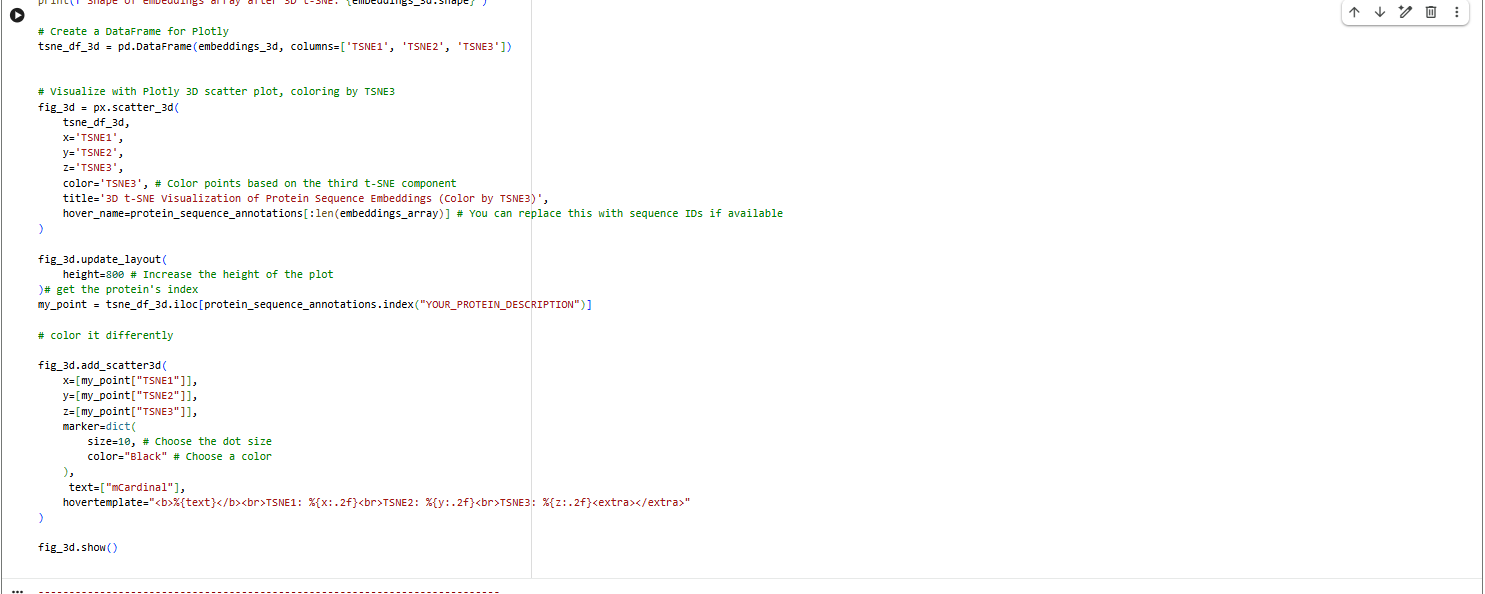
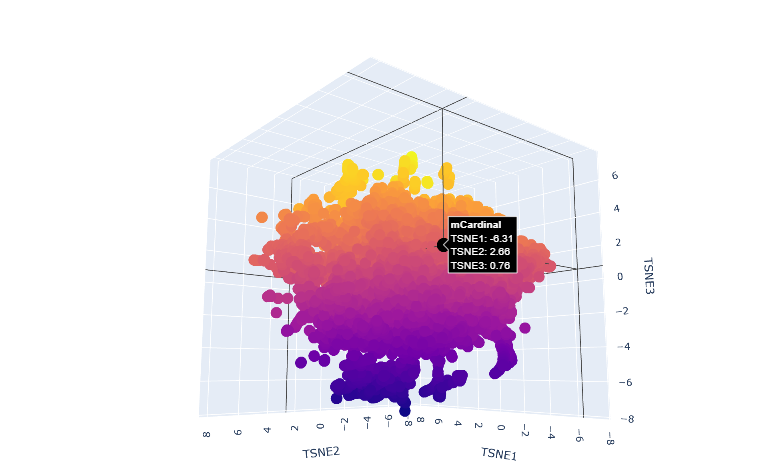
Latent Space Analysis
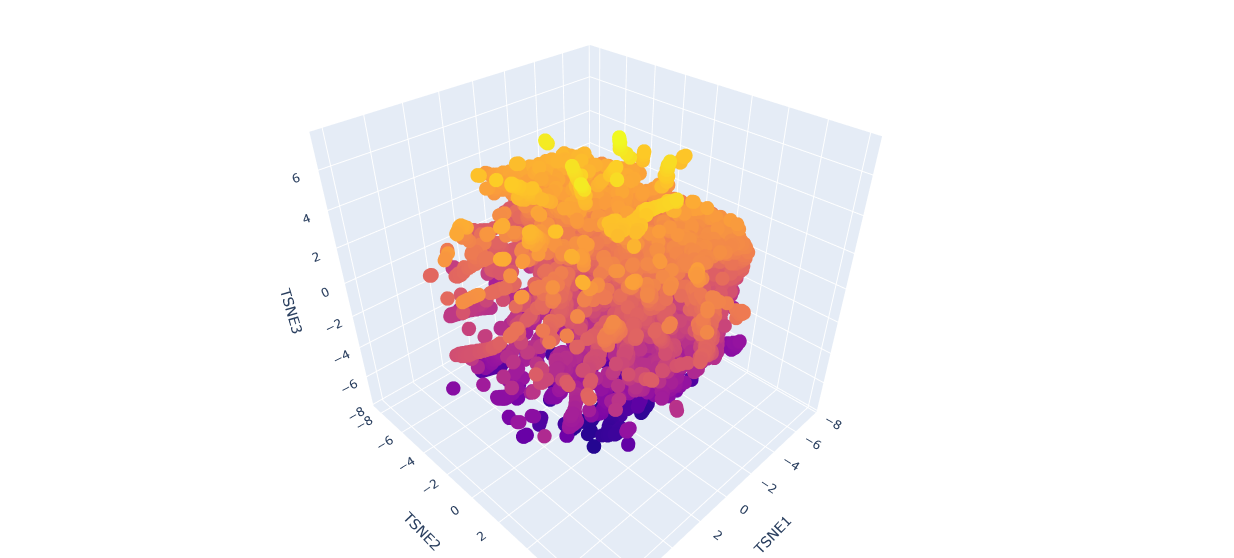
a.Use the provided sequence dataset to embed proteins in reduced dimensionality.
b.Analyze the different formed neighborhoods: do they approximate similar proteins?
The proteins in the neighbourhood approximate similar proteins.
c.Place your protein in the resulting map and explain its position and similarity to its neighbors.
The orange-to-purple gradient likely shows point density (denser orange/yellow central cloud for common mCardinal-like sequences like Vibrio cholorae.
The main orange-yellow hexagonal group (tSNE1 ≈ -1 to 3, tSNE2 ≈ -2 to 2) likely includes related far-red/red emitters like mNeptune, eqFP578, mKate2, or TagRFP, derived from similar Anthozoa.
C2 Protein Folding
Folding a protein
1.Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I faced repeated errors while folding the protein.
Yes the predicted co-ordinates match the original structure.
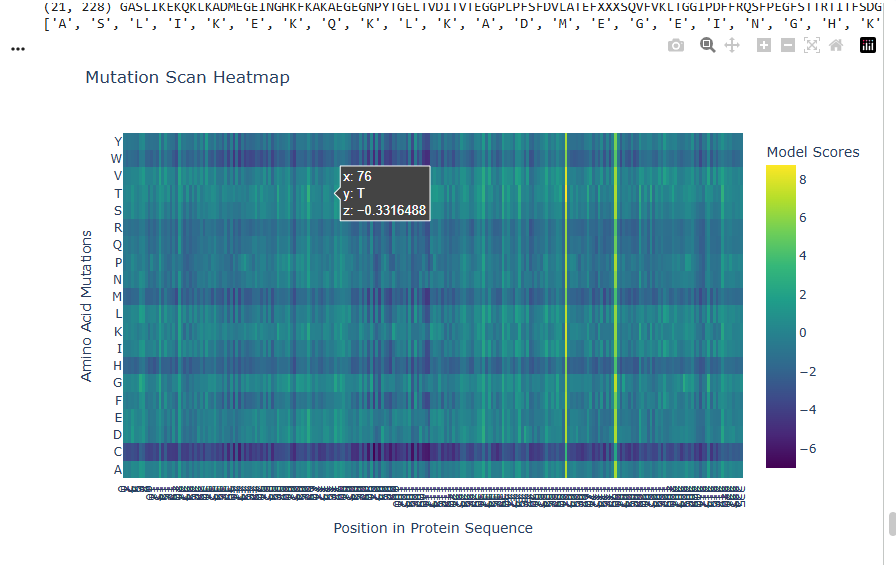
2.Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I deleted 4 residues GEEL from the beginning of sequence and did a mutational scan.
Yes the proteins still folds back into its native structure after a segment of deletion in sequences.It is resilient to mutations.
C3 Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1.Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
2.Input this sequence into ESMFold and compare the predicted structure to your original.
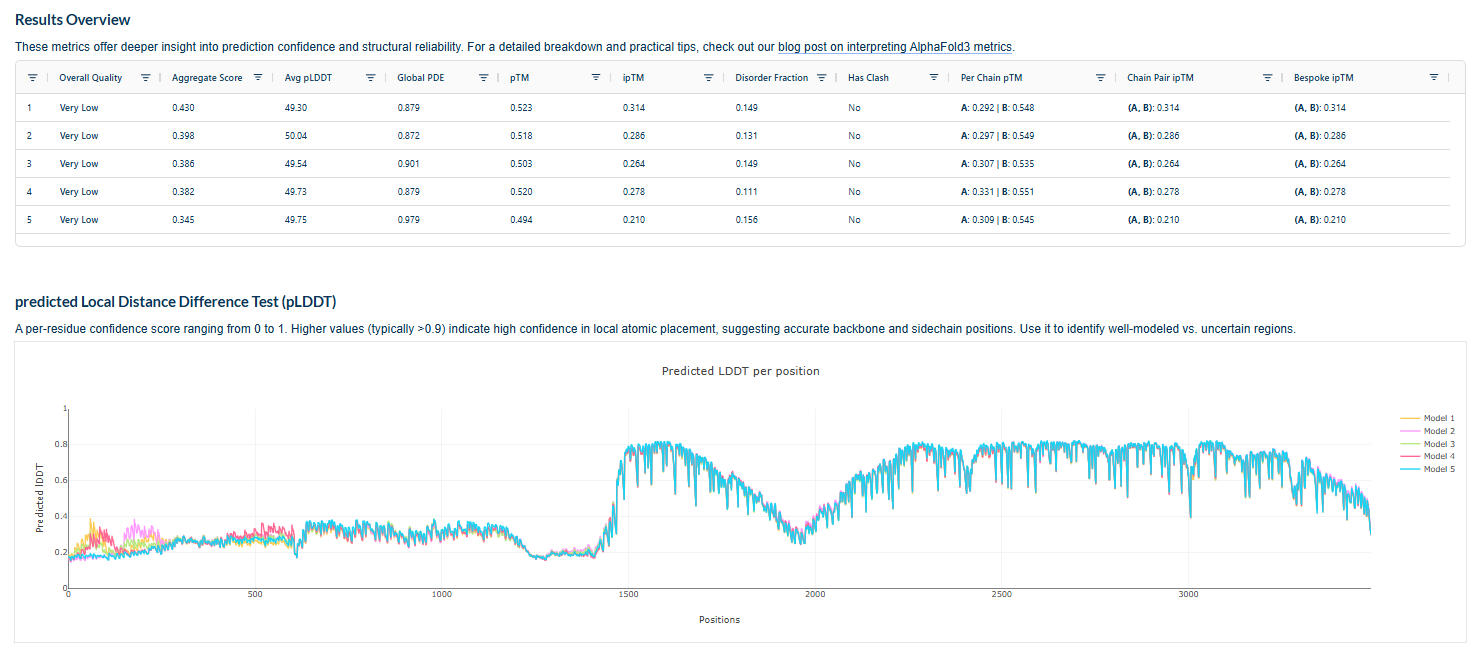
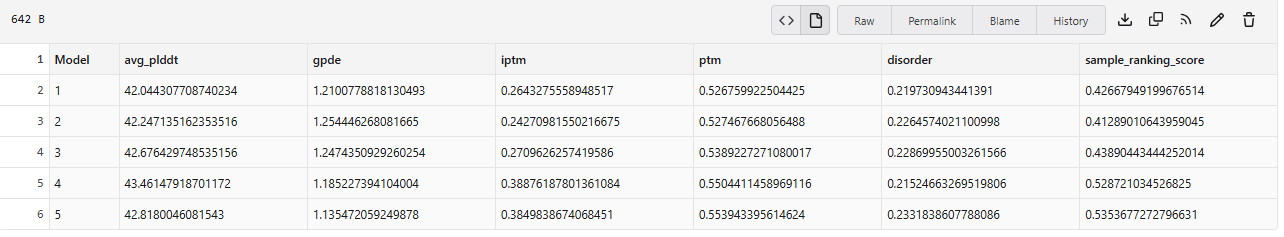
ESMFold 3D Structure: The predicted 3D structure had a ptm score of 0.785 and an average pLDDT of 73.443, indicating a generally good confidence in the protein’s fold and supporting that the designed sequence folds into a stable, well-defined structure.
Visual inspection of the 3D structure provided qualitative assessment of its compactness, secondary structure elements, and overall tertiary fold, confirming the design folds as expected.It is sinmilar to original structure.
Part D. Group Brainstorm on Bacteriophage Engineering
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
The group plans to stabilize the protein by making it fold without DnaJ chaperone protein of host.
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
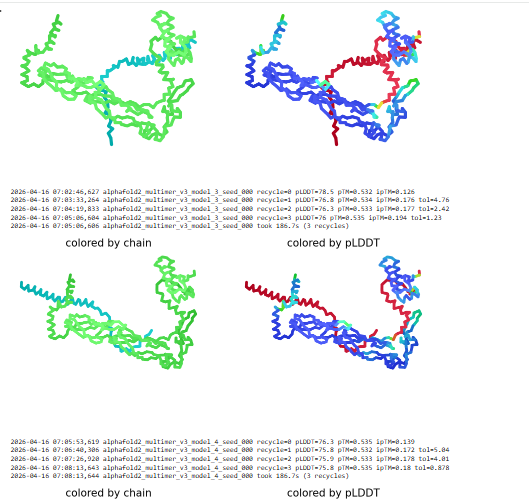
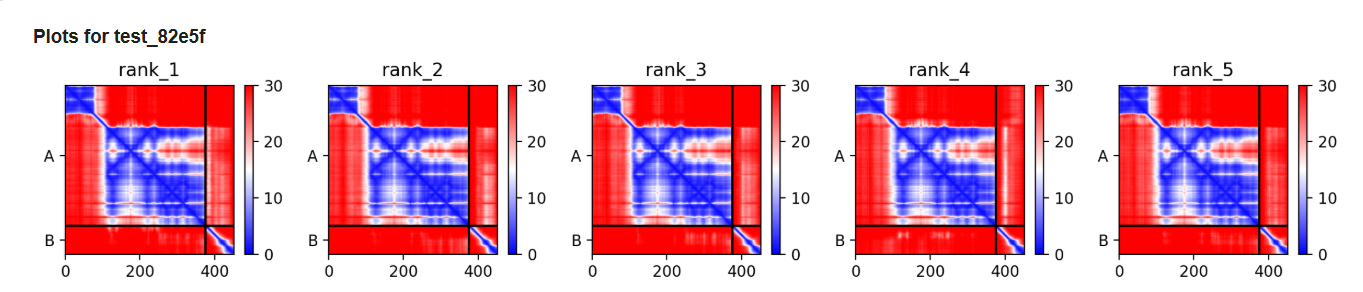
This can be done by predicting mutations using mutational scans and protein ESM2 fold to predict the fold after the mutation. This can be validated using Alphafold multimer.
Other generative models can be used to make the protein co-fold with the DnaJ protein.This can be studied using alphafold multimer.
The lysis proteins could be studied and probably a mechanism to apply to co-fold the protein with other chaperones and analyse the fold and stability of mature protein against E.coli host. pBLAST may be used to study different lysis proteins and how it has evolved and can be analysed for using similar mechanisms but folding with a different chaperone.
Why do you think those tools might help solve your chosen sub-problem?
The major goal is to prevent the interaction of DnaJ with lysis protein as E.coli is mutating this mechanism leading to antimicrobial resistance. So the aim is to either make the protein fold independent of DnaJ of host by mutations or co-folding or by adding similar chaperones for it to fold.
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”)
The protein might not fold in a desired way, may lead to loss of function of protein.
Since we are not sure about the interactions of bacteria with proteins co folded with other chaperones.
Include a schematic of your pipeline.
5.Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
I want to mutate the lysis protein at three parts N-terminal, C-terminal and the middle part and see how it co folds with DnaJ protein.I will use esmfold and Alphafold3 to validate the predicted structure.
References
1.Colab notebook to count amino acids.
2.The UniProt Consortium. (2025). UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Research, 53(D1), D609-D617.The UniProt Consortium. (2023). UniProt Tools: BLAST, Align, Peptide search and ID mapping. Current Protocols, 3(3), e697.
Schrödinger, LLC. (2025). The PyMOL Molecular Graphics System (Version 2.6.2). https://www.pymol.org/ generating the protein structure visuals
H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne, The Protein Data Bank (2000) Nucleic Acids Research 28: 235-242 https://doi.org/10.1093/nar/28.1.235. RCSB.org - for mCardinal structure
Lambert, TJ (2019) FPbase: a community-editable fluorescent protein database. Nature Methods. 16, 277–278. doi: 10.1038/s41592-019-0352-8-FP Base-mCArdinal Protein Sequence
HTGAA_ProteinDesign2026.ipynb for esm2 fold, latent space analysis, mutational heat map and inverse folding
2.Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
3.Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
4.To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
5.Record the perplexity scores that indicate PepMLM’s confidence in the binders.
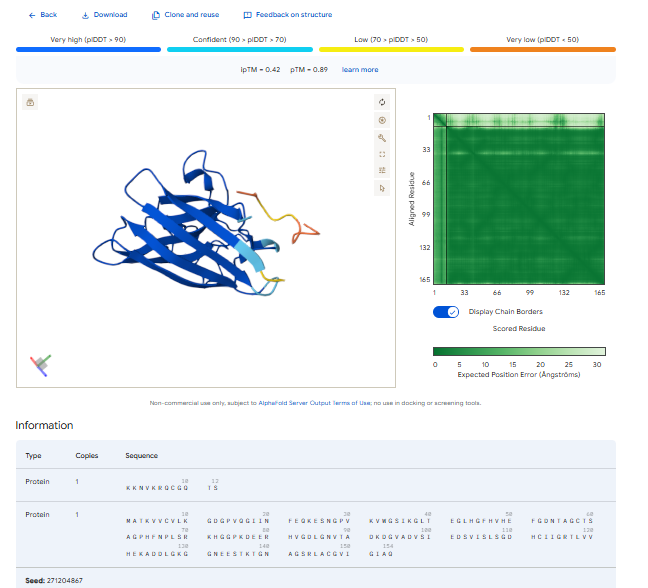
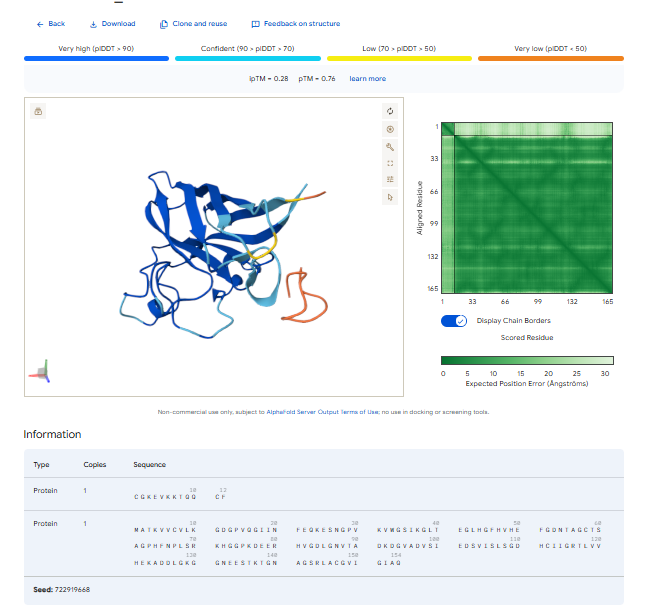
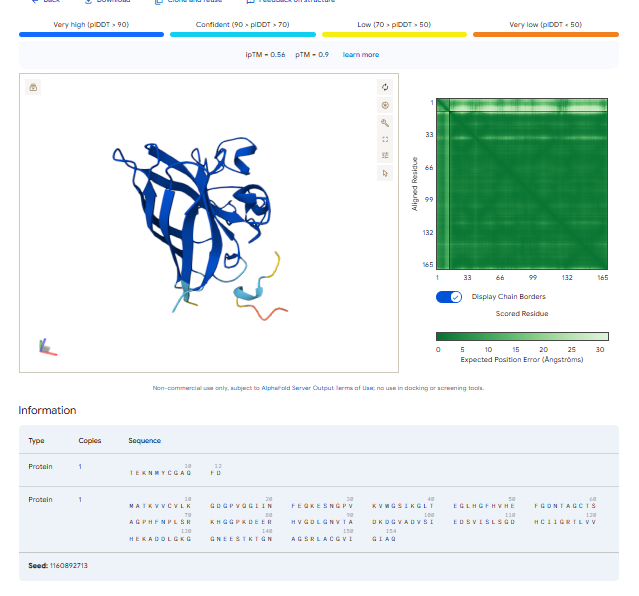
Part 2: Evaluate Binders with AlphaFold3
1.For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
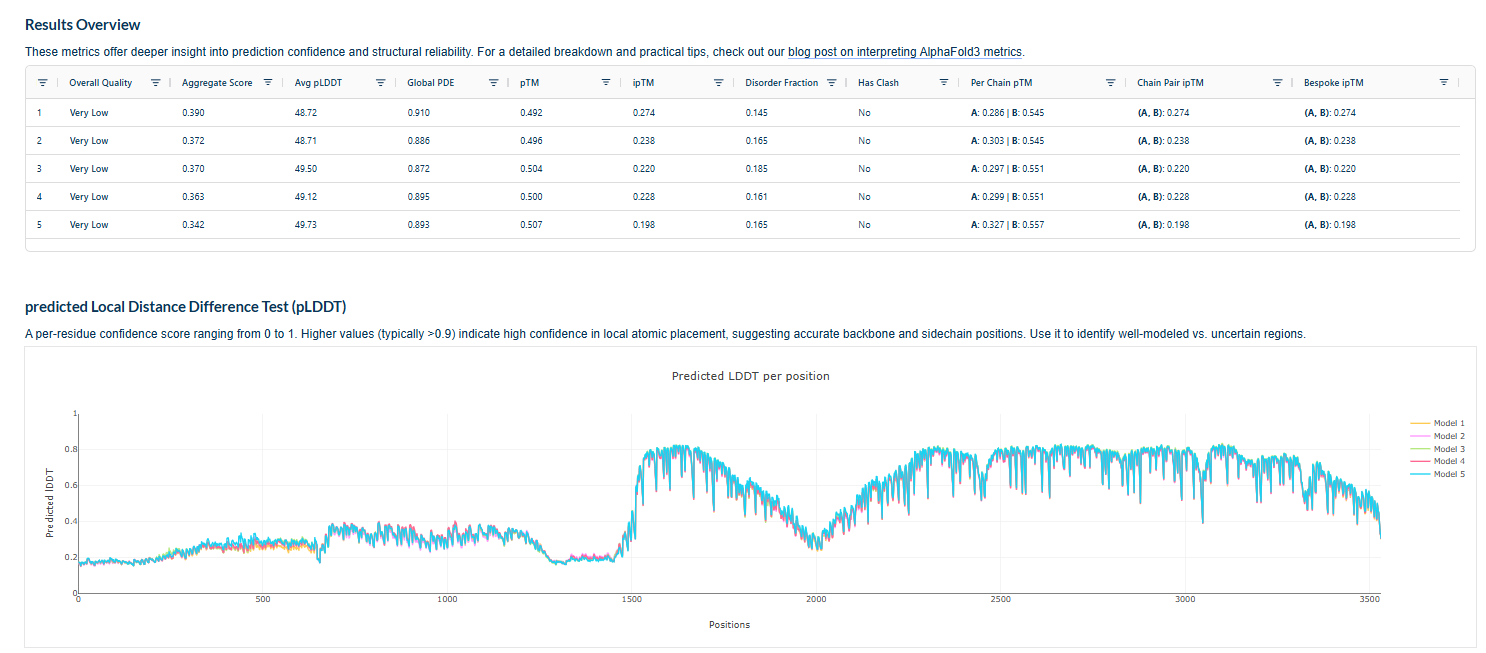
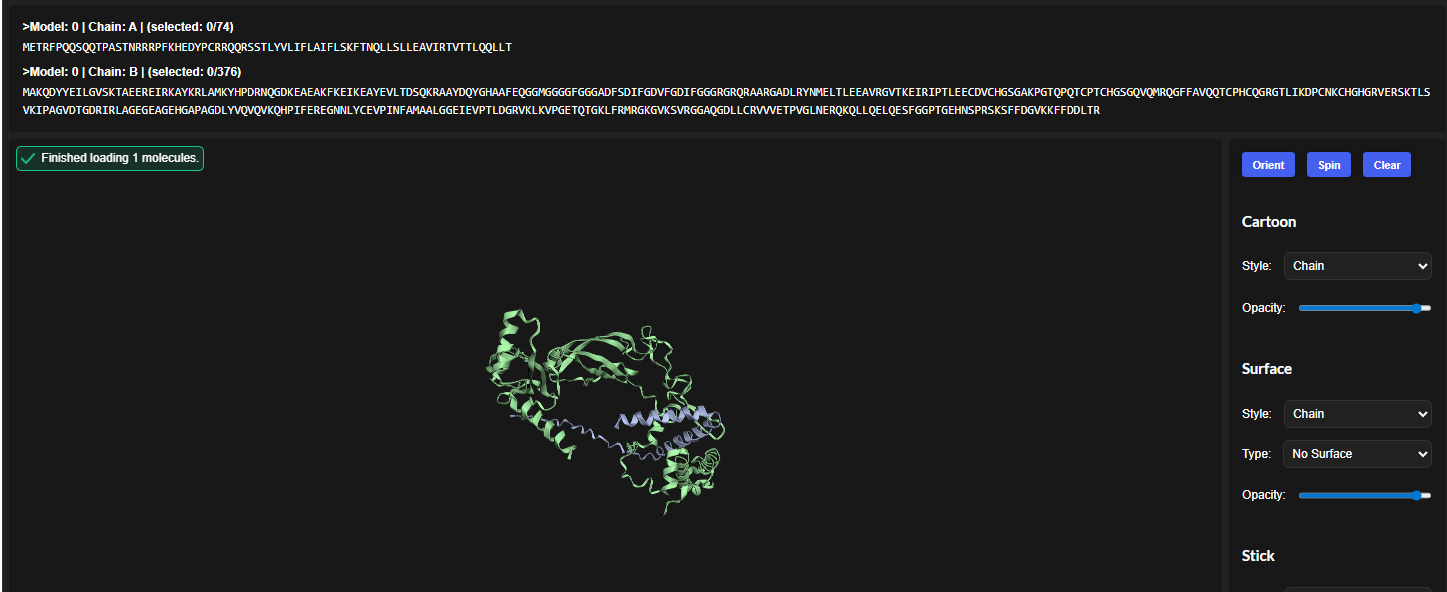
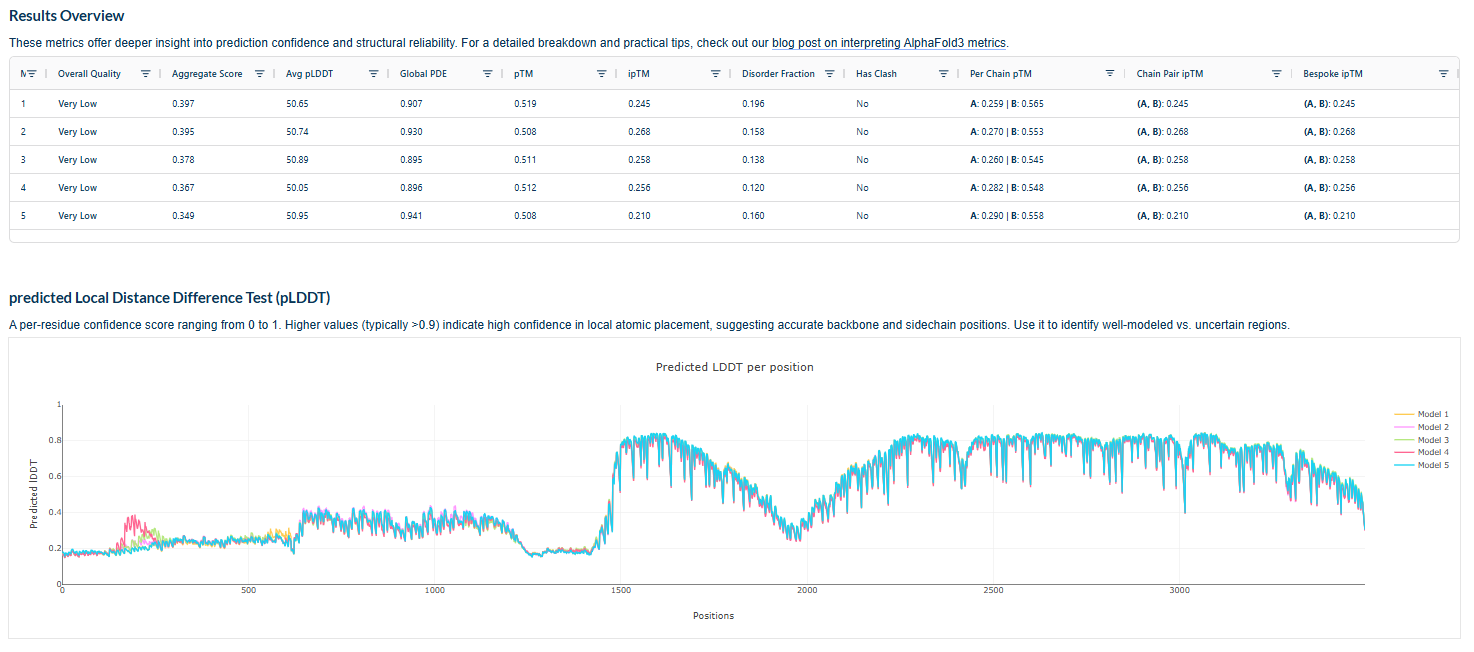
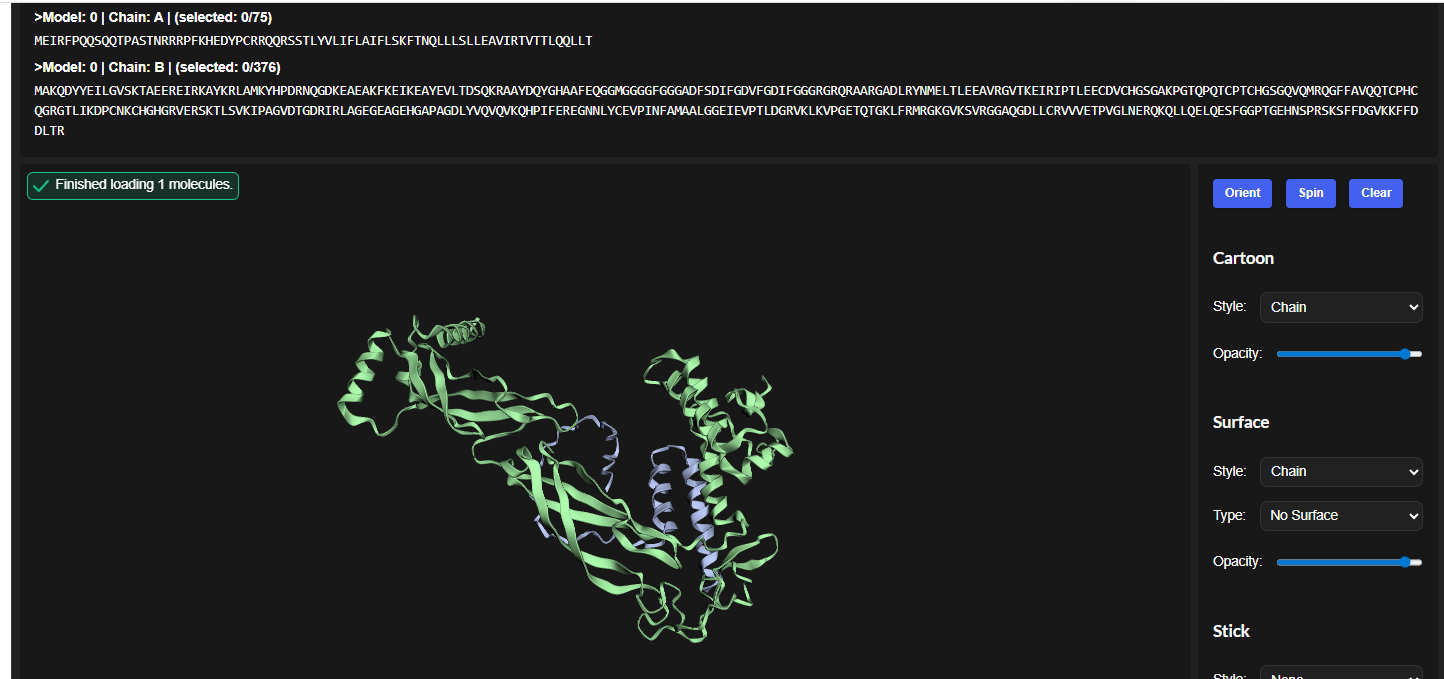
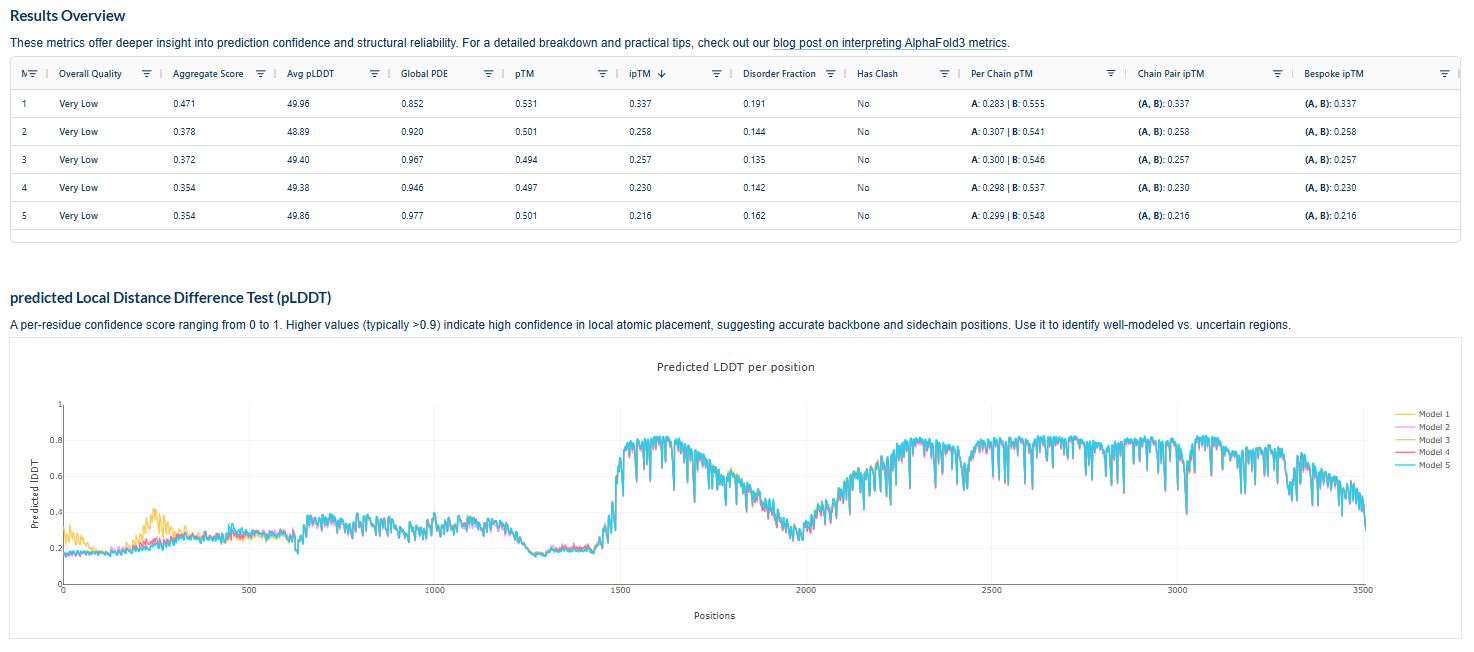
3.Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In all the figures the peptide appears to bind to the β-barrel region or approach the dimer interface. It appears surface bound.
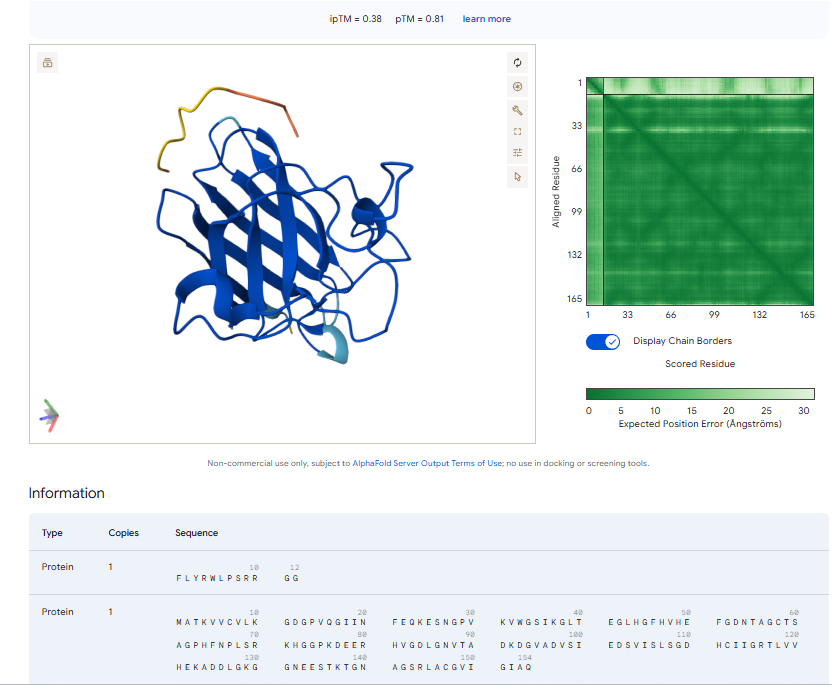
4.In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
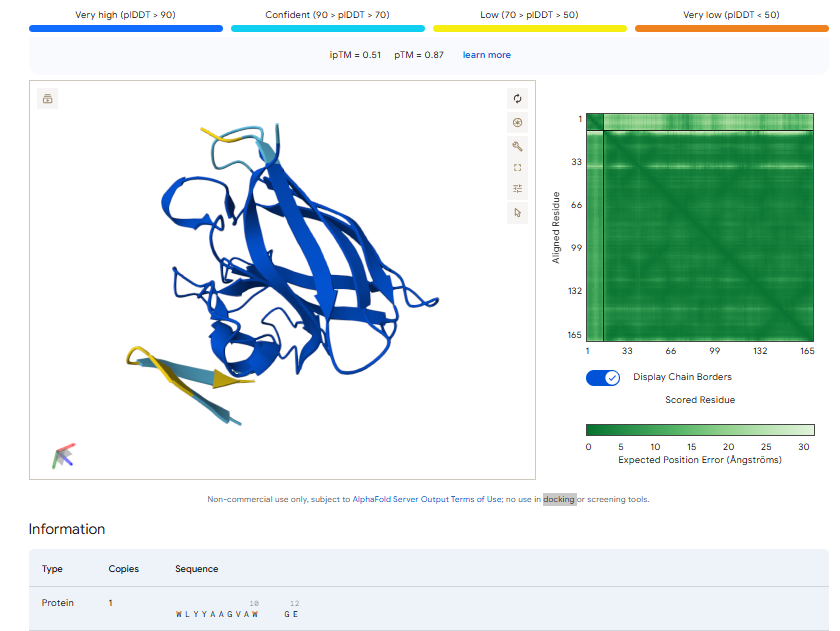
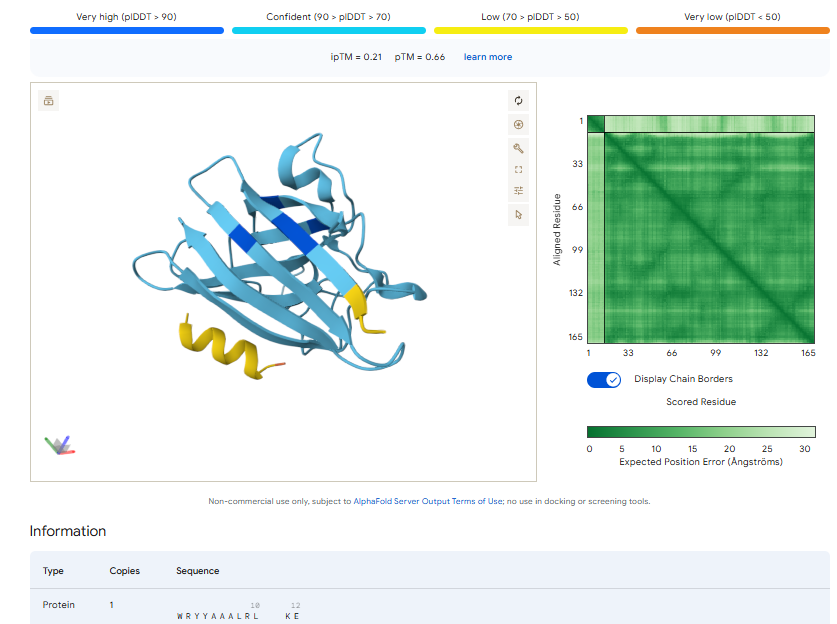
The above image of known binder shows ipTM value of 0.38 and pTm of 0.81.
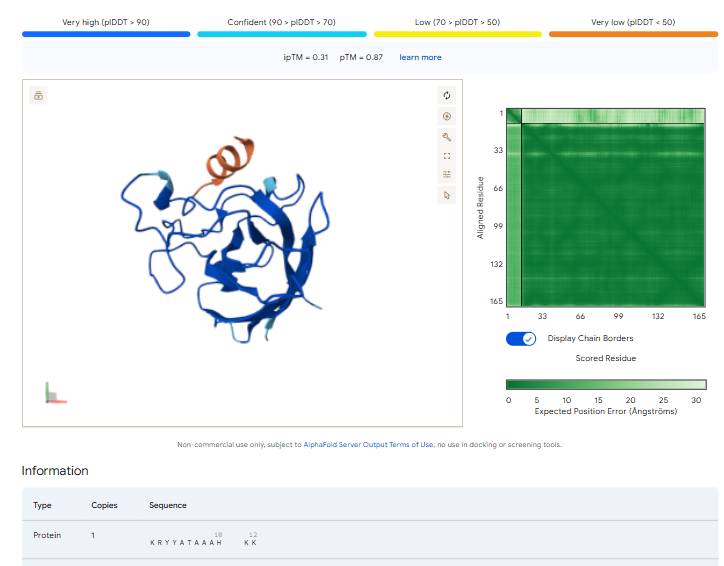
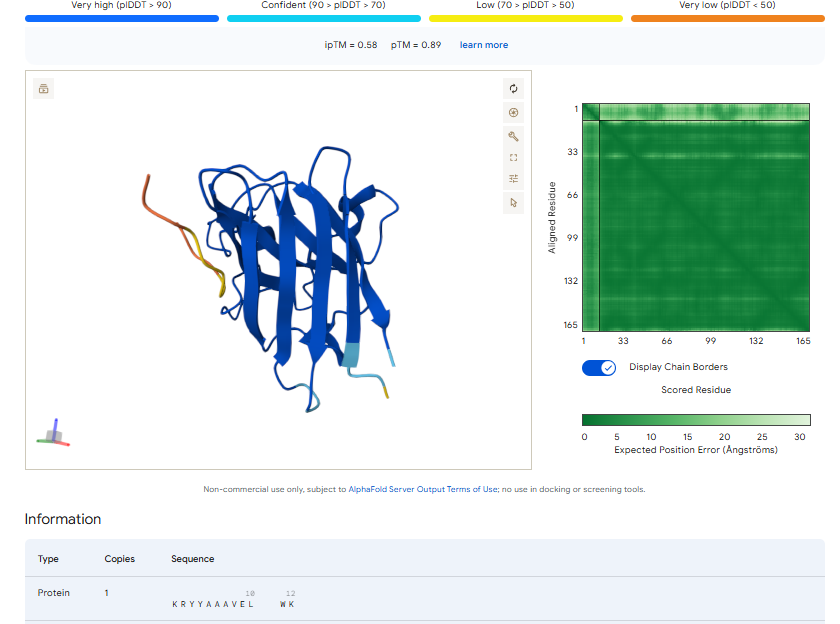
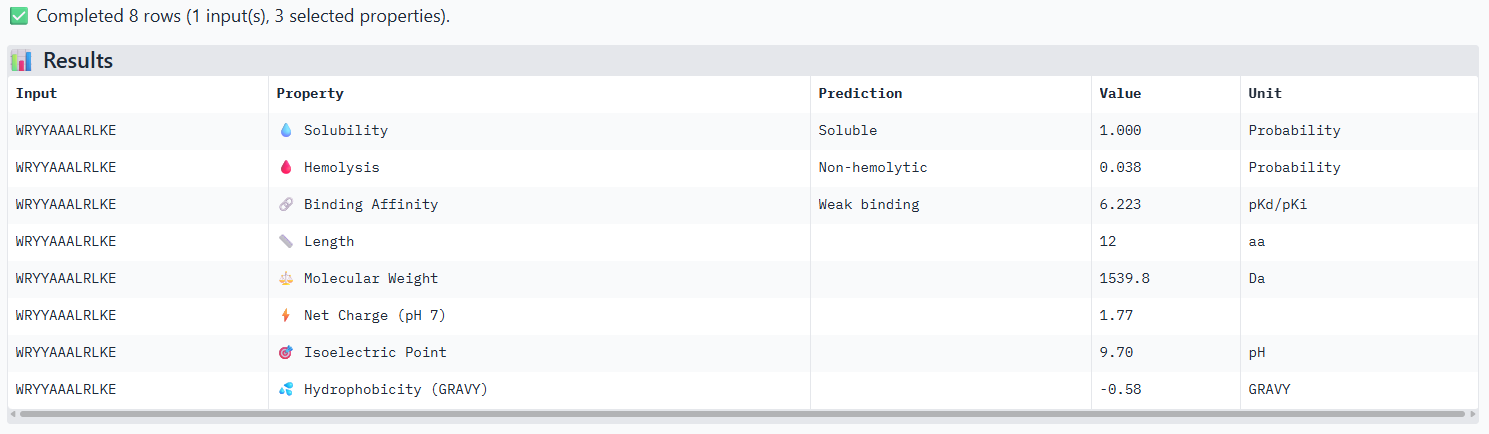
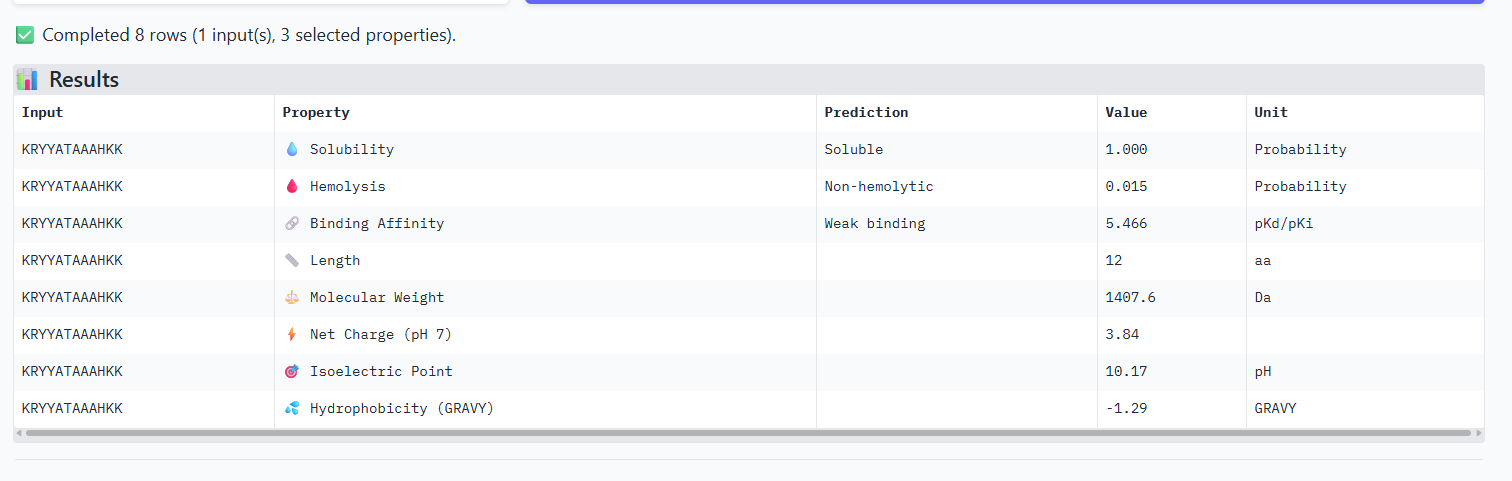
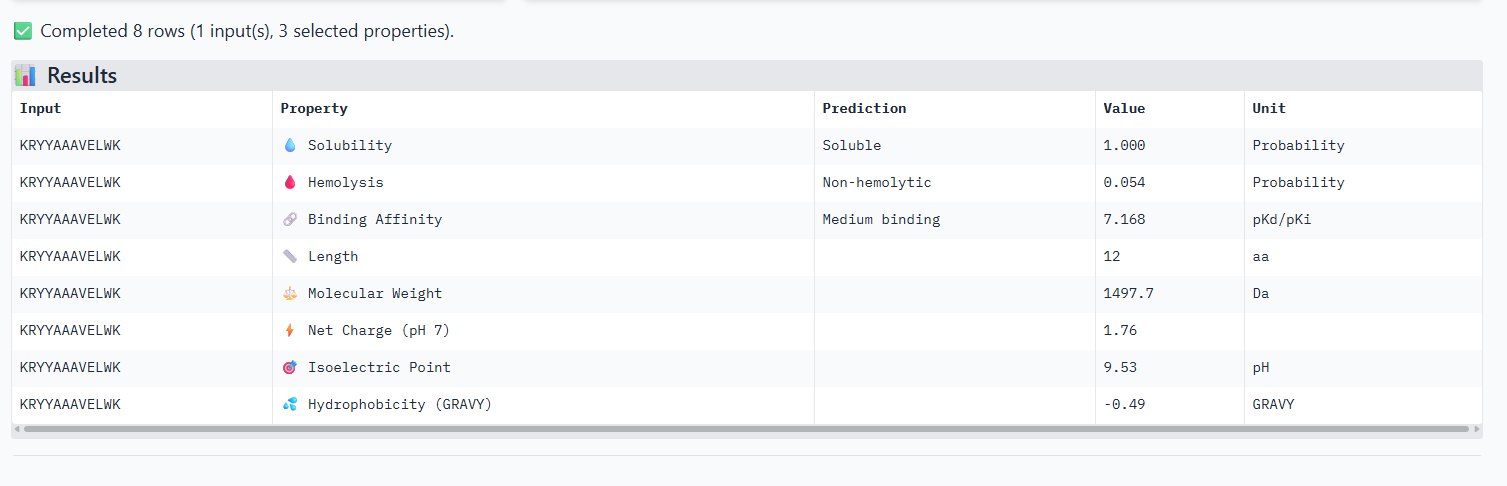
The ipTm values observed for other binders only one comes close to ipTM of 0.31 and pTm of 0.87.The rest are all binders have higher ipTM in the range of 051-0.60.The (KRYYAAAVELWK) due to its balanced high ipTM, affinity, solubility, and specificity for SOD1 A4V—ideal for therapeutic prototyping in cell-free systems or biosensor integration may be a good hit.
None deeply penetrate pockets, typical for PepMLM 12-mers on structured targets like SOD1.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
1.Paste the peptide sequence.
2.Paste the A4V mutant SOD1 sequence in the target field.
3.Check the boxes
1,Predicted binding affinity
2.Solubility
3.Hemolysis probability
4.Net charge (pH 7)
5.Molecular weight
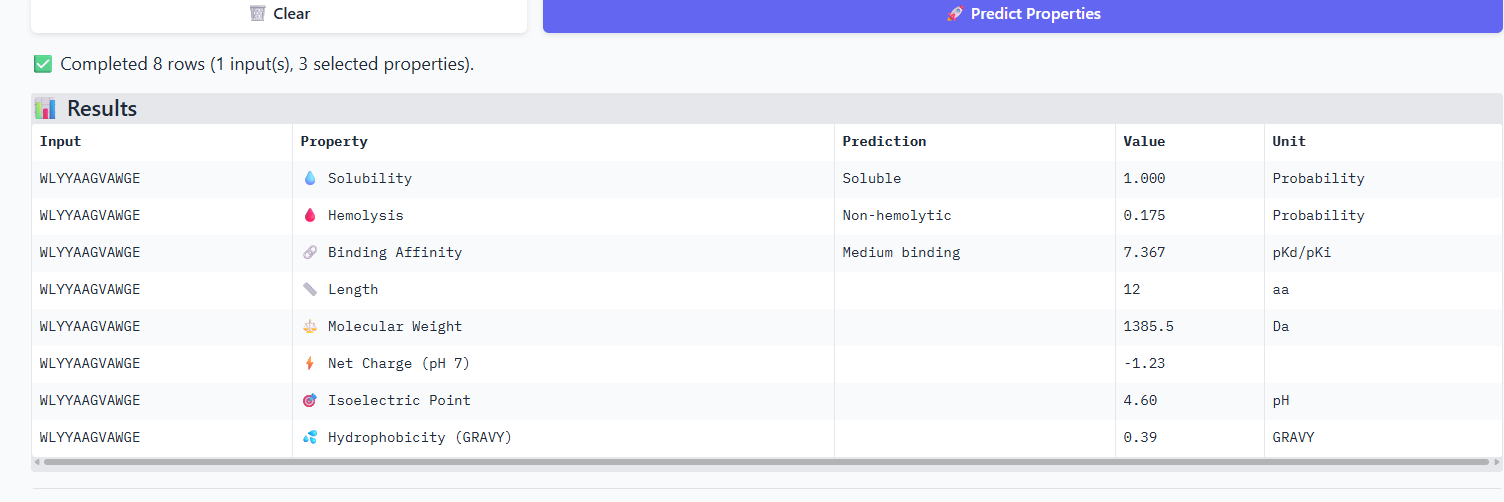
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
The iPTM scores and binding affinity are related, the highter the score the higher the bonding affinity.I could not find any strong binder only found medium binding binders. All of them were soluble and non-haemolytic.
Choose one peptide you would advance and justify your decision briefly.
I would like to go with KRYYAAAVELWK binder as it has 7.168 binding affinity, higher than all four. Has high specificity for target SOD1 mutant protein. It has high iPTM score of 0.58.
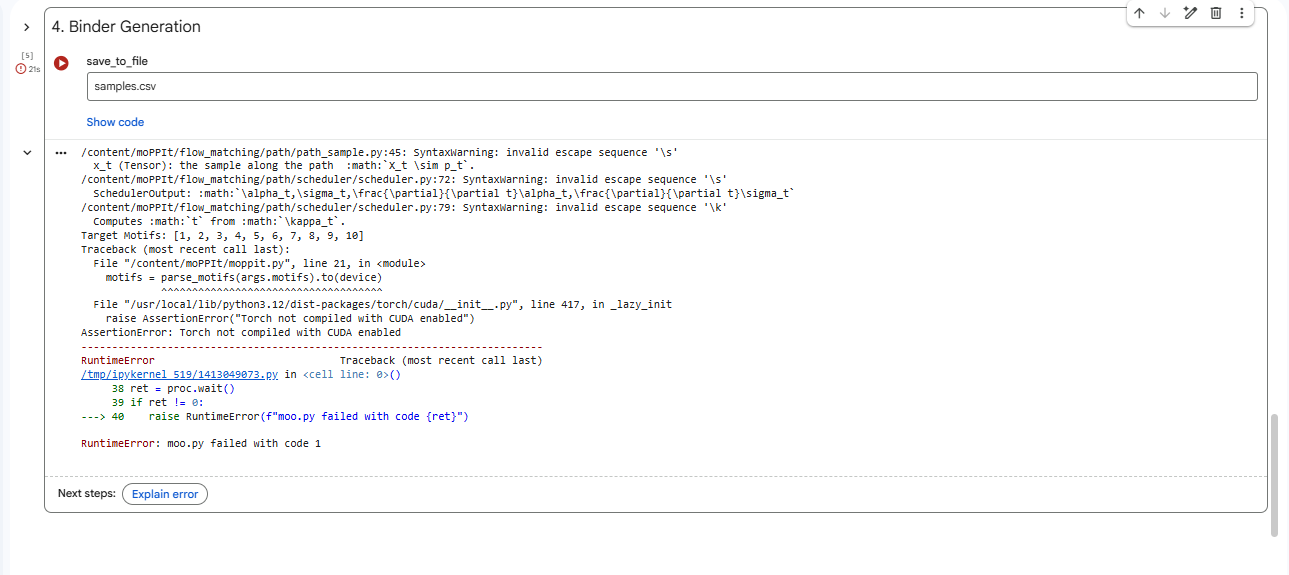
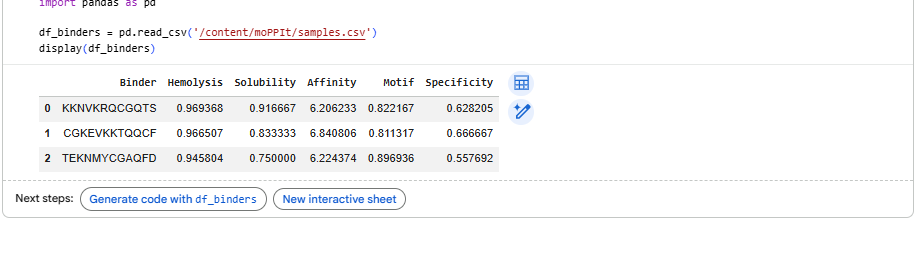
Part 4: Generate Optimized Peptides with moPPIt
4.After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Higher values (closer to 1) indicate better performance; affinity measures binding likelihood to the target motif, the specificty of binder 1 is best compared to the other two.
Evaluating peptides like KKKRGGST and TEKVQAGCF from moPPIt for clinical advancement requires a rigorous, multi-stage preclinical pipeline to confirm computational predictions and ensure safety/efficacy.Sequences can be refined using ProteinMPNN for improved affinity/specificity.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
Part 0: Sign-up to Boltz Lab
Part 1: Structural Predictions in the Sandbox
Running Your Three Predictions
Navigate to the Boltz Sandbox at lab.boltz.bio and log in to your account.
Go to Sandbox → New Prediction
Name this BRD4 binder JQ1
Select ‘Complex’, add ‘Sequence from RCSB’, and add 3MXF
Continue through Constraints (not needed for this example), and select Jq1 as the Binder
for an affinity prediction.
Submit the prediction.
Use the ‘Duplicate Prediction’ in the results review, and remove the small molecule.
Add in the SMILES for the Hit and Lead.
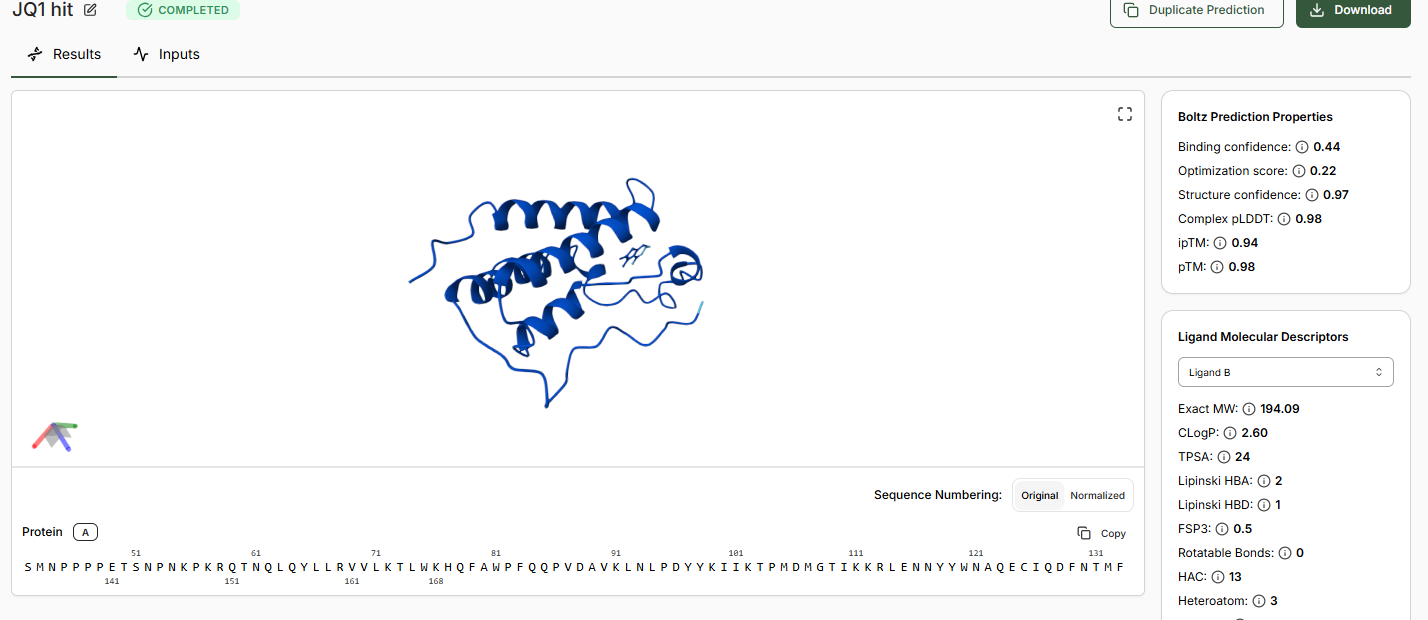
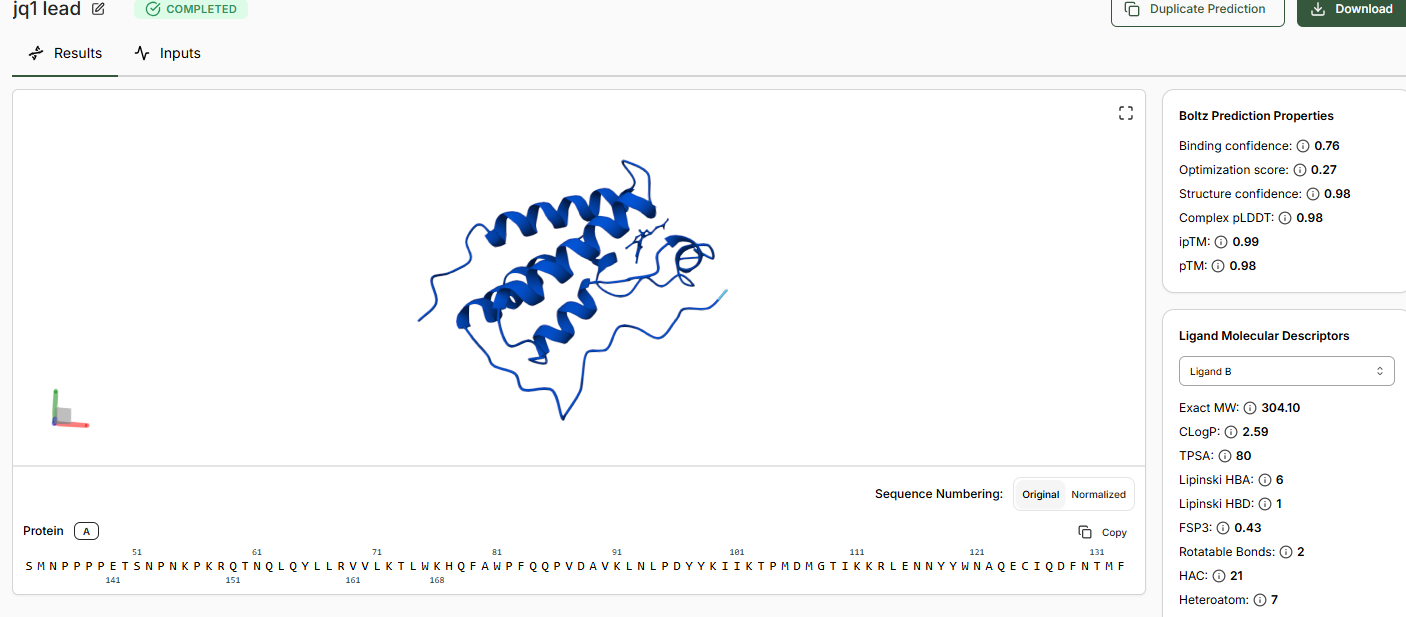
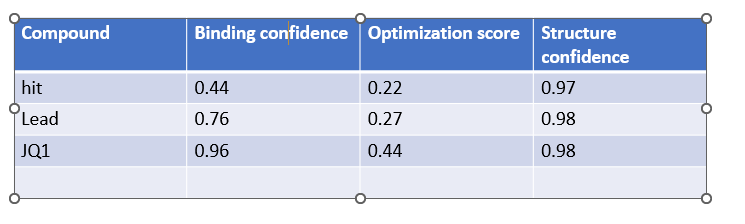
When predictions complete, record your results in the table below
Does Binding Confidence increase as you move from hit to clinical candidate? What would
you expect, and why might it deviate?
Yes the binding confidence increases from hit to clinical candidate.
The goal of drug discovery is to incraese the binding affinity and selectivity of the drug without comprimising on efficacy,potency, pharmocological properties, its a balance of all these properties.
It might deviate due to the in vitro environment assay not matching with the complex environment in body and due to the balancing of all drug properties like binding strength, potency etc.
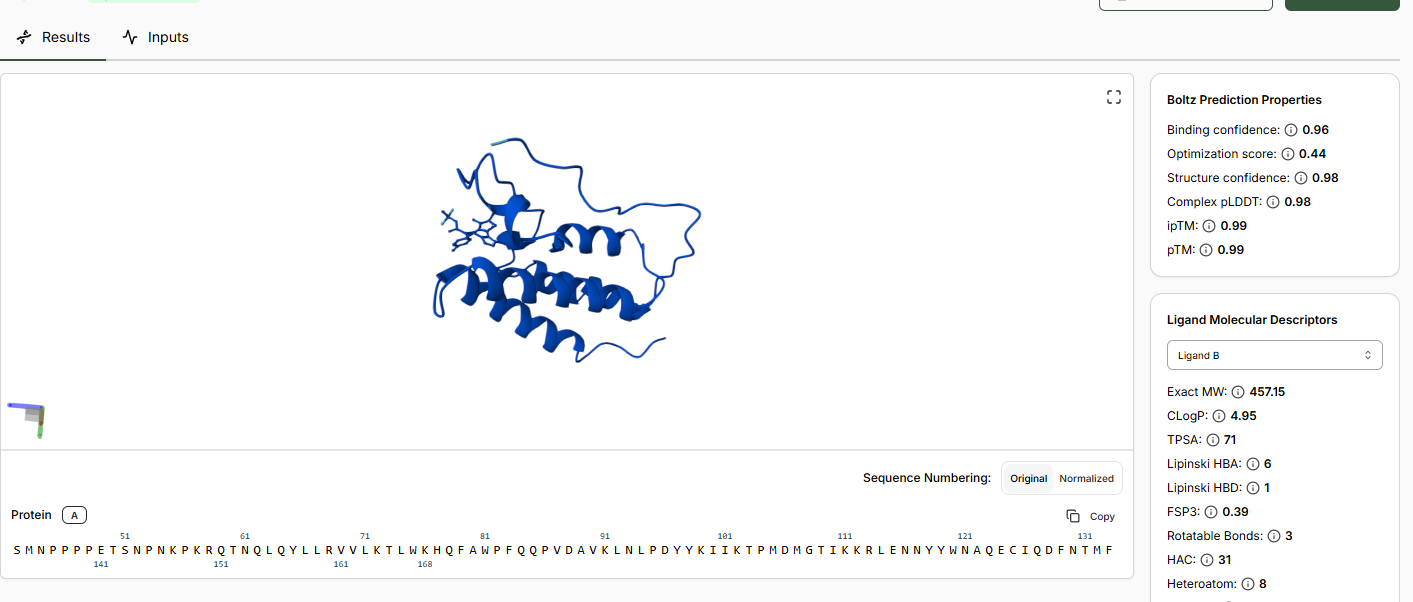
•Inspect the predicted binding pose for JQ1. Can you identify potential key binding
interactions.
The approach is there towards the helices of the BRD4 domain.
•Compare the Optimization Scores. How do the scores compare for JQ1 vs the Lead.
The optimization scores are 0.27 for lead and 0.44 for JQ1. The difference in the values tell us that the lead has initial activity but compared to JQ1 requires more modifications for binding affinity, selectivity and pharmocological properties.
Part 2: Setting Up a BRD4 Design Project
Now you will create a small molecule Design Project - the Boltz Lab workflow for virtual screening
and lead optimisation. We will set up BRD4 as a target using the clinical candidate as our structural
reference.
2.1 Creating the Target
From the dashboard, create a Design Projects via ‘New Project’
Name your project: ‘BRD4 Workshop '
Select ‘Small Molecule’
Click Add Target and add the protein structure as in the Sandbox using PDB code 3MXF
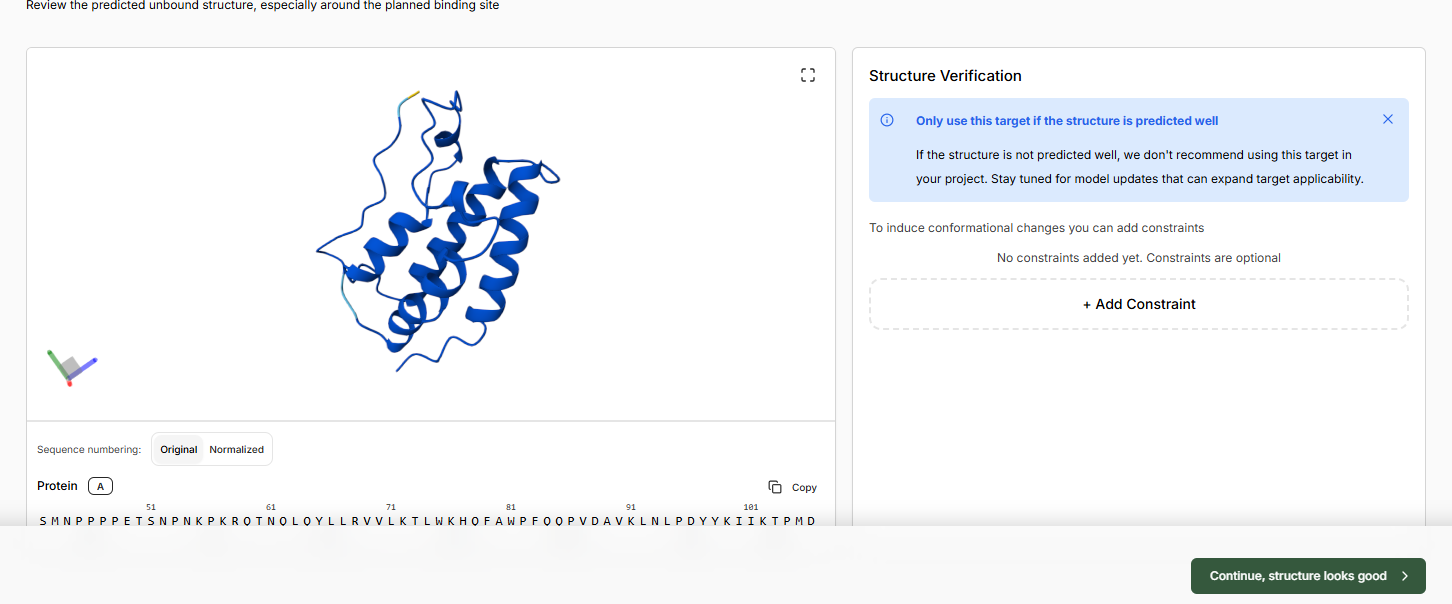
Continue and let the apo structure complete. Continue if the structure looks good.
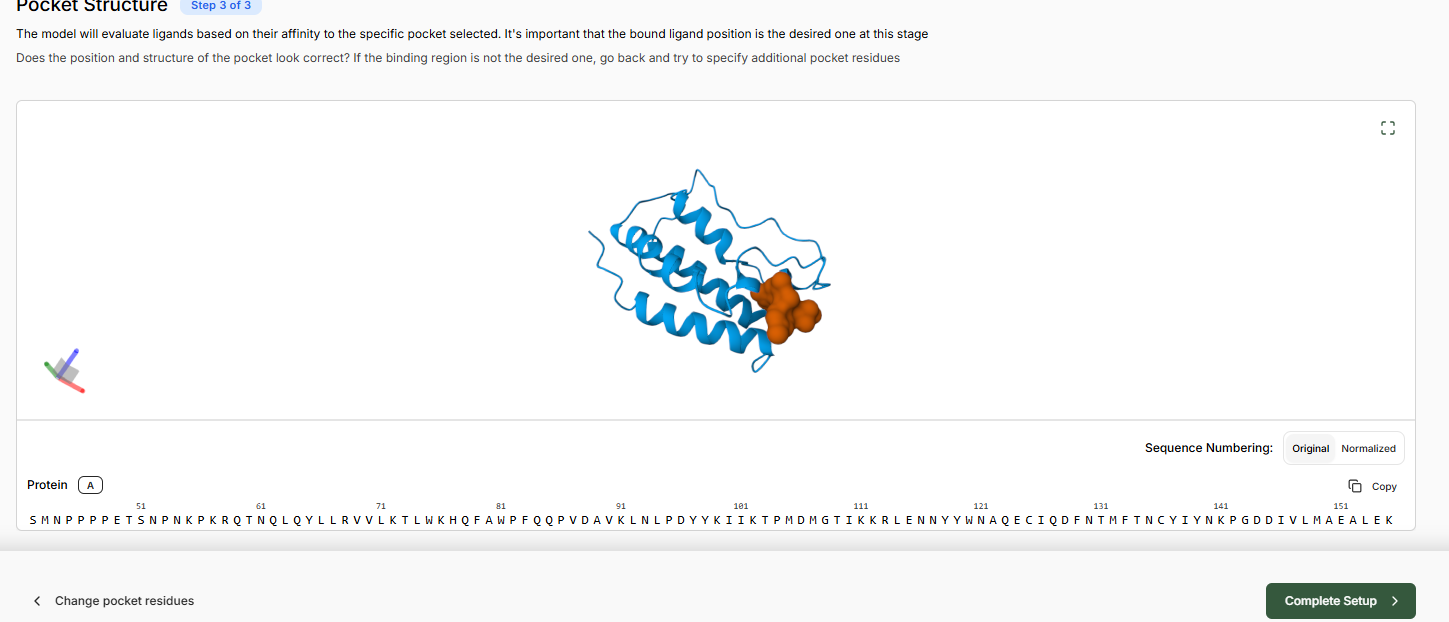
Leave binding residue selection blank, the platform will auto-detect the pocket
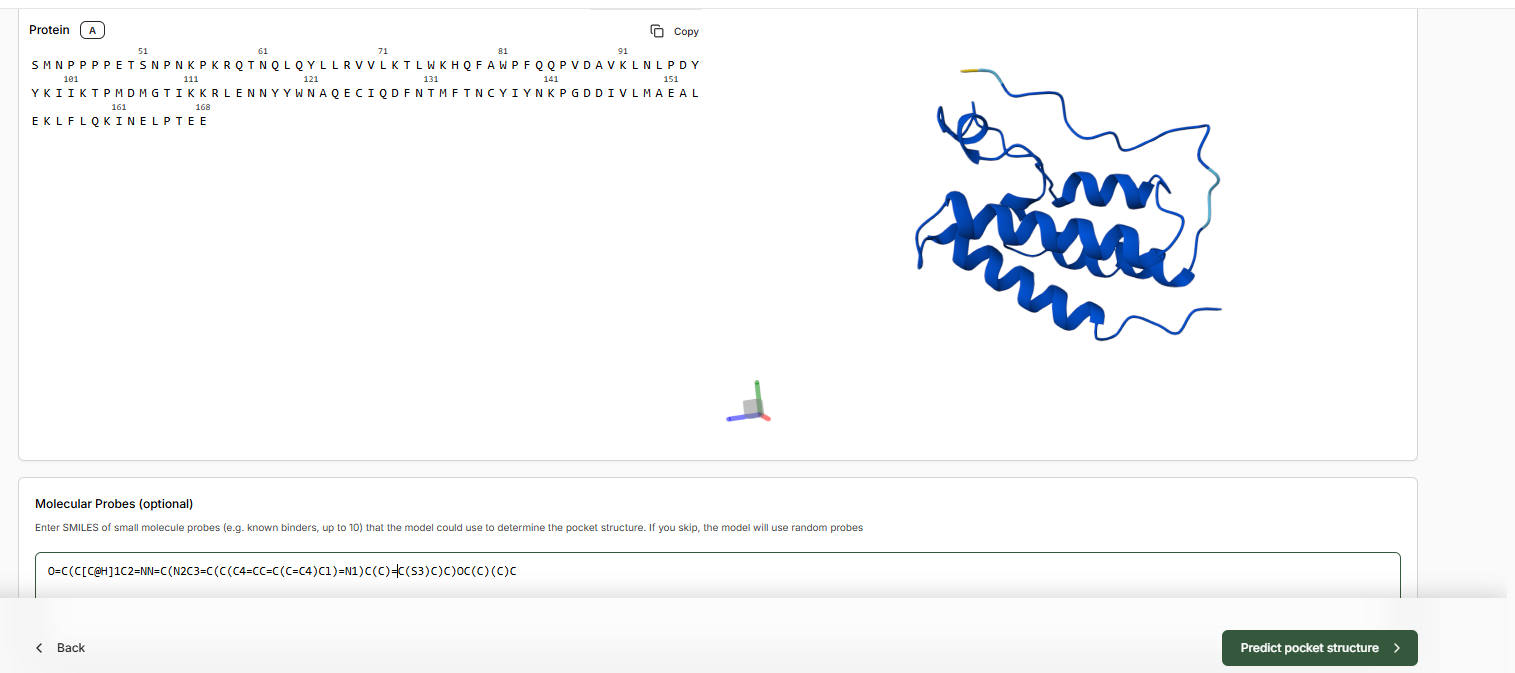
In the Molecular Probe field, paste the JQ1 SMILES.
Predict Pocket Structure and complete the Target Set-Up
Part 3: Running Your Virtual Screen
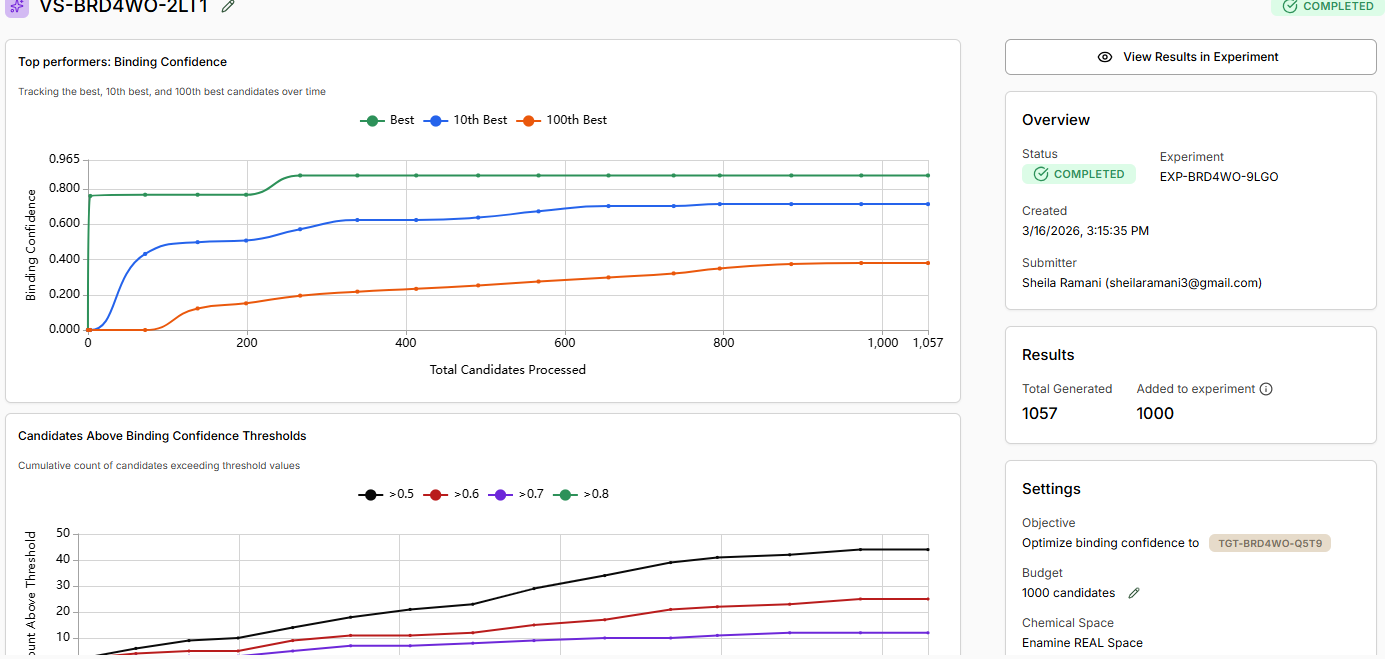
Run a Generative Design Campaign
We will utilize the Boltz Lab small-molecule generative workflow. This generates novel molecules
optimised for BRD4 binding using Boltz-2 as the scoring function.
After creating the design project, Boltz Lab will prompt you to Generate binders with AI.
Name your experiment, provide a relevant hypothesis, and Create the Experiment.
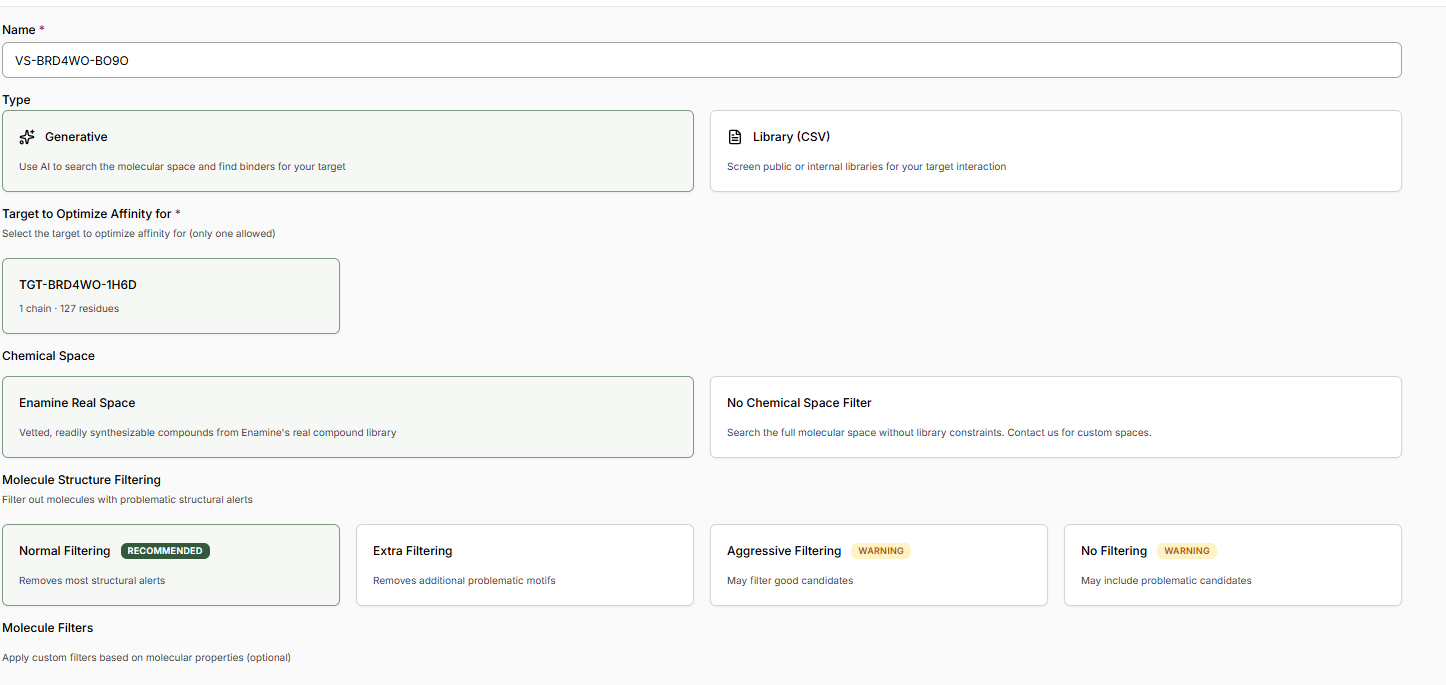
The New Virtual Screen will be pre-configured with a Generative screen using the Enamine
REAL space.
Keep ‘Normal Filtering’ selected. This will ensure we only generate molecules acceptable to
a medicinal chemist.
Decide if you would like to apply any Molecule Filters. We recommend the ‘Drug-Like’
Preset.
Select a custom number of Binders and enter 1K.
Start the Virtual Screen.
Allow binders to be generated, and View Results in Experiment
Part 4: Analysis and Discussion
As your experiment completes, use the ‘Quick Add Candidates’ on the experiment screen to add
JQ1 as a benchmark for generated designs.
Interpreting Your Results
As your experiment completes, use the ‘Quick Add Candidates’ on the experiment screen to add
JQ1 as a benchmark for generated designs.
From your screen output, identify three categories of molecules:
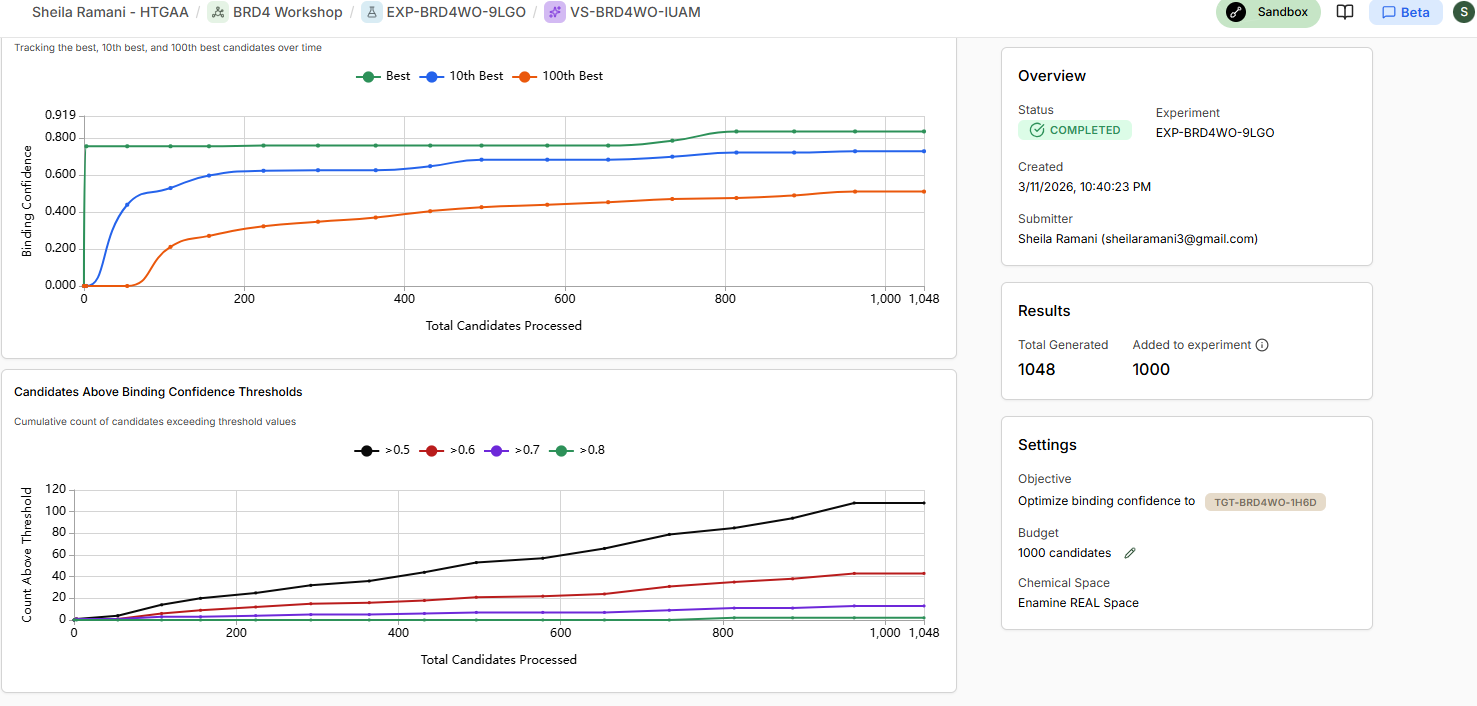
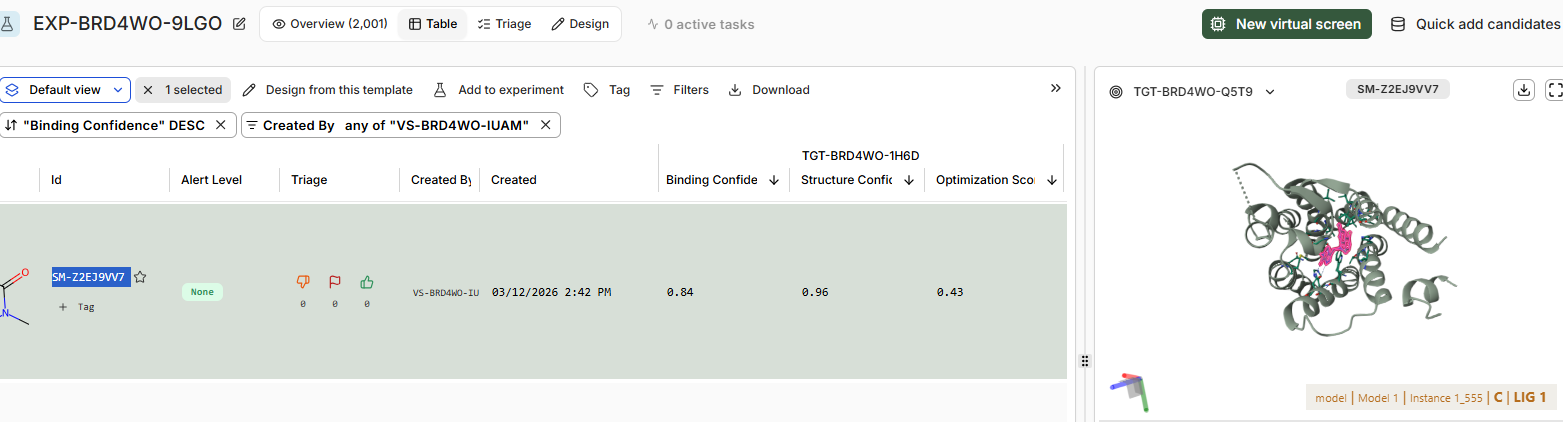
There is only 1 candiadate as high confidence binder SM-Z2EJ9VV7
22 of them are in the moderate confidence binder category, and rest in low confidence / no binders category.
Discussion:
As the virtual screen completes, assess the following:
• How does JQ1 in the Design Project screen alongside the library. Does it score as the top compound?
Binding confidence is 0.96 as compared to 0.84 of the candidate compound.
Yes it scores as the top compound.
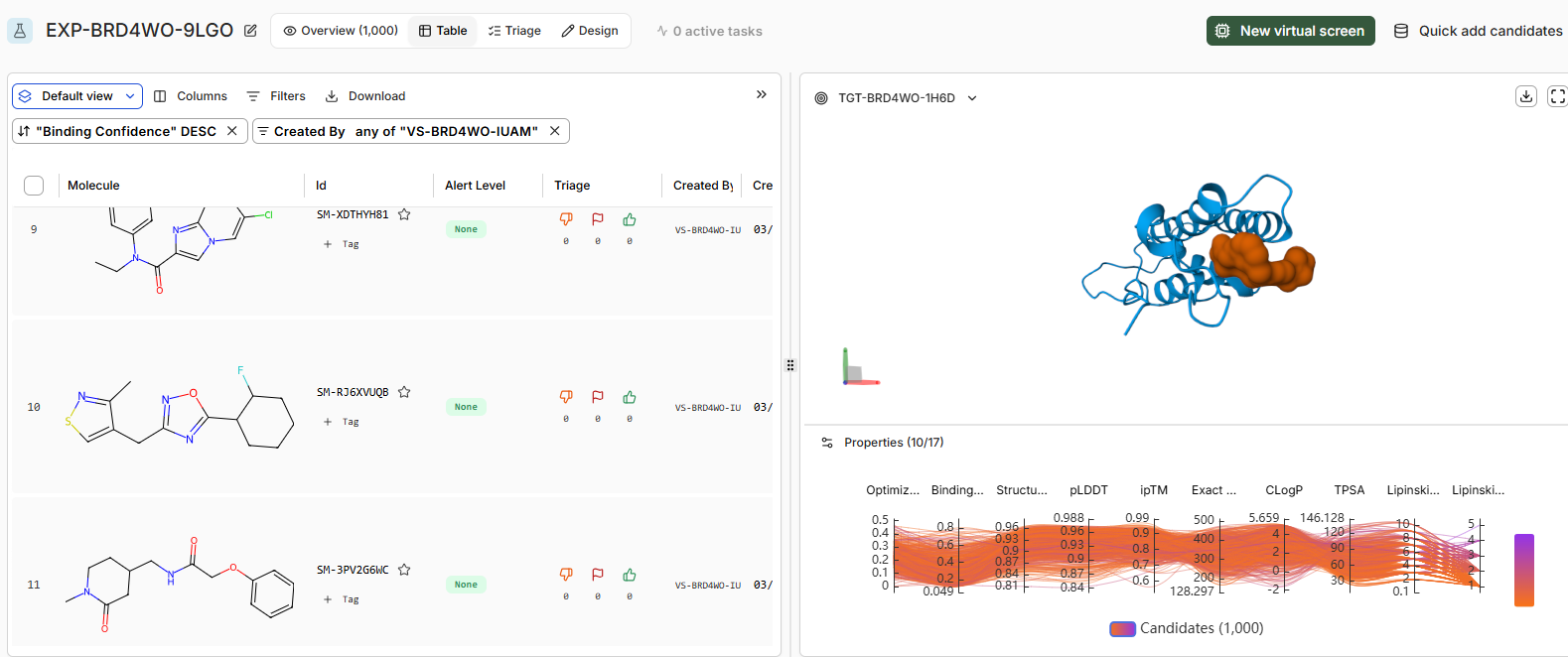
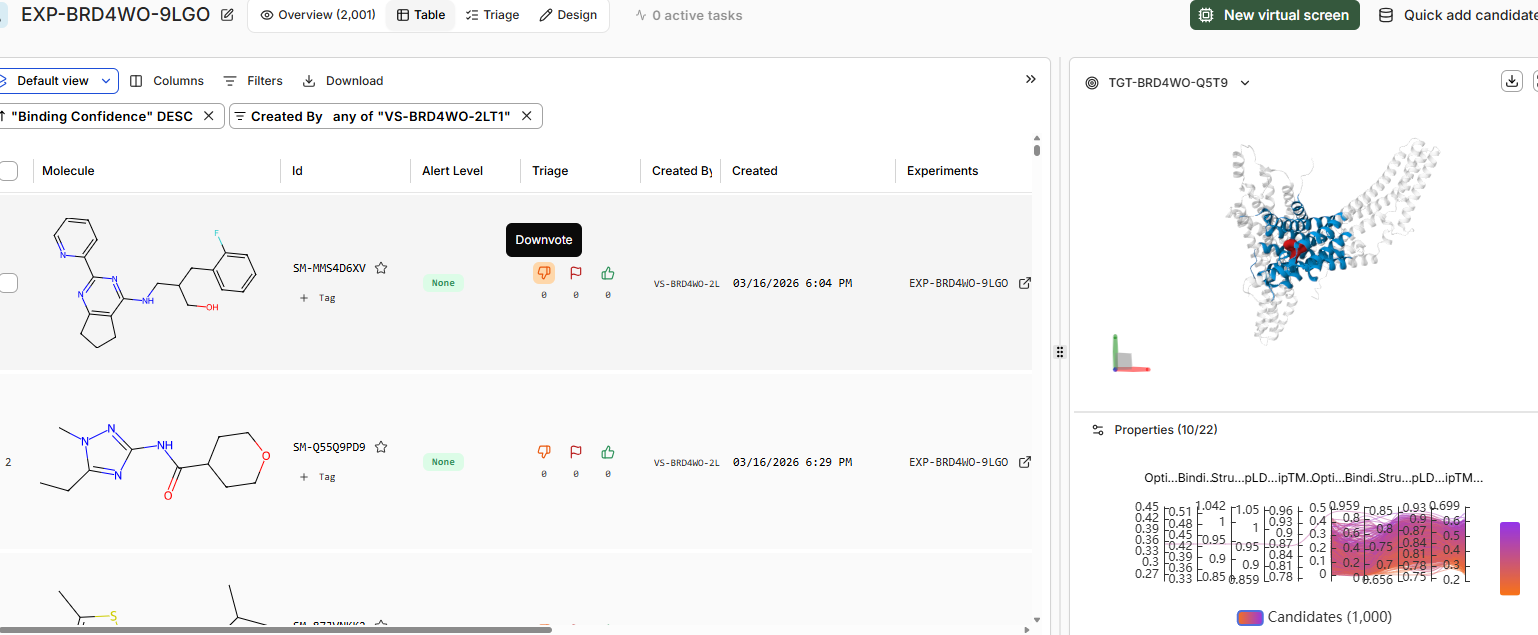
• How do the top scoring binders compare in binding pose to JQ1?
The binding pose of the top binder is similar to JQ1.Key differences include denser packing and fewer disordered loops (less yellow) in the high candidate, suggesting improved prediction reliability. Perceptual image similarity is moderate, confirming related but distinct poses.
• Try adding a second target to your project via the dropdown in the structure viewer, for
example, BRD2 (PDB: 5UEN). Re-run the top scoring binders against BRD2 and compare
which compounds score highly for BRD4 but not BRD2. This is a selectivity analysis - a key
part of real BET inhibitor programs.
The top binder for BRD4 binds with binding confidence of 0.30 to the BRD2 domain.
SM-MMS4D6XV is the top binder for BRD2 domain but binding confidence of same is 0.43 with the BRD4 domain.
Part C: Final Project: L-Protein Mutants
Evolutionary Analysis and Chaperone Generalization
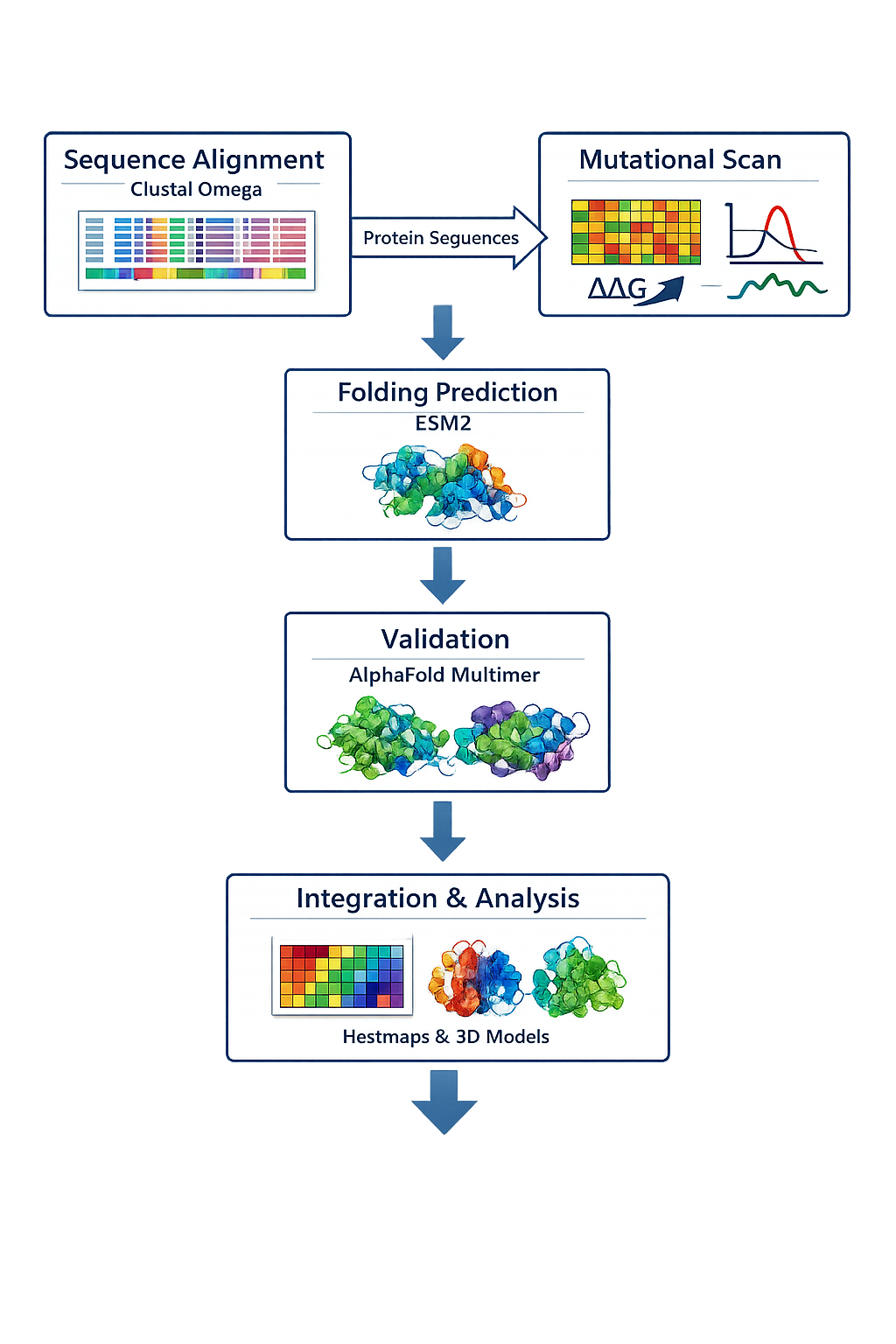
Use pBLAST to survey lysis protein orthologs across bacteria, reconstructing evolutionary trajectories of stability determinants and chaperone interactions. Top candidates will be redesigned for co-folding with alternative chaperones (e.g., DnaK or GroEL), with AF2-Multimer analysis of mature protein folds and in silico stability assays (e.g., ΔΔG predictions) against E. coli host factors to minimize proteotoxicity.
Courtesty: pBLAST tool.
Based on the Clustal omega results mutation was done on the below sites and the protein co folded with DnaJ.
Coutesy: Neurosnap and tamarindBio
Mutations at site 7 8 and 9 removed QQS and left a gap
Deletions that remove the entire N‑terminal basic region (and therefore also remove 7–9) eliminate DnaJ binding and DnaJ requirement, and actually accelerate lysis, because the N‑terminal domain is no longer there to interfere with target binding when not chaperoned by DnaJ.(Jennifer et. al)
Mutation at 61 site E reoved and a gap is left
Position 61 falls within this TM domain that drives membrane insertion, oligomerization into higher‑order complexes, and the formation of large lesions spanning the outer membrane, peptidoglycan, and inner membrane.Chosen as one mutation was to be done in the C-domain of the protein.(Mezhyrova J et.al)
Mutation at site 3 with I instead of T
Position 3 is located in the N-terminal soluble domain of the L-protein.May alter the kinetics, efficiency, or interaction with host proteins (like DnaJ) due to their location in the regulatory N-terminal domain. ( Chamakura et al.)
This shows image with all mutations.
Courtesy: ESM fold
Expected Outcome
If the hypothesis is correct, the resulting lysis protein variants should show improved predicted structural stability, decreased dependence on native DnaJ interaction, and retention of a mature fold compatible with lytic activity in E. coli. More broadly, this strategy could establish a generalizable framework for reengineering host-dependent phage lysis proteins into more robust synthetic biology modules for antimicrobial and biosensing applications.
Plots and graphs for models
The predicted model using Alphafold3 for the mutant.
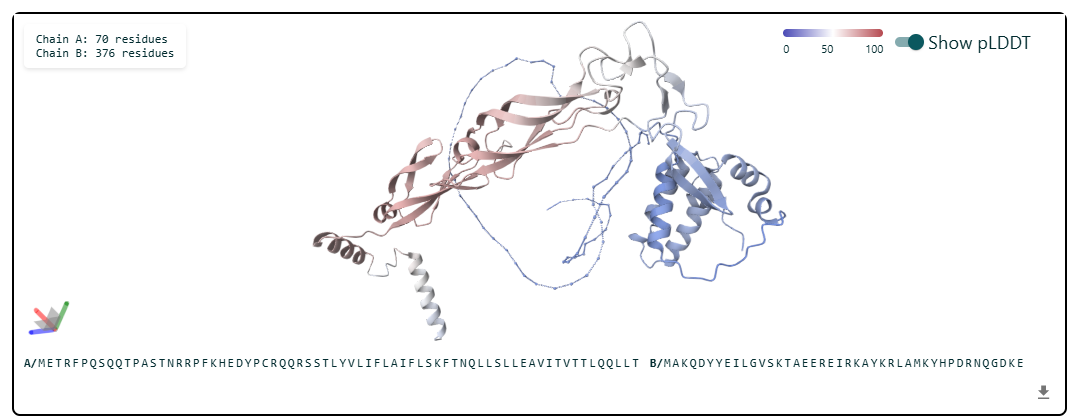
Prediction Interpretation
The image by itself does not strongly support a specific co-folded structure.possibly, but this plot alone does not show evidence that it already co-folds well.A better test is to run a complex prediction with the relevant partner and inspect whether the interface residues are consistently high confidence and whether the relative placement is stable.
References:
1.Chamakura KR, Edwards GB, Young R. Mutational analysis of the MS2 lysis protein L. Microbiology (Reading). 2017 Jul;163(7):961-969. doi: 10.1099/mic.0.000485. Epub 2017 Jul 21. PMID: 28691656; PMCID: PMC5775895.
2.Chamakura KRTran JS, Young R2017.MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ. J Bacteriol199:10.1128/jb.00058-17.https://doi.org/10.1128/jb.00058-17
3.Mezhyrova J, Martin J, Börnsen C, Dötsch V, Frangakis AS, Morgner N, Bernhard F. In vitro characterization of the phage lysis protein MS2-L. Microbiome Res Rep. 2023 Jul 20;2(4):28. doi: 10.20517/mrr.2023.28. PMID: 38045926; PMCID: PMC10688784.
4.Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). https://doi.org/10.1038/s41586-024-07487-w
5.Zeming Lin et al. ,Evolutionary-scale prediction of atomic-level protein structure with a language model.Science379,1123-1130(2023).DOI:10.1126/science.ade2574
PepMLM Colab linked from the HuggingFace PepMLM-650M model card- generating peptide binders
Abramson, J et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (2024).alphafoldserver.com to evaluate structure of binder
PeptiVerse-Zhang, Y., Tang, S., Chen, T., Mahood, E., & Chatterjee, P. (2026). PeptiVerse: A unified platform for therapeutic peptide property prediction. arXiv. doi.org
moPPit Colab linked from the HuggingFace moPPIt model card- to generate optimised peptides
Passaro, S., et al. (2025). Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. bioRxiv, 2025.06.14.659707. https://doi.org/10.1101/2025.06.14.659707- for drug discovery
16.Lysis protein mutated structure predictions were generated using OpenFold3, an open-source reproduction of the AlphaFold 3 architecture (Abramson et al., 2024; OpenFold Consortium, 2024), implemented on the Neurosnap platform (Neurosnap Inc., 2024).
17.Lytic protein complete 5 point mutated structures and complex interactions were modeled using the OpenFold3 architecture (Abramson et al., 2024; OpenFold Consortium, 2024), deployed via the cloud-based Tamarind Bio interface (Tamarind Bio, Inc., 2024).
18.Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. doi.orgCamacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10(1), 421. doi.org
1.What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The components of Phusion High-Fidelity PCR Master Mix contains Phusion DNA Polymerase (high-accuracy, 50x higher than Taq), dNTPs (nucleotide building blocks), and optimized reaction buffer with MgCl2(essential for enzyme activity).
2.What are some factors that determine primer annealing temperature during PCR?
The annealing temperature depends on the melting temperatures of primers.It is generally 3–5°C lower than the lowest temperature of the primer-template pair.
-The GC content of primers and length of primers increase the melting temperature, require higher annealing temperature.
-Higher magnesium ion concentration tends to satbilize teh duplex tehreby increasing annealing temperature.
-Higher concentration of primers require higher annealing temperature.
-DMSO or glycerol decrease melting temperature of Dna so lower annealing temperatuer is required.
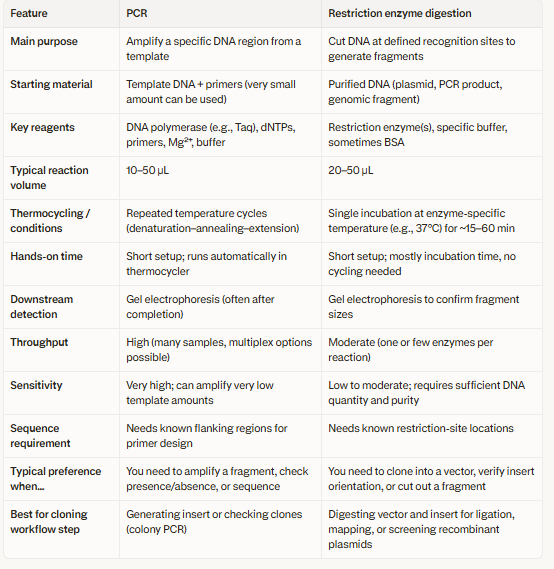
3.There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests.Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
4.How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The designed primers should be of 15-40 bp in length, terminal overlaps matching adjacent fragments, mutations can be minimised by using high fidelity polymerases and purify PCR products.This will remove templates and primers. Gel electrophoresis can be used to verify the PCR or restriction digest products by linearizing the vectors.
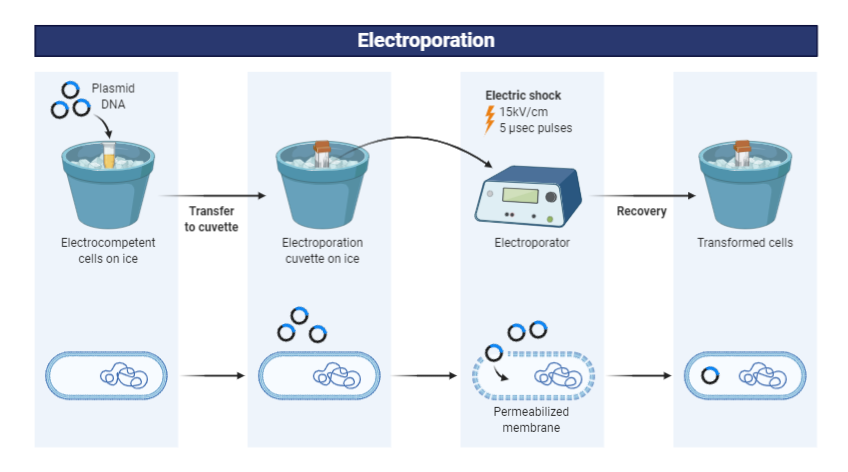
5.How does the plasmid DNA enter the E. coli cells during transformation?
The plasmid DNA can enter via the electroporation method where a brief electric pulse is applied to a cell. This creates pores in the membrane and allows the plasmid DNA to enter the cell.
The other method is to treat the cell to Heat shock method. The cells are treated with cold CaCl2,making it permeable and neutralizes charge on DNA. A sudden increase in tempertaure to 42°C helps create pores in the membrane and allows DNA to enter the cell.
Heat shock method
Electroporation method
Courtesy: BioRender.com
6.Describe another assembly method in detail (such as Golden Gate Assembly)
1.Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a precise, “one-pot” cloning method that utilizes Type IIS restriction enzymes (e.g., BsaI, BsmBI) and T4 DNA ligase to directionally assemble multiple DNA fragments simultaneously. It is “scarless,” meaning no extra, unwanted sequences remain between fragments, making it superior to traditional restriction cloning.
Type IIS enzymes recognize asymmetric DNA sequences but cleave outside of them, leaving unique 4-base pair overhangs. These overhangs are designed to be complementary, allowing multiple fragments to ligate in a specific order.
The restriction digestion and ligation occur in a single tube, typically using thermal cycling to alternate between optimal digestion and ligation temperatures.
The Type IIS recognition sites are positioned at the very ends of the DNA fragments, designed so they are removed from the final assembled product. Because the product lacks these sites, it cannot be re-digested, making the reaction irreversible and highly efficient. If internal Type IIS sites exist within the DNA fragments, they must be removed via silent mutations (a process called domestication) to prevent premature digestion.
2.Model this assembly method with Benchling or a similar tool!
Courtesy: Benchling
Assignment: Asimov Kernel
1.Create a Repository for your work
2.Create a blank Notebook entry to document the homework and save it to that Repository
4.Create a blank Construct and save it to your Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
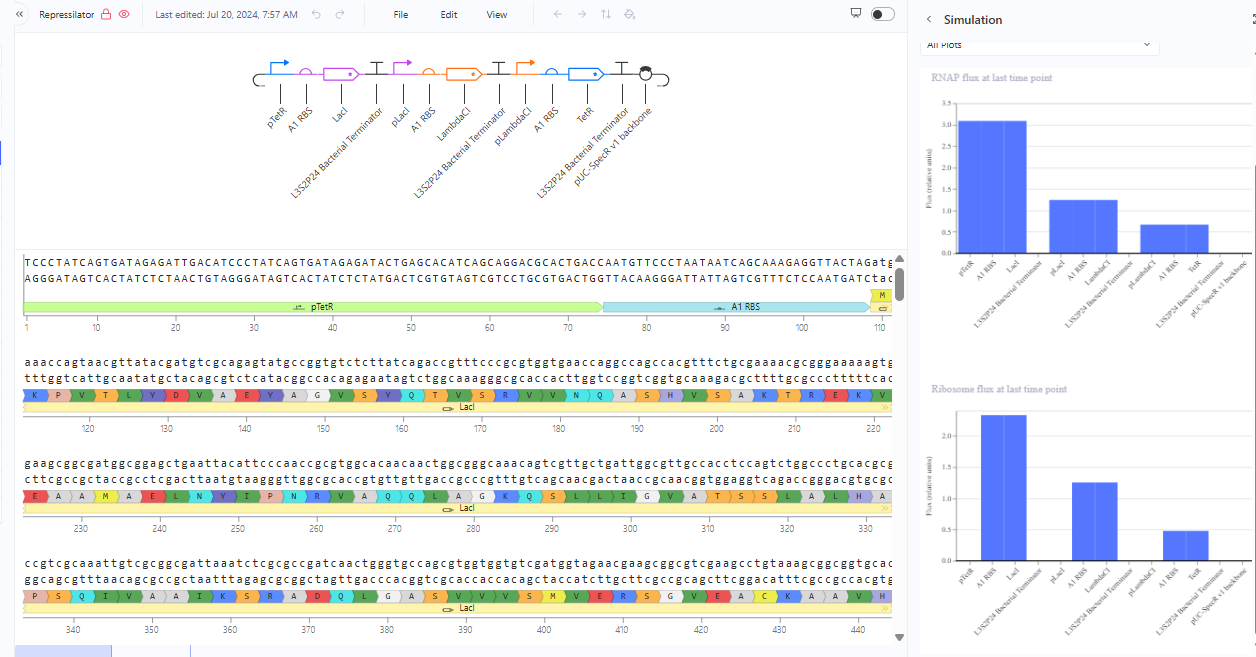
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
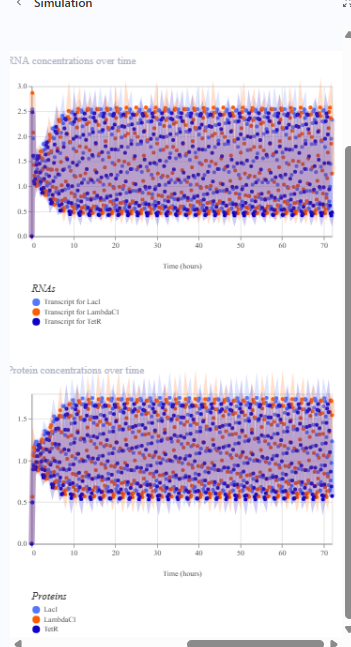
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
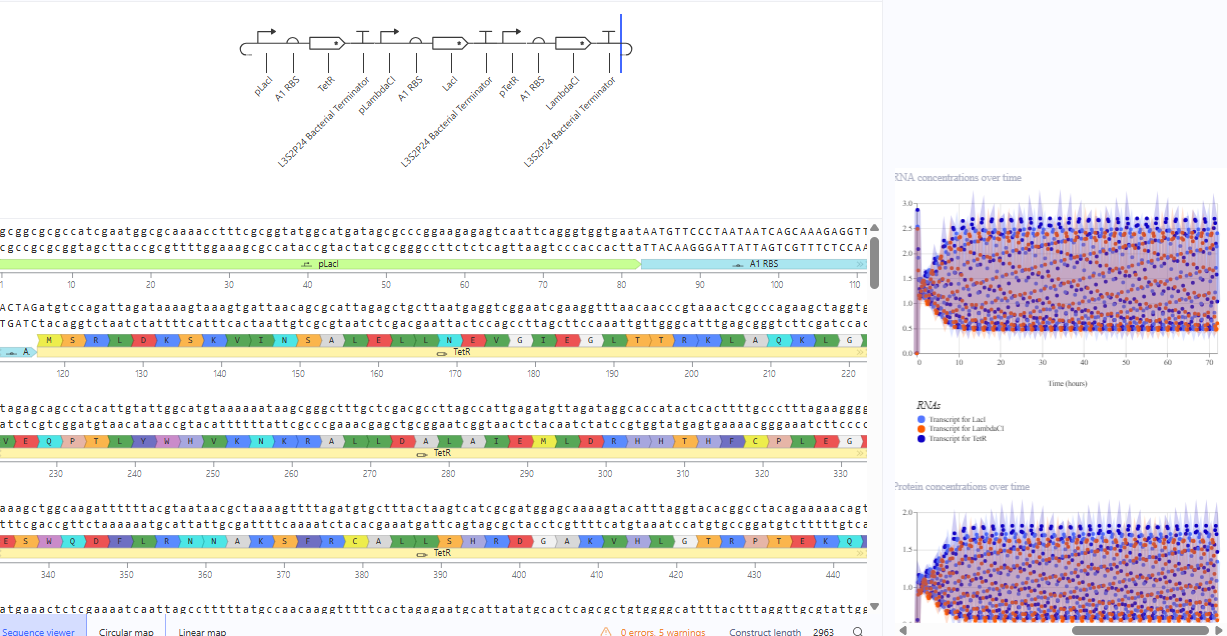
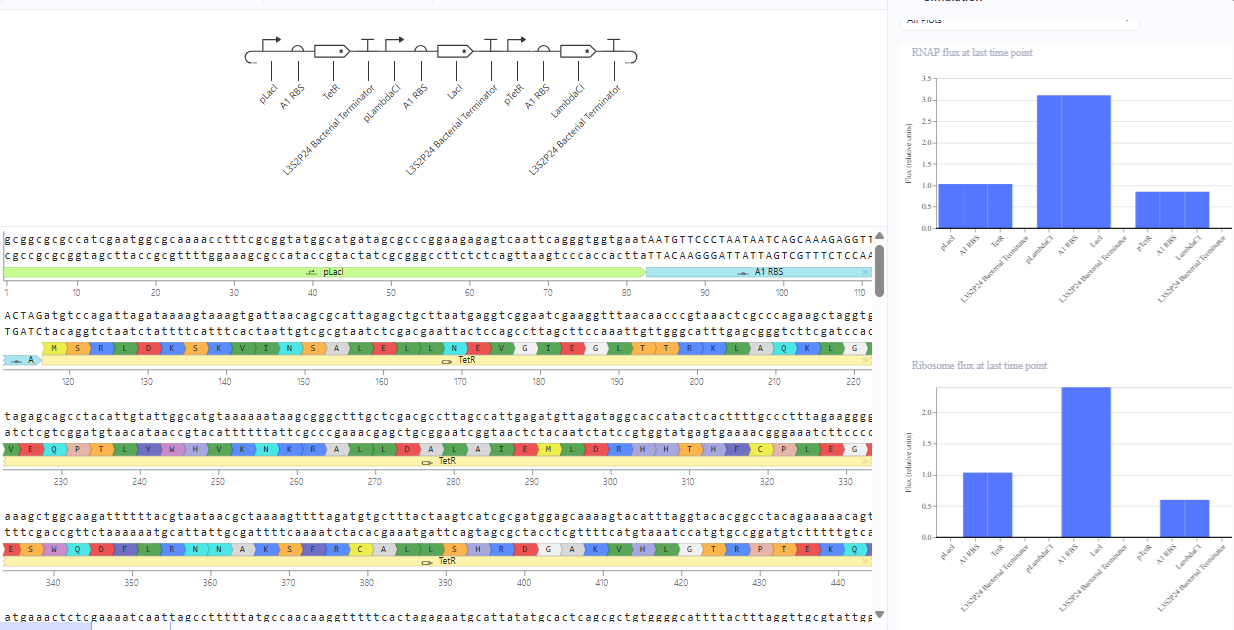
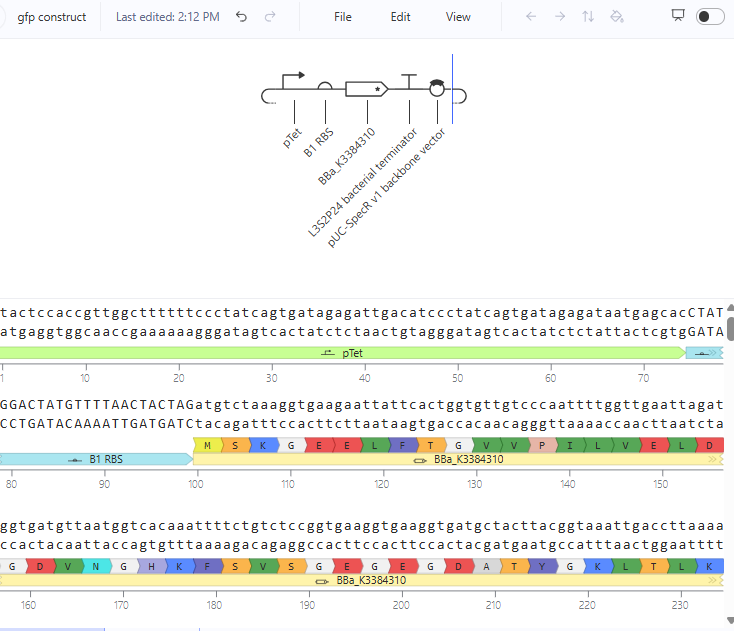
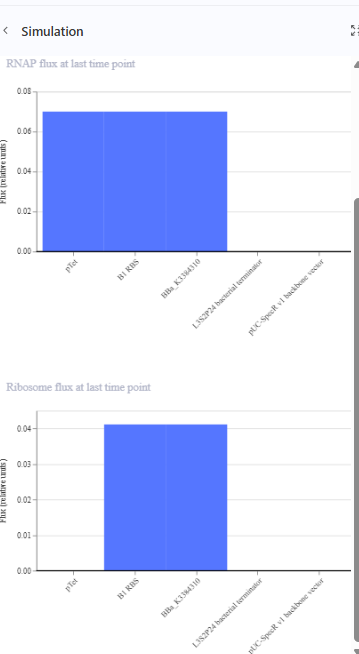
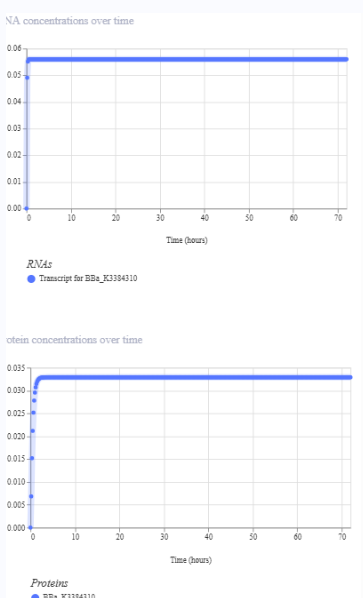
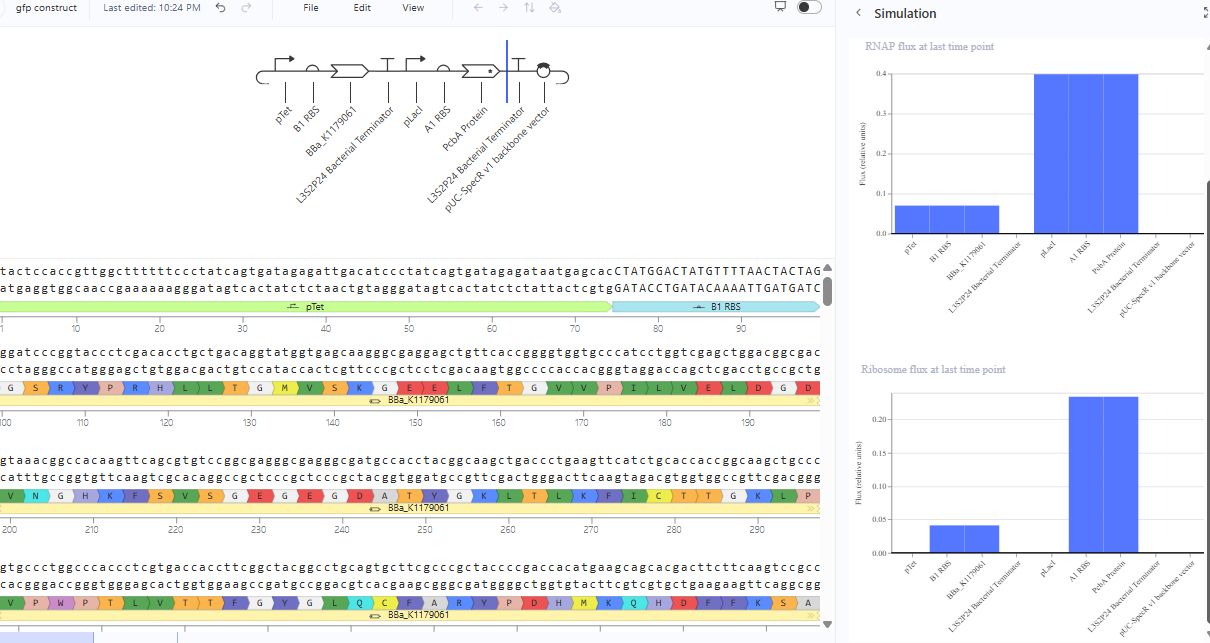
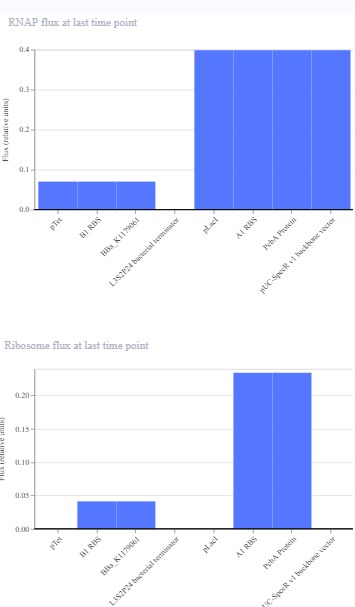
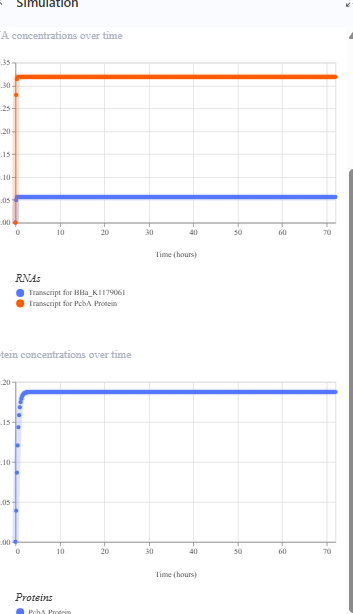
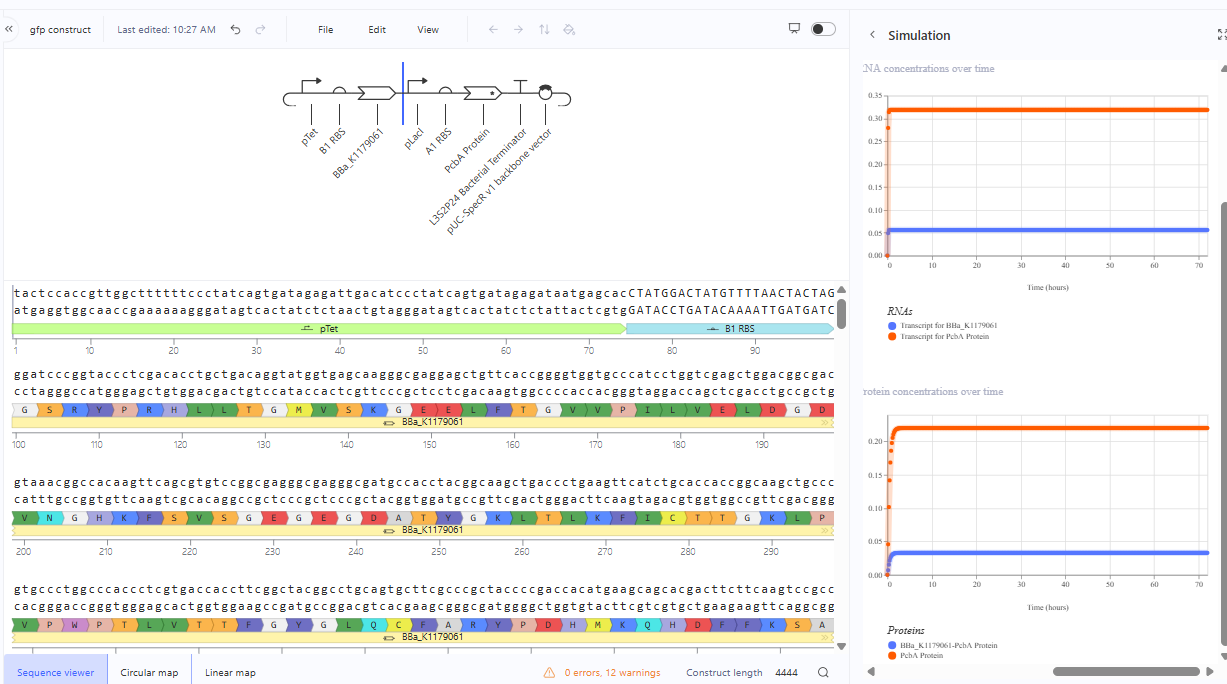
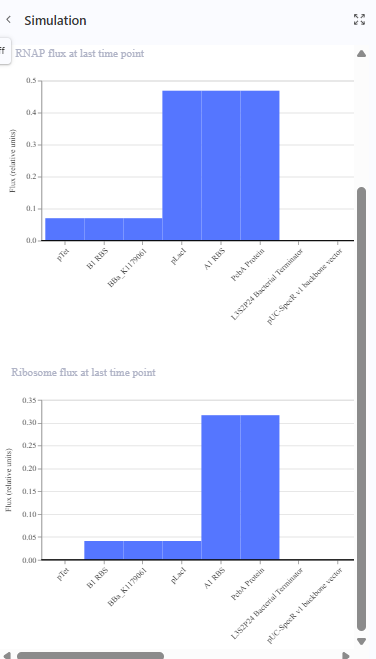
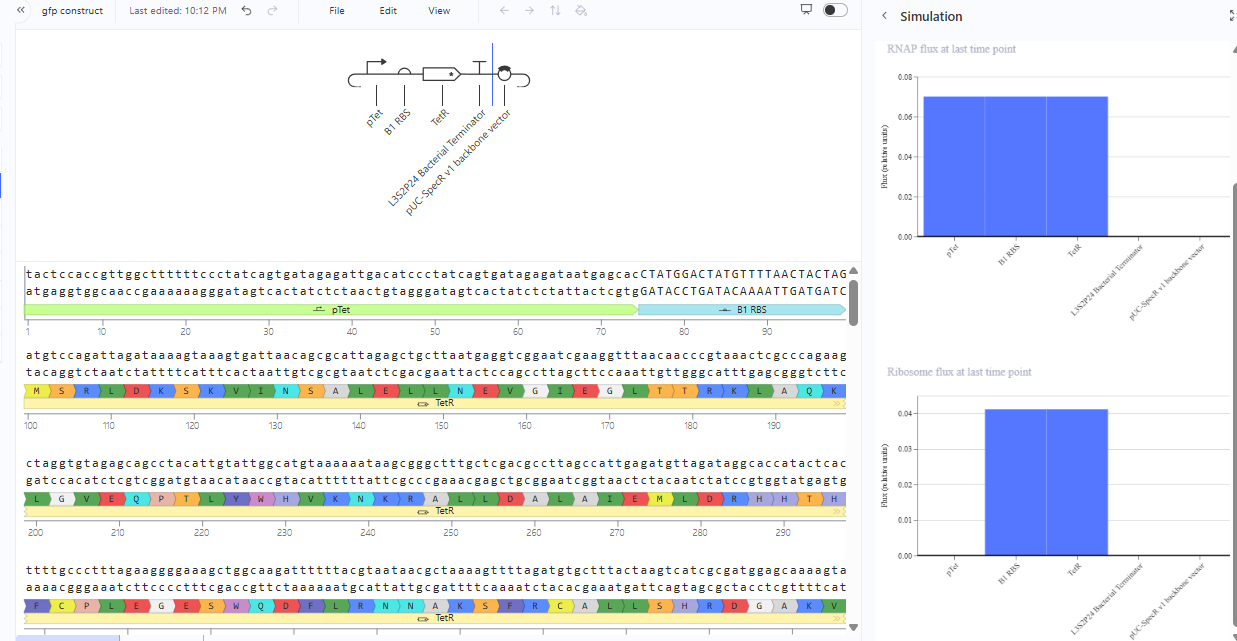
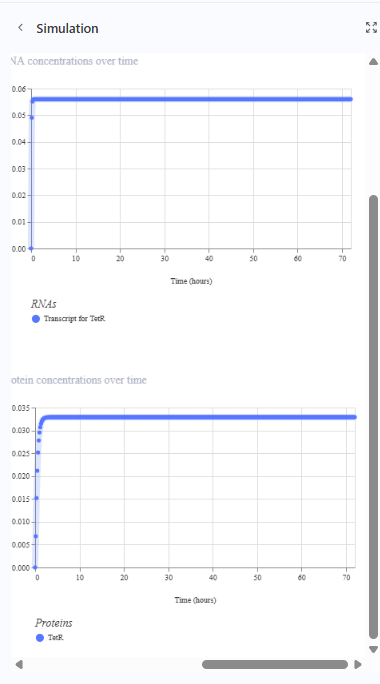
The repressilator cretaed by me does work as expected. In comparison to the construct found in Bacterial demos, the graph obtained is similar.
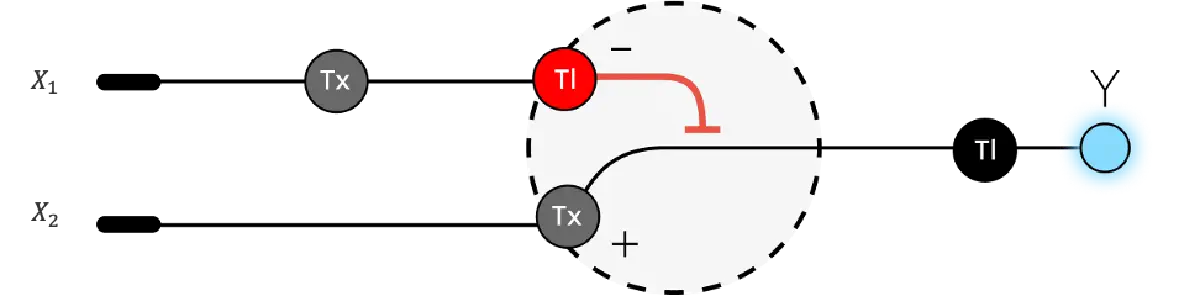
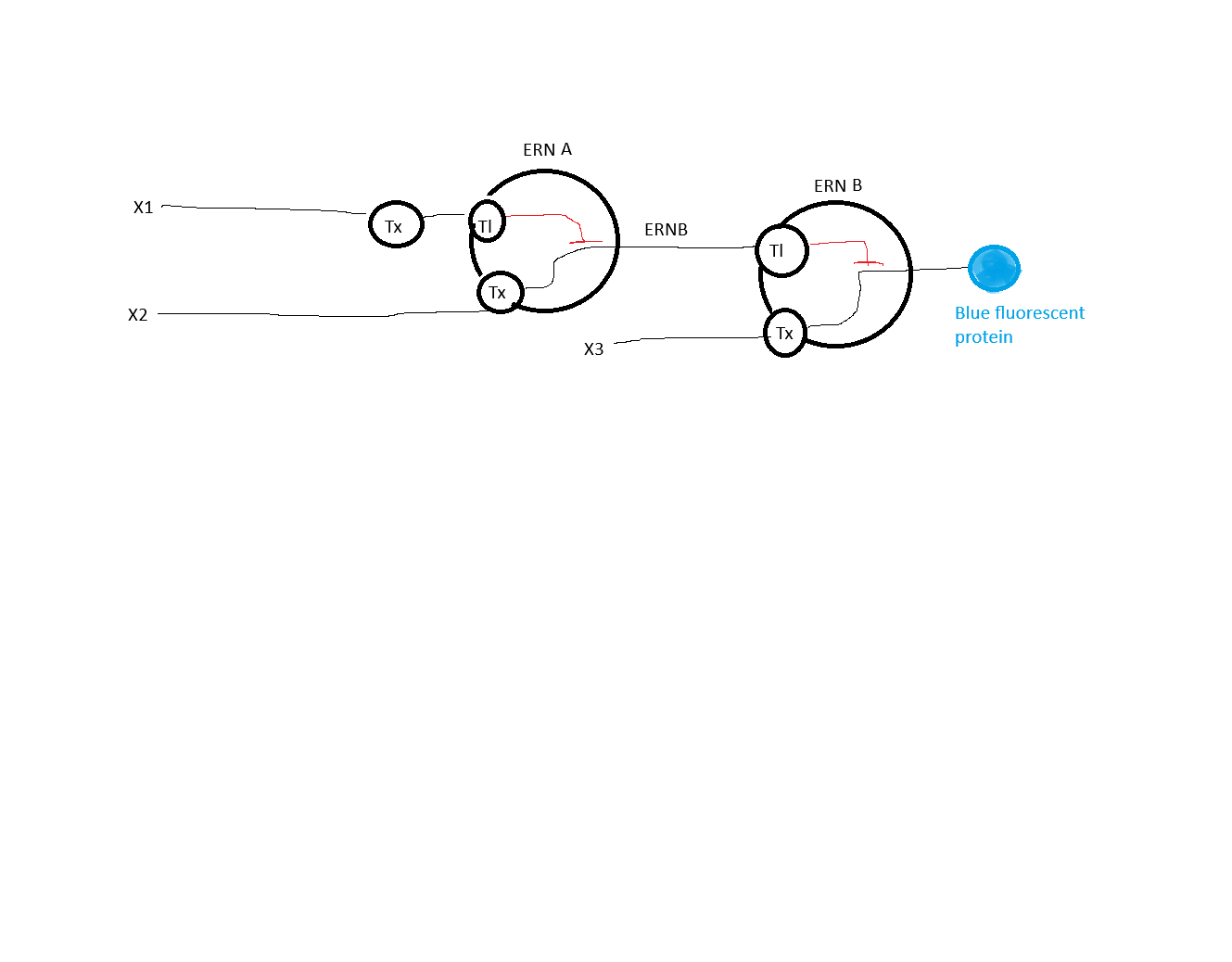
The RNA flux (rate of RNA production) for the plac promoter (often ) in the original repressilator is generally higher than that of pTet and pLambda
due to higher basal or un-repressed promoter strength.The pLacI promoter, which regulates lacI, is a stronger promoter compared to other two when not fully repressed.The lacI system is inherently leakier than tetR or cI, allowing higher basal expression and faster overall accumulation of its mRNA when the repressive factor (TetR) is not present at high concentrations.
5.Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
Construct 1 : Expressing yeGFP gene
The construct should express the protein yeGFP.
Construct 2: expression of eYFP and PcbA gene
The two genes should be expressed independently.
In a two-gene construct, particularly if they are in close proximity, the activation of the strong promoter pLacI might cause the RNA polymerase to interfere with the transcription of the weaker promoter pTetR, resulting in no mRNA for eYFP.
The same construct when the terminator for the eYFP was removed gave the following result.The transcription continues into the next downstream gene.
Construction 3: expression of autoregulatory TetR
The production of TetR by its promoter pTetR is autoregulated meaning it represses its own production. The amount of TetR controls the construct.
The results match my expectations.
References
1.Benchling [Biology Software]. (2026).- Golden Gate Assembly construct
BioRender.com- heat shock and electroporation method
Nielsen, A. A., Der, B. S., Shin, J., Vaidyanathan, P., Paralanov, V., Strychalski, E. A., Ross, D., Densmore, D., & Voigt, C. A. (2016). Genetic circuit design automation. Science, 354(6310), aac7341. doi.org
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1.What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs allow biological systems to act as analog processors, mimicking neural network learning and offering higher versatility, precision, and robustness in synthetic biology applications.
Context-dependent responses rather than simple “on/off” outputs.
Mimic artificial neural networks using transcriptional regulators, allow for complex,
Nonlinear processing of multiple inputs,
Higher fault tolerance, and the ability to perform regression analysis, which is not possible with traditional digital genetic circuits.
2.Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful application of IANN would be to detect early stage breast cancer. early stage breast cancer has elevated levels of mir-155 and mir-21.
Input : mir-21 (X2 )
ERN-(X1)
output: killer protein
Increasing translation rate of endoribonuclease translation rate:
The negative element increases (X1)so we have more endoribonuclease and lower would be the output(killer protein) so decision boundary shifts left.
Increasing translation rate of output:killer protein
The positive element increases(X2), the decision boundary moves down.
With Bias:
Both inputs are high (X1 and X2) bias has a fixed value, output will also be high.( killer protein). This happens because X1 and X2 are both higher than the bias. Bias is negative.
In the case when X1 and X2 are low the output is low bercause both X1 and X2 are lower than the bias.
Only when X1 or X2 or combination of both are higehr than bias we get output(killer protein). This is called the high pass.
Low pass: The X1 And X2 are low and output is high.
The bias is positive and X1 and X2 are repressors so they are negative weights.
The X1 and X2 are lower than the bias so the output is high.
only when X1 and X2 in combiantion exceeds the bias will the output be low.
Two neurons would be required to create the dual region where the X1 and X2 would be positive weights in one neuron and negative weights in the other. the bias would also be positive and negative in the two neurons based on the positive and negative weights of inputs.
Bandpass: Three neurons would be required. X1 and X2 would be positive weights in one neuron and negative weights in the other, the bias would also be positive and negative in the two neurons based on the positive and negative weights of inputs. The output of this would be the input for the third neuron, which would have anegative weight and the bias would be positive. the output which is the negative weight if above the bias then teh final output is low otherwise the output is high.
Limitations
The intracellular environment is noisy and highly crowded. The IANN needs to be robust against degradation by cellular enzymes (nucleases).
The IANN components incorporated in vivo should not pose toxicity and generate immune response from patient.
The IANN produces output based on specific known cancer markers, what if the cancer cell mutates - they may fail.
3.Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Neuron 1
X1- DNA encoding the endoribonuclease A (ERNA) which is the negative weight
X2- DNA encoding the endoribonuclease B(ERNB) which is the positive weight as it controls the output.
Neuron 2
X3- DNA encoding blue fluorescent protein which is the positive weight
The output of neuron 1 (ERNB) is the input for neuron 2 and is the negative weight.
Assignment Part 2: Fungal Materials
1.What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts
Fungal materials, primarily derived from mycelium (the root structure of fungi), are transforming industries by providing eco-friendly, biodegradable alternatives to plastics, leather, and construction materials. These materials are grown on agricultural or industrial waste, turning low-value byproducts into high-value, customizable composites.
Mycelium Packaging (Packaging Foam): Used to replace expanded polystyrene (EPS) or Styrofoam. It is molded into shapes to protect fragile goods during shipping.
Myco-leather (Fungal Leather): A flexible, non-woven mat derived from mycelium (e.g., Mylo™, Mylea™, Reishi™) used in fashion for handbags, wallets, shoe soles, and clothing.Mycelium Insulation Boards: Panels created for thermal and acoustic insulation in building construction, providing fire resistance and soundproofing.
MycoTree (Structural Material): Dense mycelium composites used to create structural components, such as self-supporting structures, furniture, and door cores.
Mycoprotein Food Analogues: Filamentous fungi (e.g., Fusarium venenatum) are fermented to produce meat substitutes (e.g., Quorn™), providing a high-protein, low-fat sustainable food source
Chen, Jianyue. (2025). ReishiTM Mushroom Leather: Pioneering Sustainable Leather in the Fashion Industry. Frontiers in Business, Economics and Management. 18. 24-32. 10.54097/nzds8q40.
Fungal materials are eco-friendly, sustainable,biodegradable.
Production cost is low and lower carbon footprint compared to plastic, ceramic, steel.
They can be engineered to be surprisingly strong, with some pressed mycelium surpassing wood in tensile strength.
Higher fire resistance than synthetic foams, emitting fewer toxic fumes and taking longer to reach flashover.
Fungi can grow into customized molds and even heal small damages in the material matrix.
Disadvantages
Untreated fungal materials have a high tendency to absorb moisture, which can compromise structural integrity and make them unsuitable for high-humidity environments.
They often exhibit poor tensile and flexural strength, making them brittle or weak under pulling/bending stress compared to traditional wood, plastics, or metals.
Certain fungal species can produce toxic metabolites (mycotoxins) if contamination occurs during the manufacturing process, requiring strict regulatory and safety controls.
Growing a specific fungus requires a sterile, highly controlled environment. During the slow incubation/growth phase, unwanted airborne molds or bacteria can easily contaminate the batch, resulting in high failure rates and material waste.
Extra read: Luo D, Yang J, Peek N. 3D-Printed Mycelium Biocomposites: Method for 3D Printing and Growing Fungi-Based Composites. 3D Print Addit Manuf. 2025 Apr 14;12(2):98-111. doi: 10.1089/3dp.2023.0342. PMID: 40308659; PMCID: PMC12038323.
2.What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I would want to genetically engineer fungi to produce marine derived secondary metabolites against cancer.Their mechanism involves targeting multiple pathways simultaneously, inducing apoptosis, and inhibiting angiogenesis and metastasis with lower toxicity to healthy cells.
Fungi are eukaryotic, can be used to make eukayotic protein with proper folding and post translational modifications.
Fungal fermentation occurs in acidic environments as compared to bacteria and can prevent bacterial contamination.
Fungi have Biosynthetic gene clusters which they use to produce secondary metabolites like polyketides,terpenes.This capability enables the production of pharmaceuticals, pigments, and complex specialized chemicals.
The filamentous fungi possess remarkable secretory pathways, helpful in secreting proteins at 10-100 fold higher quantities.
Assignment Part 3: First DNA Twist Order
This the sequence of interest to be incorporated in the plasmid.
The Twist Bioscience Order plasmid
References
1.Chen, Jianyue. (2025). ReishiTM Mushroom Leather: Pioneering Sustainable Leather in the Fashion Industry. Frontiers in Business, Economics and Management. 18. 24-32. 10.54097/nzds8q40.
5.Luo D, Yang J, Peek N. 3D-Printed Mycelium Biocomposites: Method for 3D Printing and Growing Fungi-Based Composites. 3D Print Addit Manuf. 2025 Apr 14;12(2):98-111. doi: 10.1089/3dp.2023.0342. PMID: 40308659; PMCID: PMC12038323.
6.Benchling [Biology Software]. (2026) lysostaphin gene construct in pTwist plasmid
“Living material assembly of bacteriogenic protocells” by Can Xu, Nicolas Martin, Mei Li, and Stephen Mann, 14 September 2022, Nature.
DOI: 10.1038/s41586-022-05223-w
Homework Part A: General and Lecturer-Specific Questions
General homework questions
1.Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Advantages: Proteins that are toxic to cells can be produced in cell free systems as there is no fear of damage to cell.
Metabolic pathways can be engineered to bypass unwanted steps and can increase the yield of proteins and allowing the removal of waste products.
The additives like chaperones or lipososmes or detergents can be added directly and conditions like pH and temperatutre optimized as the reactions are taking plavce outside the cell.
Cell-Free Protein Synthesis bypasses time-consuming cloning, cell transformation, and expression optimization steps, allowing for direct protein production from linear DNA (PCR products) in hours rather than days.
Proteins that are toxic to cell like antimicrobial peptides are produced because there is no cell system that can be damaged by these toxic proteins. The production of antimicrobial peptides (AMPs), such as human -defensin-2 and magainin 2, which are designed to kill bacteria, can be produced in high-concentration cell-free systems without damaging the production machinery.
Incorporation of Non-Canonical Amino Acids (NCAAs): Cell-free systems make it easy to incorporate unnatural amino acids site-specifically into proteins for drug discovery, as they do not have to cross a cell membrane.These are amino acids that are incorporated by genetic code expansion and modified tRNA synthetases that recognise amber codon TAG.
Courtesy:Carlson, Erik & Gan, Rui & Hodgman, Charles & Jewett, Michael. (2011). Cell-Free Protein Synthesis: Applications Come of Age. Biotechnology advances. 30. 1185-94. 10.1016/j.biotechadv.2011.09.016.
2.Describe the main components of a cell-free expression system and explain the role of each component.
Cell Extract (Lysate or Reconstituted): Contains the machinery required for transcription and translation, including ribosomes, tRNAs, aminoacyl-tRNA synthetases, and initiation/elongation factors.
Role: Provides the fundamental molecular machinery to transcribe DNA to mRNA and translate mRNA into protein.
Plasmid DNA, PCR product, or linear DNA encoding the gene of interest.
Role: Provides DNA to be transcribed and translated by the cell-free system.
Nucleoside triphosphates (NTPs: ATP, GTP, CTP, UTP), often supplemented with molecules like phosphoenolpyruvate (PEP) or creatine phosphate.
Role: Powers the high energy demands of translation (peptide bond formation) and transcription.
Amino Acids: The 20 essential building blocks for protein synthesis.
Role: Utilized by ribosomes to assemble polypeptide chains.
RNA polymerase: T7 polymerase added in the mix if transcription and translation has to be carried out.
Role: Transcription of DNA to mRNA.
Reaction Buffer and Cofactors: Contains salts (e.g., Mg, K), pH buffers, and sometimes RNase/protease inhibitors.
Role: Optimizes the environment for enzymatic activity (e.g., proper magnesium concentration for ribosome function) and protects the RNA and protein products.
Chaperones may be added for protein folding.
Courtesy:Brookwell, August, Javin P. Oza, and Filippo Caschera. 2021. “Biotechnology Applications of Cell-Free Expression Systems” Life 11, no. 12: 1367. https://doi.org/10.3390/life11121367
3.Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
The energy regenaration is critical as continuous transcription and translation require high ATP concentrations that has to be maintained. The protein synthesis stops in absence of ATP depletion, reducing yield and reaction.
Phosphoenolpyruvate (PEP) System
A highly effective method is using the PEP/pyruvate kinase (PK) system to continuously regenerate ATP from ADP.
Mechanism: Pyruvate kinase catalyzes the transfer of a high-energy phosphate group from phosphoenolpyruvate (PEP) to ADP, forming ATP and pyruvate.
Implementation: Supplement the reaction mixture with 10-30mM PEP and a catalytic amount of Pyruvate Kinase (>10U/ml) along with necessary magnesium and potassium ions.
Result: This system provides a high phosphate potential, significantly extending the duration of protein synthesis compared to simply adding ATP
Figure courtesy:ChatGPT
4.Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Courtesy: perplexity
Prokaryotic Cell-Free System Production (e.g., E. coli lysate)
I choose the protein Renilla luciferase as it is used as reporter in many experiments.
Bacterial Cell-Free Protein Synthesis (CFPS) is a widely used, in vitro, open-system technique for producing luciferase proteins (such as Renilla or Firefly luciferase) by using crude cell extracts (typically E. coli) instead of living cells. It enables high-yield production, rapid prototyping of genes, and the ability to produce proteins that might be toxic to living cells.
Eukaryotic Cell-Free System Production
Protein Choice: Human Erythropoietin (EPO)
Why: Eukaryotic systems possess chaperones and machinery for complex folding and post-translational modifications (e.g., glycosylation). Eukaryotic, particularly mammalian or plant-based, systems are necessary to produce active, properly folded transmembrane or glycoproteins, which would otherwise become insoluble aggregates in prokaryotic systems.
5.How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Optimizing Expression of a G-Protein Coupled Receptor (GPCR)
Optimizing G-protein coupled receptor (GPCR) expression involves using codon-optimized genes, N-terminal/C-terminal tagging, and selecting high-expressing mammalian (CHO-K1, HEK293) or insect (Sf9) cell clones. Techniques like using stabilized receptors (STARs), lower culture temperatures (20-27°C), adding ligand or DMSO to increase functional folding, and using detergent-screened membranes ensure high-yield, functional production.
Key Challenges in GPCR Expression
Low Expression Yields: Native GPCRs are present in low abundance; heterologous expression often results in poor yields.
Instability & Aggregation: GPCRs are highly dynamic and flexible, leading to rapid unfolding, degradation, or aggregation once extracted from the lipid bilayer as it is a 7-transmembrane protein and highly hydrophobic.
Toxicity: Overexpression of membrane proteins can overwhelm the host cell’s translocation machinery, leading to cell death.
Incorrect Folding/Post-translational Modifications (PTMs): Prokaryotic systems lack necessary PTMs (e.g., glycosylation), while eukaryotes may incorrectly fold the receptor.
Poor Membrane Targeting: Receptors may be trapped in the Endoplasmic Reticulum (ER) or mislocalized within the cell.
A successful setup combines codon-optimized synthetic genes, fusion partner engineering, and low-temperature, inducible expression in mammalian cells supplemented with pharmacological chaperones.
Courtesy: Generated by Copilot
6.Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
1.Template degradation by nucleases
Problem: Exogenous DNA/mRNA gets degraded by nucleases in lysate, leading to truncated or absent protein.
Fix: Add broad-spectrum RNase/DNase inhibitors (e.g., RNasin) or switch to engineered nuclease-deficient extraction strains (such as endA- E. coli strains)or employ highly stabilized linear expression templates.
2.mRNA secondary structure issues
Problem: Strong secondary structures near the 5’ end can block ribosome binding or cause pauses, reducing translation.
Fix: Introduce synonymous mutations to weaken hairpins without changing the protein sequence, or add chaperones to support more efficient translation.
3.Premature energy depletion
Problem: Rapid consumption of ATP/GTP in cell-free systems exhausts energy regeneration, stopping translation early.
Fix: Optimize energy regeneration components (e.g., PEP, creatine phosphate, feeding buffers) and/or reduce incubation temperature to extend system lifetime.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Courtesy:Monck, C., Elani, Y. & Ceroni, F. Genetically programmed synthetic cells for thermo-responsive protein synthesis and cargo release. Nat Chem Biol 20, 1380–1386 (2024). https://doi.org/10.1038/s41589-024-01673-7
1.Pick a function and describe it.
A time-lapse video showing cells of the synthetic organism JCVI-syn3A growing and dividing under a light microscope, from a research collaboration between the J. Craig Venter Institute, the National Institute of Standards and Technology and the Massachusetts Institute of Technology Center for Bits and Atoms. The scale bar represents 50 micrometers.
Credit: E. Strychalski/NIST and J. Pelletier/MIT
a.What would your synthetic cell do? What is the input and what is the output?
The synthetic cell should sense the bacterial biofilm produced and then devour the film formation. Synthetic cells should beengi neered with genetic circuits that sense chemical signals (autoinducers) produced by bacteria within a biofilm. Once a specific thtreshold is sensed, the synthetic cells activate, producing enzymes to break down the biofilm formed.
Input:Autoinducers like N-Acyl homoserine lactones (AHLs) or Autoinducer-2 (AI-2), which are produced by pathogenic bacteria to communicate and form protective biofilms.
Output:Lactonase or acylase to degrade the Quorum sensing signals, which halts the biofilm maturation.
b.Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes it can be realized in cell free systems except that devouring the biofilm cannot be performed in these systems instead these systems release enzymes like lactonases directly into the environment that degrades the extrapolymeric substances thet form the biofilm.
Susceptible to sample interference (contamination) in complex environments, unlike compartmentalized synthetic cells.
c.Could this function be realized by genetically modified natural cell?
Yes this function can be realised in genetically modified natural cells. Natural cells like E.coli cells can be engineered to sense the presence of the biofilms and this can trigger the promoter to produce lactonases or other enzymes that degrade biofilm.
d.Describe the desired outcome of your synthetic cell operation.
The use of synthetic cells helps to sense biofilm production and degrade the biofilm protecting the bacteria underneath. These bacteria are again susceptible to antibiotic treatment. The harmful bacteria is destroyed and the beneficical ones are not harmed thereby preventing off target effects.
2.Design all components that would need to be part of your synthetic cell.
a.What would be the membrane made of?
Membrane would be made of phospholipids and cholesterol. The cell meembrane is also modified to express receptors specific to Acylhomoserine lactones(AHL)), signal molecules produced during biofilm formation.
b.What would you encapsulate inside? Enzymes, small molecules.
The cell encapsulates a cell-free transcription-translation (TX-TL) system, which contains necessary enzymes (like T7 RNA polymerase, ribosomes), a DNA circuit activated by AHL signalling molecules, bound to the transcriptional regulator Lux R and expresses the enzyme encoded by DNA and a-haemolysin for pore formation.
c.Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
Bacterial Tx/Tl systems are best because the input is AHL that can be sensed by Lux R and initiate gene expression in synthetic cells.
d.How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The membrane is permeable to AHL, synthetic cell expresses a-haemolysin to create pores in membrane for release of lactonases to degrade the biofilm.
3.Experimental details
a.List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: Phospholipids, cholesterol
Enzymes: bacterial cell-free Tx/Tl, lactonases, Lux R
Genes: a-hemolysin (aHL)
b.How will you measure the function of your system?
ATP bioluminescence is a highly sensitive method to measure the efficiency of biofilm degradation. Since living cells contain ATP, the degradation of the biofilm directly correlates with a reduction in ATP levels.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Courtesy:Ho G, Kubušová V, Irabien C, Li V, Weinstein A, Chawla S, Yeung D, Mershin A, Zolotovsky K and Mogas-Soldevila L (2023) Multiscale design of cell-free biologically active architectural structures. Front. Bioeng. Biotechnol. 11:1125156. doi: 10.3389/fbioe.2023.1125156
1.Write a one-sentence summary pitch sentence describing your concept.
A wall hanging made using freeze dried cell free systems that can sense the quality of air within the house.
Courtesy: ChatGPT
2.How will the idea work, in more detail? Write 3-4 sentences or more.
The freeze dried cell free systems expressing B-galactosidase is embedded in the paint.A DNA-based sensor is designed using specific transcription factors (TFs) or toehold switches that respond to target indoor pollutants. When a specific pollutant (e.g., formaldehyde from furniture) reaches a certain concentration in the room, it acts as an environmental signal.
The Output: The biosensor activates and produces a colorimetric change (e.g., changing from white to blue).
The lyophilized cell free reaction powder is mixed with The powder is mixed with a bio-compatible, permeable binder (like specialized gels or alginate) and applied directly to the canvas or paper using screen printing or inkjet printing, allowing air to reach the sensor. The system requires moisture to function. To initiate the sensor, the user sprays a fine mist of water onto the painted area.
Reaction: If a pollutant is present, it diffuses into the spot, triggering the genetic circuit to produce the blue dye, thus changing the color.
3.What societal challenge or market need will this address?
These are programmable materials that contribute to sensing pollutants on rehydation with water and do not contain living, genetically modified organisms, making them safe for indoor environments.These kinds of products are made accesible to general public making tehm aware of the scientific advancements.
These meet the current market needs for a smart, interactive, and sustainable home decor. Cell-free systems allow for the creation of biodegradable smart materials, reducing reliance on conventional synthetic electronics and plastics.
4.How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The sensors have a limited lifespan (hours to days after rehydration) and need to be replaced, meaning the art piece must allow for new sensor spots to be applied over time.
This can be mitigated by using art designs in such a way that the lyophilized cell free systems are placed in certain pieces of the design that are removable probably that can be replaced with another new set of button of same shape in the design so that the wall hanging and its function is retained for years.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
1.Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Musculoskeletal decline in microgravity is a rapid, serious consequence of spaceflight, causing astronauts to lose 1–2% bone mineral density per month and substantial muscle mass, particularly in weight-bearing legs and back.The slow-twitch muscle fibres atrophying the most in the lower limb.The bones become brittle as the microarchitecture degrades.
The absence of gravity relates to no work required by bones and muscles to support the body so there is no bone formation and increased degradation.Mitochondrial stress and oxidative stress is more in space that leads to muscle degradation.
These help us understand better serious musculoskeletal diseaes on earth and how bone and muscle cells undergo damage on earth. This deterioration is a major health risk for long-term missions to Mars, as it can cause significant loss of function, increased fracture risk, and kidney stones from high calcium levels.
2.Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Pax7(Paired box 7)is a crucial transcription factor and the primary marker for muscle stem cells, known as satellite cells (SCs).It is involved in maintenance, self-renewal, and regenerative capacity of skeletal muscle.
3.Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
There is a decrease in expression of Pax7 in muscle tissues in spaceflight. This leads to reduced myogenic potential, decreased cell growth, and overall sarcopenia-like muscle weakening.Microgravity, leads to impairment of the normal functioning of these cells, directly impacting muscle regenerative capacity.The reduction in Pax7 is accompanied by an increase in muscle-wasting markers, such as myostatin, which further drives the degradation of muscle tissue in the absence of gravity.Microgravity causes muscles to experience significantly reduced mechanical load, leading to a decrease in Pax7+ cells and a corresponding decrease in muscle fiber size.
4.Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
The research goal is to analyze the Pax7 expression in samples from crew members by amplifying it in miniPCR cycler and then administer the pax7 expression sytems lyophilized as BioBits, rehydrated, injected into teh muscle of interest.
Pax 7 expression is crucial for muscle regeneration. Space microgravity and the infrequent usage of muscles in space leads to musculoskeletal decline that can be tried to combat by expressing the Pax7 in cell free systems and injecting them in affected area to regenerate the muscle mass.The DNA sampling using miniPCR cycler can enable a specific dosage of a Pax7-based therapy is effective in promoting regeneration or mitigating atrophy in space.
BioBits® pellets are freeze-dried and contain the essential cellular machinery (enzymes, ribosomes) needed for protein synthesis, activated simply by adding water and the appropriate DNA, making them ideal for space-based, refrigerated-free, on-demand drug synthesis.
The dosage of BioBits and number of times to be administered will depend on the muscle mass loss and time spent in space.These would require further experimental validation.
5.Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
miniPCR Analysis (Space-Based)
Blood samples are collected and analysed for muscle loss biomarkers before, during, and after spaceflight.
DNA is extracted using a commercial blood DNA extraction kit.
Amplify DNA using mini PCr cycler to analyse the markers for muscle loss.
Controls: Include positive control (known high-expression sample) and negative control (water).
BioBits Pax7 expression
Lyophilized cell-free extract (BioBits) containing E. coli transcription/translation machinery.
The BioBit pellets are rehydrated using DNA from samples of crew members.
Pax7 promoter with GFP is used as construct.
Incubate at 30-37°C for 4-12 hours in the miniPCR machine or heat block.
Visualize fluorescence on smartphone, indicating expression of the construct,to confirm that the synthetic construct is functional in microgravity.
Controls: Negative control (no cDNA), positive control (known muscle tissue sample).
This can be then administered via injections to affected areas.
Courtesy: Generated by Copilot
Homework Part B: Individual Final Project
My DNA Twist Order
References
“Generate image for PAX mini PCR.” Microsoft Copilot,22-5-2026, copilot.microsoft.com.
Ho G, Kubušová V, Irabien C, Li V, Weinstein A, Chawla S, Yeung D, Mershin A, Zolotovsky K and Mogas-Soldevila L (2023) Multiscale design of cell-free biologically active architectural structures. Front. Bioeng. Biotechnol. 11:1125156. doi: 10.3389/fbioe.2023.1125156
Monck, C., Elani, Y. & Ceroni, F. Genetically programmed synthetic cells for thermo-responsive protein synthesis and cargo release. Nat Chem Biol 20, 1380–1386 (2024). https://doi.org/10.1038/s41589-024-01673-7
Brookwell, August, Javin P. Oza, and Filippo Caschera. 2021. “Biotechnology Applications of Cell-Free Expression Systems” Life 11, no. 12: 1367. https://doi.org/10.3390/life11121367
Carlson, Erik & Gan, Rui & Hodgman, Charles & Jewett, Michael. (2011). Cell-Free Protein Synthesis: Applications Come of Age. Biotechnology advances. 30. 1185-94. 10.1016/j.biotechadv.2011.09.016.
“Optimised GPCR mebrane protein CFPS Image” Microsoft Copilot,22-5-2026, copilot.microsoft.com.
“Phosphoenol pyruvate system image” prompt. ChatGPT, GPT-4o version, OpenAI, 10 Mar. 2026, https://chatgpt.com
1.Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
In the final project the presence of specific mec A mRNA is detected using aptamer fluorescence.The fluorescence intensity is directly proportional to the amount of the mRNA present at the site.
The system relies on the COF acting as a quencher for a fluorophore-labeled aptamer. The efficiency of the “turn-on” signal upon target binding must be measured to calculate the detection limit.
The mec A mRNA knockdown is also measured along with protein depletion using RT-qPCR and Western Blot respectively.The phenotypic Minimal Inhibitory Concentration (MIC) shift assay is also measured to measure thesensitivity of the bateria after treatment.
The cell wall binding domain specificity to MRSA bacteria is also measured by flow cytomeery of the pellet and pellet fold change of MRSA, MSSa and E.coli cells.
2.Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Measure the fluorescence recovery upon target binding (mecA mRNA)
Determine the lowest concentration of mec A mRNA that can be detected.
Ensure the aptamer only binds to the targeted mecA mRNA.
The cell wall binding specificity to bacteria is also studied to measure FAM fluorescence in pellet and supernatant by Flow cytometry.
The mRNA silencing is measured by RT-qPCR and protein depletion by Western blot.
3.What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Smartphone-based Handheld Readers: 3D-printed attachments for smartphones that integrate a violet light source (LED) and a specialized emission filter, allowing them to serve as portable, low-cost scanners for quantifying fluorescence intensity.
The COF surface acts as a proximity quencher, holding the FAM-tagged aptamer in a dark OFF state through the inner filter effect and photoinduced electron transfer. Upon binding mecA mRNA, the aptamer undergoes a conformational change that separates the FAM fluorophore from the COF surface, generating a bright turn-on fluorescent signal detectable by the handheld device.
Courtesy: Claude.ai
Homework: Waters Part 1 — Molecular Weight
1.We will be analyzing an eGFP standard onto a BioAccord LC-MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the denatured (unfolded) state. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
1.Based only on the predicted amino acid sequence of eGFP (see below), what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
2.Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation.
Select two charge states from intact LC-MS data (Figure 1) and:
3.Calculate the mass accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using these formulae:
3.Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No we cannot see it.To determine the charge state ($z$) directly from a single zoomed-in peak envelope, you must be able to observe the spacing between the individual isotopic peaks ($\Delta m/z$). In the inset the peaks are not baseline resolved.We cannot observe it directly from osotopes in inset , we can calculate it.
1. Main Spectrum Charge State ($z_2$) Determination:
Using two adjacent, highly abundant peaks:
$m_1 = 848.9758$
$m_2 = 824.0635$
Formula for the charge state of the lower $m/z$ peak:
$$z_2 = \frac{m_1 - 1.0078}{m_1 - m_2}$$
Using the $1\text{ Da}$ approximation:
$$z_2 = \frac{848.9758 - 1}{848.9758 - 824.0635} \approx 34$$
2. Intact Mass ($M$) of eGFP Protein:
Knowing the charge is $+34$ for the $824.0635$ peak:
$$M = (34 \times 824.0635) - 34$$
$$M \approx 27,984.16\text{ Da}$$
3. Zoomed-In Peak Charge State Determination:
Using the intact mass ($M \approx 27,984\text{ Da}$) to find the charge state for a peak centered around $m/z \approx 1474$:
$$z = \frac{M}{m/z}$$
$$z = \frac{27984}{1474} \approx 18.98 \rightarrow z = 19+$$
Conclusion:
The peak at $m/z \approx 1474$ represents the $+19$ charge state.
Homework: Waters Part 2 — Secondary/Tertiary structure
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Native proteins maintain their folded, biologically active structures, while denatured proteins unfold into extended, disordered chains due to disruption of non-covalent interactions like hydrogen bonds and hydrophobic effects.Denaturation (e.g., via acid like formic or heat) breaks the highly ordered tertiary or quarternary structure, yielding random coils with exposed residues. Unfolding increases solvent-accessible surface area, often shifting charge statesNative proteins typically show narrow, high-charge distributions (e.g., +4 to +7 here), while denatured ones display broader, lower-charge envelopes (+3 to +5).
Mass spectrometers distinguish via charge state distributions (CSD) in ESI-TOF spectra: native MS yields narrow, symmetric peaks at higher m/z (lower z, intact fold limits adduction); denatured MS shows wider CSD at lower m/z (higher z, flexible chain binds more ions). Isotope resolution sharpens in native due to stable conformers.
Top Spectrum (Green, Denatured): This spectrum shows a broad distribution of many peaks across a wide range of mass-to-charge ratios (m/z), particularly at lower (m/z) values. This is characteristic of a highly charged species, as the charge (z) is high and the mass (m) is the same.
Bottom Spectrum (Red, Native): This spectrum shows fewer, more intense peaks clustered at higher (m/z) values. This indicates a lower charge state distribution, consistent with a compact, folded protein where fewer sites were available for protonation.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 Q-Tof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
2. Charge State Calculation ($z$):
Using the monoisotopic mass difference of carbon-13 ($\Delta m \approx 1.00335\text{ Da}$):
$$z = \frac{1.00335}{\Delta m/z}$$
Substituting the value:
$$z = \frac{1.00335}{0.0918} \approx 10.93$$
Final Result:
Calculated charge: $z \approx 11$
The charge state of the peak at approximately (2800,m/z) is approximately 11. This is determined by measuring the mass-to-charge difference between adjacent isotopic peaks in the high-resolution inset and dividing the known isotopic mass difference (approx. 1.00335 Da) by the observed (m/z) difference.
Homework: Waters Part 3 — Peptide Map Work - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
The lysine residues are marked in yellow and arginine residues are marked in light blue.
1.How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
There are 20 lysines(K) and 6 Arginines(R) in eGFP.
2.There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (bioinformatics resource portal of the SIB Swiss Institute of Bioinformatics) to predict a list of tryptic peptides from eGFP.
How many peptides will be generated from Tryptic digestion of eGFP?
3.Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes?You may count all peaks that are >10% relative abundance.
The maximum peak height is at 1.2e7 so all peaks that are >10% relative abundance, should have 0.1*1.2e7 = 1.2e6. There are 21 such peaks.
4.Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from Step 2.3 and 2.4? Are there more peaks in the chromatogram or fewer?
There are only 19 peptides predicted. So there are more peaks in the chromatogram. These may be due to impurities in the sample or sample degradation.
5.Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide based on its m/z and z ([M+H]+).
The most abundant peak in Figure 5b (GFP digest spectrum) is at m/z 525.76712, representing the monoisotopic m/z of the peptide’s most abundant charge state.
From the zoomed-in inset in the image, the m/z values of two adjacent major isotopic peaks are approximately 525.76712 and 526.25918.
The difference between these values is 526.25918-525.76712 = 0.492
1. Charge State ($z$) Determination:
$$\Delta m/z = 0.492$$
$$z = \frac{1}{0.492} \approx 2.03 \rightarrow z = 2+$$
2. Mass-to-Charge Relation Formula:
$$\frac{m}{z} = \frac{M + z \cdot \text{H}^+}{z}$$
7.What is the percentage of the sequence that is confirmed by peptide mapping (Figure 6)?
The image shows a peptide mapping result for GFP (Chain 1), explicitly labeling the identified sequence coverage as 88%.
Bonus Questions
8.Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html.
What is the sequence of the eGFP peptide that best matches the MS/MS fragmentation spectrum in Figure 5c?
9.Do the Peptide Map data make sense and do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, showing the % amino acid coverage of peptides positively identified by their calculated mass and fragmentation pattern.
The results make sense because they are from the fragments of GFP protein as they map to different protions of GFP sequence and it identified 88%.The rest of the sequences were not properly digested probably. There could be presence of post translational modifications that interfered with the expected mass and could not be matched with theoretical sequence.
Homework: Waters Part 4 — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Oligomer Identifications
The peak at ~4.01 MDa corresponds to a decamer of 7FU subunits (10 × 340 kDa = 3.4 MDa; observed shift due to glycosylation/adducts common in CDMS).
The peak at ~7.52 MDa aligns with a decamer of 8FU subunits (10 × 400 kDa = 4 MDa; higher oligomers or mixtures may shift observed mass).
The peak at ~8.33 MDa likely represents a di-decamer (20mer) of 7FU (20 × 340 kDa ≈ 6.8 MDa; adducted/glycosylated).
The peak at ~12.67 MDa corresponds to a di-decamer of 8FU (20 × 400 kDa = 8 MDa; further adducted) or larger assemblies.
Smaller peaks (e.g., ~3.4 MDa, ~0.98 MDa) may indicate monomers or fragments, but primary oligomeric species match decameric and didecameric forms typical of KLH
Homework: Waters Part 5 — Did I make GFP?
References
1.https://web.expasy.org/compute_pi/- for molecular weight calculation.
2.“Measurement technologies” prompt. Claude, version used (e.g., Claude 3.5 Sonnet version), Anthropic, Date accessed (e.g., 22 May 2026), claude.ai.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
2.Make a note on your HTGAA webpages including:
a.What you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
I made the upper triangle part before it was modified.9 pixels.
The number of wells I contributed towards the design.
b.What you liked about the project
I liked the way the 1536 plate has been converted for bioart purpose instead of a test validation plate, i also liked the teamwork from people all around the world come together to get a unified artwork done. The best part was involving different fluorescent protein for colouring the pixel.
c.What about this collaborative art experiment could be made better for next year.
Increase the number of plates if possible, so that we can make a bigger design!! I contributed very little, missed the fun.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1.Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Salts/Buffer
Potassium Glutamate
HEPES-KOH pH 7.5
Magnesium Glutamate
Potassium phosphate monobasic
Potassium phosphate dibasic
Energy / Nucleotide System
Ribose
Glucose
AMP
CMP
GMP
UMP
Guanine
Translation Mix (Amino Acids)
17 Amino Acid Mix
Tyrosine
Cysteine
Additives
Nicotinamide
Backfill
Nuclease Free Water
Lysate and Buffer System
E. coli Lysate (BL21 (DE3) Star): Contains the essential cellular machinery, including ribosomes, tRNAs, and native enzymes, required to drive the translation of proteins from a DNA template.
Potassium Glutamate: Acts as the primary salt to maintain optimal osmotic pressure and ionic strength for efficient enzyme activity and protein folding.
HEPES-KOH pH 7.5: Functions as a buffering agent to maintain a stable physiological pH throughout the reaction.
Magnesium Glutamate: Provides essential Mg 2+ions, which are critical cofactors for ribosomes, RNA polymerase, and the stabilization of nucleic acid structures.
Potassium phosphate (monobasic/dibasic): Acts as a secondary buffer and a crucial source of inorganic phosphate to support metabolic energy regeneration.
Energy and Nucleotide System
Ribose and Glucose: Serve as sustainable metabolic substrates that are broken down by endogenous enzymes in the lysate to regenerate ATP and other required nucleotides.
AMP, CMP, GMP, UMP, and Guanine: Provide the essential nucleotide precursors that are phosphorylated or converted in situ into triphosphates (ATP, CTP, GTP, UTP) necessary for transcription and translation.
Translation Mix and Additives
17 Amino Acid Mix, Tyrosine, and Cysteine: These serve as the fundamental building blocks required by ribosomes to assemble polypeptide chains during translation.
Nicotinamide: Often acts as a modulator of cellular metabolic pathways and enzyme activity, which can help support extended reaction longevity and stability.
Nuclease Free Water: Provides the solvent medium for the reaction while ensuring the absence of enzymes that would otherwise degrade the DNA or RNA templates.
2.Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
1-Hour Optimized PEP-NTP Master Mix
Energy Source: Uses Phosphoenolpyruvate (PEP) to directly regenerate NTPs (Nucleoside Triphosphates) from Nucleoside Monophosphates (NMPs).
Components: Contains high-energy intermediates designed for immediate enzymatic conversion.
Fast turnover for high-throughput screening, short duration cell - free protein synthesis.
PEP is expensive
20-Hour NMP-Ribose-Glucose Master Mix
Energy Source: Relies on Ribose (likely Ribose-5-phosphate) and Glucose, which fuel slower, more complex metabolic pathways (like the pentose phosphate pathway and glycolysis) to generate precursors and energy.
Components: Contains base substrates (NMPs, sugars) that require enzymatic conversion to reach the final product, suitable for cell-free systems relying on metabolic regeneration.
Extended for maximal yield accumulation
Cost-effective substrates; sustained energy
Courtesy: Perplexity
3.How can transcription occur if GMP is not included but Guanine is?
Transcription occurs if GMP is not included but Guanine is present via the salvage pathway.
In this pathway,cells can recycle free guanine or convert it into guanine nucleotides using enzymes like Hypoxanthine-guanine phosphoribosyltransferase (HGPRT). This enzyme converts Guanine and PRPP (5-phosphoribosyl-1-pyrophosphate) into GMP.GMP is then phosphorylated by guanylate kinase into GDP, which is subsequently converted into GTP by nucleoside-diphosphate kinase (NDK).
While GTP (not GMP) is the actual substrate required for RNA synthesis,the GTP synthesized via the salvage pathway from guanine can then be used in transcription.
Courtesy: Copilot
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1.Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP
mRFP1
mKO2
mTurquoise2
mScarlet_I
Electra2
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
sfGFP (Superfolder GFP):
Property: Extremely fast folding and enhanced solubility.
Effect: sfGFP folds correctly even when fused to poorly folding proteins, making it an ideal, reliable reporter for protein synthesis in E. coli cell-free lysates. It matures quickly, allowing for rapid readout of gene expression.
mRFP1 (Monomeric Red Fluorescent Protein 1):
Property: Rapid maturation compared to its parent protein (DsRed).
Effect: Rapid maturation allows for faster detection of protein synthesis. As a monomer, it does not form tetramers that could hinder the functional activity of fused proteins, allowing for more accurate, quantitative readouts of protein expression levels.
mKO2 (Monomeric Kusabira Orange 2):
Property: High brightness and fast maturation.
Effect: mKO2 offers a bright, orange readout that enables strong signals in cell-free systems, enabling high sensitivity in biosensor applications. Its high brightness is critical when measuring low-level synthesis.
mTurquoise2 (mTq2):
Property: High quantum yield (QY=0.93) and excellent photostability.
Effect: Because mTurquoise2 is one of the brightest cyan fluorescent proteins available, it is an excellent FRET donor. In cell-free systems, it provides a very high signal-to-noise ratio and, due to its mono-exponential lifetime, it is optimal for FLIM (Fluorescence Lifetime Imaging) readouts.
mScarlet_I:
Property: High molecular brightness (product of extinction coefficient and quantum yield).
Effect: As a bright red monomeric FP, mScarlet_I provides high-intensity, red-shifted fluorescence (good for avoiding auto-fluorescence) and is commonly used for quantifying protein production in eukaryotic cell-free systems, maturing rapidly to report on translation.
Electra2:
Property: Good solubility and stability with specific sensitivity to acidity.
Effect: Electra2 behaves as a stable blue fluorescent protein (BFP) suitable for tracking in cell-free systems. However, like other DsRed-derived proteins, it can form aggregates (puncta) in certain conditions and its readout can be affected by the pH of the cell-free reaction.
2.Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
sfGFP
Hypothesis: For sfGFP, increasing Mg²⁺ slightly and maintaining adequate amino-acid supply plus an energy-regeneration system will increase the time-integrated fluorescence over 36 hours because sfGFP benefits from efficient translation and robust folding into its fluorescent β-barrel.
Expected effect: More mature, functional sfGFP accumulates, so fluorescence rises faster and stays higher for longer.
mRFP1
Hypothesis: For mRFP1, adjusting the mastermix to favor a mildly oxidizing environment and sufficient tyrosine / precursor availability will improve chromophore maturation efficiency and increase fluorescence over 36 hours because red fluorescent proteins rely strongly on correct chromophore formation.
Expected effect: A larger fraction of synthesized mRFP1 becomes fluorescent rather than remaining non-matured protein.
mKO2
Hypothesis: For mKO2, increasing energy-regeneration components and maintaining stable pH buffering will improve peak brightness and sustained fluorescence because orange fluorescent proteins are sensitive to prolonged expression conditions and benefit from continued synthesis without pH-driven signal loss.
Expected effect: Higher and more stable orange fluorescence across the 36-hour period.
mTurquoise2
Hypothesis: For mTurquoise2, optimizing Mg²⁺ and potassium glutamate to support ribosome performance will improve folding efficiency and total fluorescent output over 36 hours because cyan proteins can be limited more by expression yield than by intrinsic chromophore formation.
Expected effect: More correctly folded cyan protein accumulates, raising fluorescence amplitude and duration.
mScarlet_I
Hypothesis: For mScarlet_I, supplementing the mastermix with robust energy-regeneration substrates and a slightly oxidizing buffer condition will improve chromophore maturation and ong-term signal retention over 36 hours because bright red proteins often depend on maintaining productive folding while avoiding late-stage signal decay.
Expected effect: Stronger red fluorescence appears earlier and remains detectable longer.
Electra2
Hypothesis: For Electra2, tuning redox balance and ionic strength in the mastermix will improve functional fluorescence yield over 36 hours because this protein’s mature fluorescent state is likely sensitive to folding environment and prolonged incubation stability.
Expected effect: A higher fraction of Electra2 reaches and maintains its fluorescent conformation, increasing total fluorescence.
3.The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
This is just a trial and error attempt for the cell-free mix.
I have designed wells based on above hypothesis for each fluorescent protein in each wells.How the composition varies for each fluorescent protein is my aim and the effect it produces on teh protein properties.
Composition of each well
Here I have adjusted the well with potassium glutamate as it is responsible for maintaing ionic strength,and the reagents related to redox balance like glucose, ribose as that improves the fluorescence of Electra2 fluorescence in a 36 hour incubation.
In this well the potassium glutamate is again varied for maintaining ionic strength. The HEPES_KOH pH composition is varied to 46.250 mM, ribose and GMP is also varied, guanine and glucose are varied with potassium phosphate monobasic to ,maintain rdox balance and energy composition in the well and study its effect on Electra2 fluorescence.
In this well the mScarlet-I fluorescence is studied by varying the composition of HEPES-KOH pH , ribose, AMP,GMP, glucose, potassium phosphate monobasic and dibasic and guanine, which is increased to produce robust energy-regeneration substrates and a slightly oxidizing buffer condition for increase in its fluorescence.
In this well sfGFP fluorescence is studied bby varying the composition of magnesium glutamate, tyrosine, t7 amino acid mix, ribose, guanine, glucose, which are increased,as Mg² and adequate energy regeneration will help it fluoresce better.
In this well mKO2 fluorescence is studioed by varying composition of HEPES-KOH pH, potassium glutamate, ribose, GMP, guanine, which is increased.As increasing energy regeneration components and maintaining pH will help it fluoresce better.
In this well the fluorescence of mTurquoise2 is studied by varying composition of potassium glutamate, magnesium glutamate and GMP, which are increased.Optimising Mg2+ and potassium glutamate will help it fluoresce better.
In this well mRFP1 fluorescence is studied by varying composition of tyrosine, t7 amino acid master mix, potassium phosphate monobasic and dibasic, GMP and ribose, which is increased.The mild oxidising environment and tyrosine sufficiently available increases fluorescence.
In this well again Electra2 fluorescence is studied by varying the composition of potassium glutamate, HEPES-KOH pH to 47.500 mM, ribose , GMP , glucose, potassium phosphate monobasic and nicotinamide, which are increased.
4.The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
This part of the data is yet to be generated.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!