Week-04-hw-protein-design-part-1
https://bio.libretexts.org/Workbench/Bio_11A_-_Introduction_to_Biology_I
Homework: Protein Design I
Part A. Conceptual Questions
1.Why are there only 20 natural amino acids?
The 20 natural amino acids evolved as optimal sets very early, during the RNA world (4 billion years ago). The format was not changed and became frozen because it would disrupt all proteins and also due to tRNA recognition limitations further expansion was prohibited.
2.Where did amino acids come from before enzymes that make them, and before life started?
The amino acids were formed by abiotic processes on early Earth(4.5 billion years ago) using gases, minerals and energy sources present at that time.
Miller-Urey experiment simulated the similar environment in their experiment and created glycine,alanine and 33 other amino acids by condensation and reduction.
The paper cited gives a good insight into this chicken and egg theory Singh, J., Thoma, B., Whitaker, D. et al. Thioester-mediated RNA aminoacylation and peptidyl-RNA synthesis in water. Nature 644, 933–944 (2025). https://doi.org/10.1038/s41586-025-09388-y
3.If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
L-amino acids form righthanded α-helices because their chirality favours such formation to prevent steric clashes in the side chains. In contrast the D-amino acids should prefer left handed helices to prevent steric clashes in the side chains.
4.Can you discover additional helices in proteins?
There are other helix types like the 3₁₀-helices, π-helices, and polyproline II (PPII) helices. They are formed by specific hydrogen bonding patterns and amino acid sequences.

Coutesy:Perplexity Pro prompt alpha helices
5.Why most molecular helices are right-handed?
The molecular helices in biology are right handed because of the L-chirality of amino acids and D-sugars. These molecular conformations sterically favour the right handed twist for stability and folding efficiency.
6.Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheet aggregate due to hydrogen bond donors/acceptors at their edges, promote edge to edge interactions with other sheets or unfolded chains.Hydrophobic side chains on edges prefer being buried by intermolecular contacts, leading to intermolecular associations that extend sheets into fibrils or amyloids.
The primary driving force for β-sheet aggregation is thermodynamics. The hydrogen bonds and Van der Waal’s forces lower free energy, further by cooperativity by dimerization. Aggregation occurs when the hydrophobic residues bury themselves in a compact core-this “collapse” reduces solvent-exposed area and drives entropy gain from released water molecules.
7.Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many amyloid diseases occur because of misfolding of proteins and adoption of β-sheet conformation and then self assembly into insoluble fibrils. Destabilization of native protein structure occurs first, then partial unfolding leading to exposure of β-strand regions that stack via hydrogen bonding into cross-β-sheet architectures. The fibrils formed are highly ordered parallel or antiparallel β-sheets,aggregate in a prion like manner, leading to plaque formation that disrupt tissue function in conditions like Alzheimer’s and type II diabetes.
Amyloid β-sheets as materials can be used as biomaterials because of their exceptional mechanical strength,biocompatibility, and nanoscale self-assembly. Non-pathogenic or engineered amyloid fibrils form robust scaffolds for tissue engineering, drug delivery, and biosensors. They mimic extracellular matrices to support cell adhesion and growth. They allow fabrications with bioplastics, hydrogels, and functional coatings for tunable properties via genetic modification or hybridization with nanoparticles.

Courtesy:Fallot LB, Natarajan C, Anderson CA, Nagelli EA, Burpo FJ, Limbocker R. From Pathology to Materials Science and Engineering: Harnessing the Amyloid State for Biotechnological Applications. ACS Appl Mater Interfaces. 2025 Nov 19;17(46):62839-62859. doi: 10.1021/acsami.5c11847. Epub 2025 Nov 10. PMID: 41211864; PMCID: PMC12635977.
8.Can you make other non-natural amino acids? Design some new amino acids.
Yes we can make.
The side chain of the amino acid has to be modified by methylation or some other functional group, or with another side chain that is bulky. Advantages: Green, selective; challenges: Low yield, stability issues.
9.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We eat beef or fish whuch is later broken down by the enzymes in our body into the building blocks of the biomolecules present in each. The whole genome of the the fish or cow is not integerated within our DNA so that we become cow or fish.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
mCardinal is the far red fluorescent protein I have chosen. It is a bright, monomeric,derived from Entacmaea quadricolor, with an emission peak around 656 nm.
I chose this because its excitation at 604 nm and emission at 659 nm, is the optimal far-red range for deep-tissue penetration. It is far brighter than mKate2 and other early-generation far-red variants.The monomeric form of the fluorescent protein, minimizes toxicity and can be used as fusion tags with target proteins without causing aggregation. Highly photostable so can be used for long term imaging.
2.Identify the amino acid sequence of your protein.
MVSKGEELIK ENMHMKLYME GTVNNHHFKC TTEGEGKPYE GTQTQRIKVV EGGPLPFAFD ILATCFMYGS KTFINHTQGI PDFFKQSFPE GFTWERVTTY EDGGVLTVTQ DTSLQDGCLI YNVKLRGVNF PSNGPVMQKK TLGWEATTET LYPADGGLEG RCDMALKLVG GGHLHCNLKT TYRSKKPAKN LKMPGVYFVD RRLERIKEAD NETYVEQHEV AVARYCDLPS KLGHKLNGMD ELYK
Courtesy:FPBase
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.

The protein is 268 amino acids long. The most common amino acid is G, it occurs 25 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
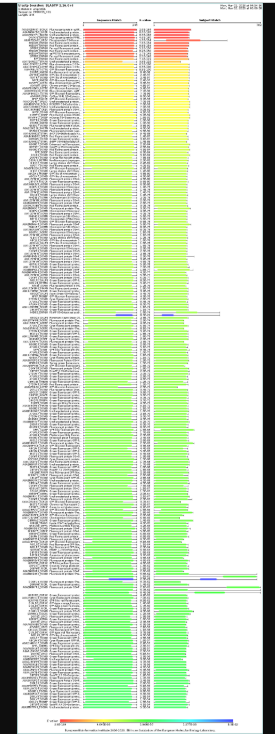
courtesy:The UniProt Consortium UniProt: the Universal Protein Knowledgebase in 2025 Nucleic Acids Res. 53:D609–D617 (2025)
It has many homologs and some of them are uncharacterised proteins too. Mostly the homologs belong to the red fluorescent protein family.
Does your protein belong to any protein family?
mCardinal belongs to the GFP-like protein family (specifically the Green Fluorescent Protein superfamily)
3.Identify the structure page of your protein in RCSB
Reference: H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne, The Protein Data Bank (2000) Nucleic Acids Research 28: 235-242 https://doi.org/10.1093/nar/28.1.235.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure was solved in 2014.it a good quality structure. its resolution is 2.21Å.
Are there any other molecules in the solved structure apart from protein?
No.
Does your protein belong to any structure classification family?
It belongs to family of Fluorescent proteins.
4.Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands) citation: Schrödinger, LLC. (2026). The PyMOL Molecular Graphics System, Version X.X. https://www.pymol.org/support.html.
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
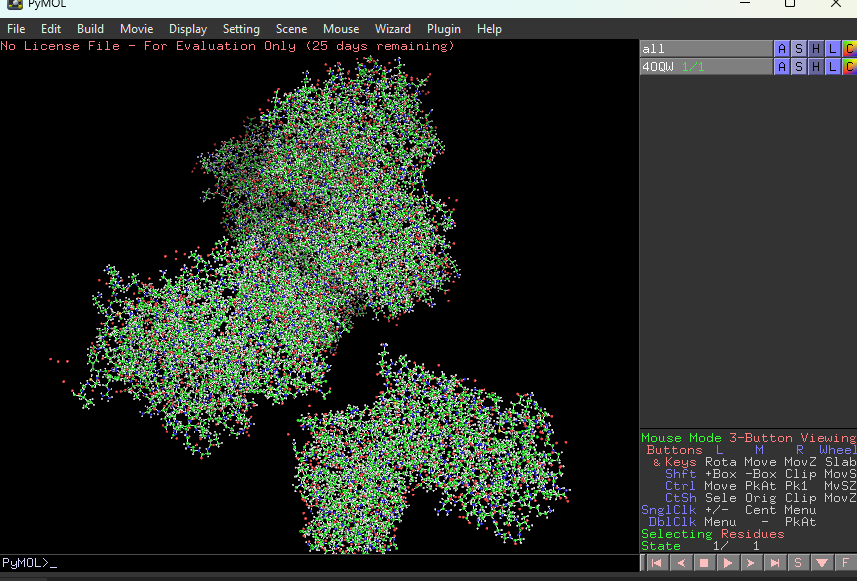
This represents ball and stick model of protein
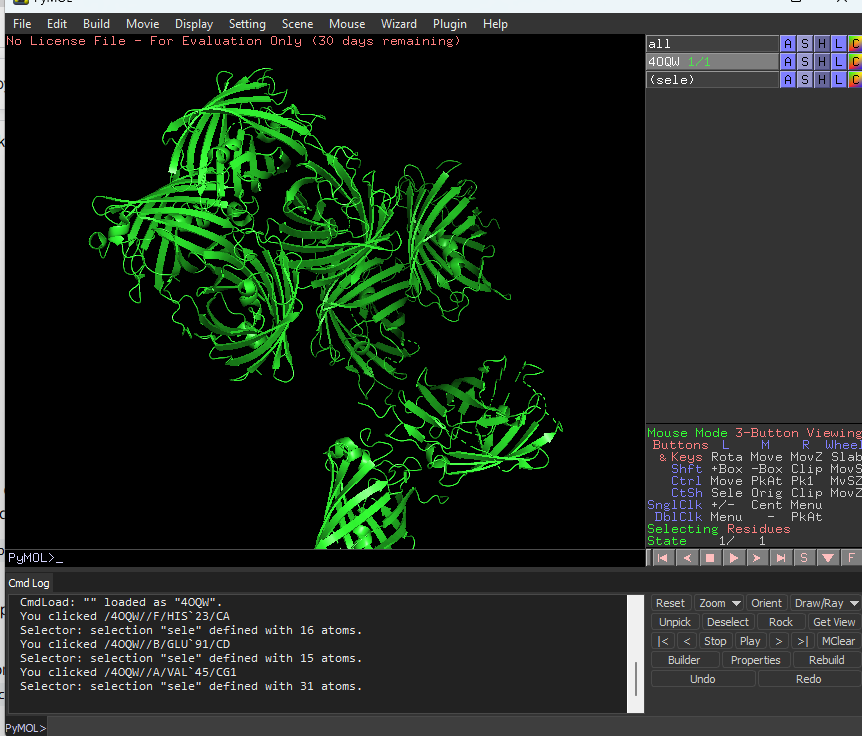
This is cartoon represenattion of protein
This is ribbon representation of protein
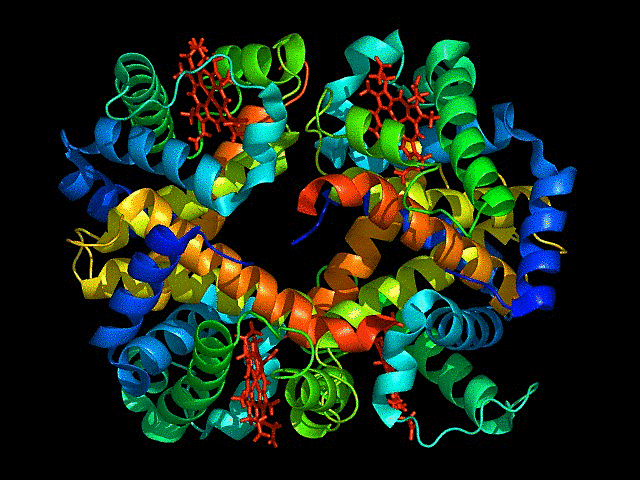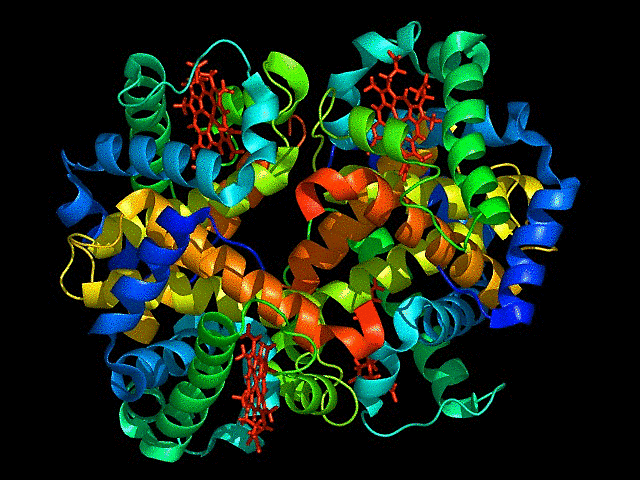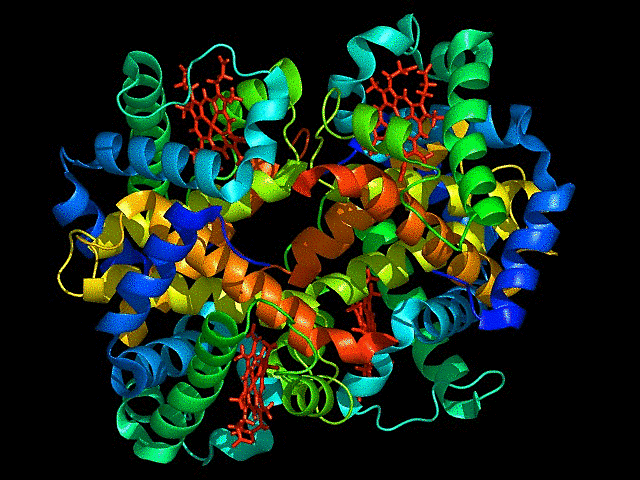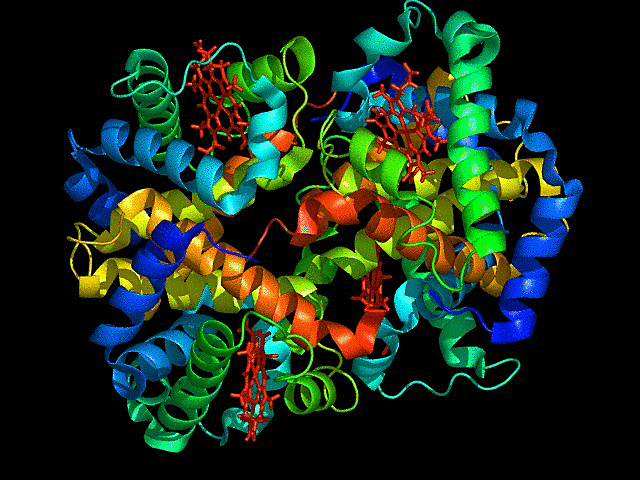
Color the protein by secondary structure. Does it have more helices or sheets?
It has more sheets.Helices are red, sheets are yellow and loops are green.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The hydrophobic residues are yellow in colour and hydrophilic are gray in colour. This colour combination tells us that hydrophilic residues are more towards the outer side of protein and hydrophobic residues lie within the molecule buried inside.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes the protein has binding pockets for chromophore binding.The darker regions near the rainbow region is the cavity or pocket.
Part C. Using ML-Based Protein Design Tools
1.Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
2.Choose your favorite protein from the PDB
I am choosing the mCardinal far red fluorescent protein.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1 Protein Language Modeling
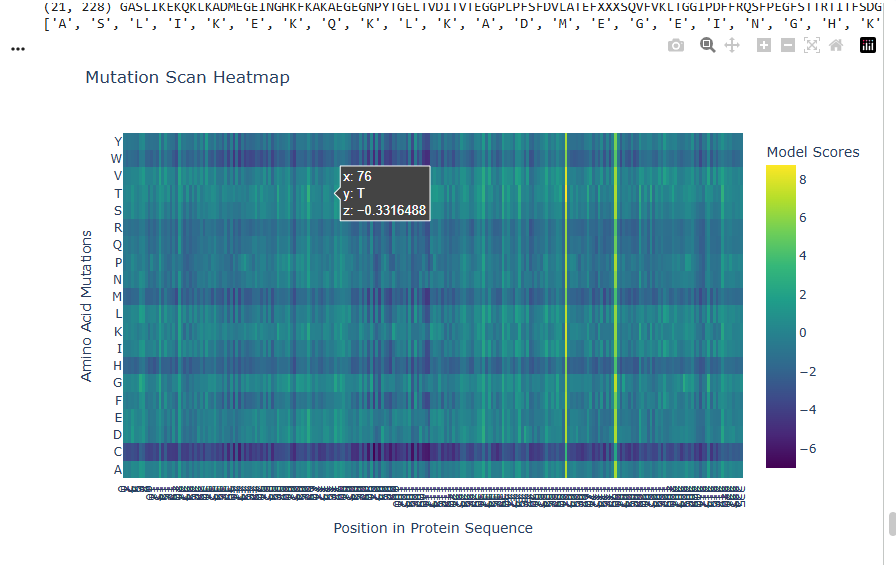
1.Deep Mutational Scans
a.Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
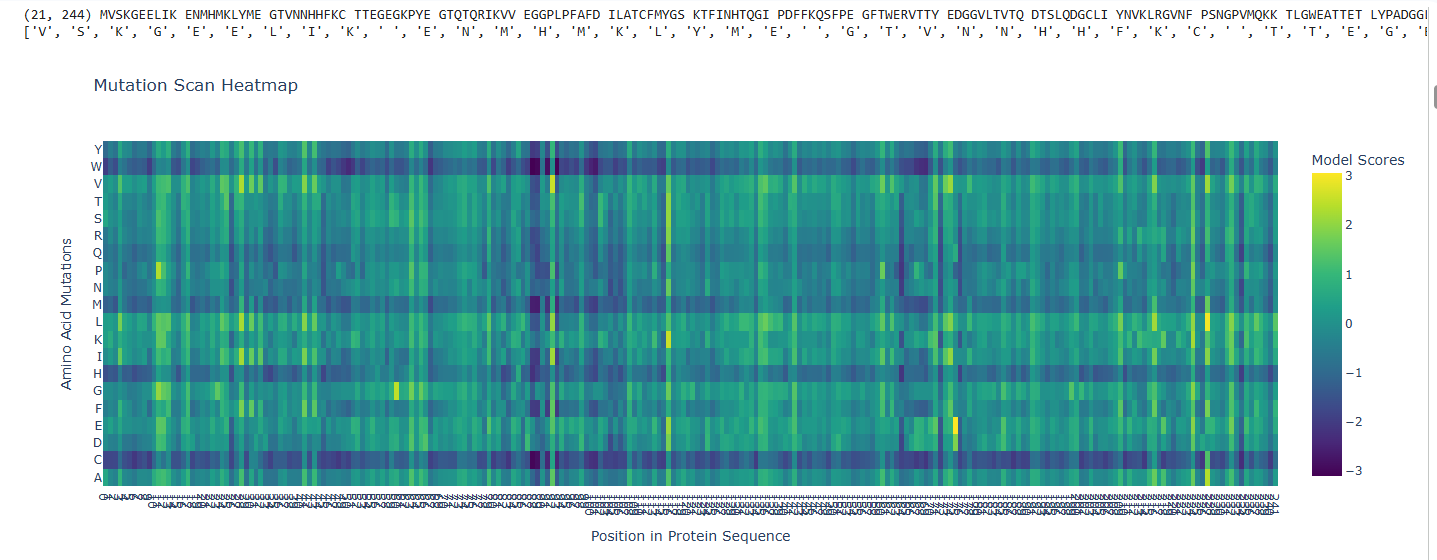
b.Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The dark blue region corresponding to w on y- axis and m- axis. thes lead to probably disruptive mutations. This region is highly conserved. The darkest puple seen here has a negative value of -3.11.
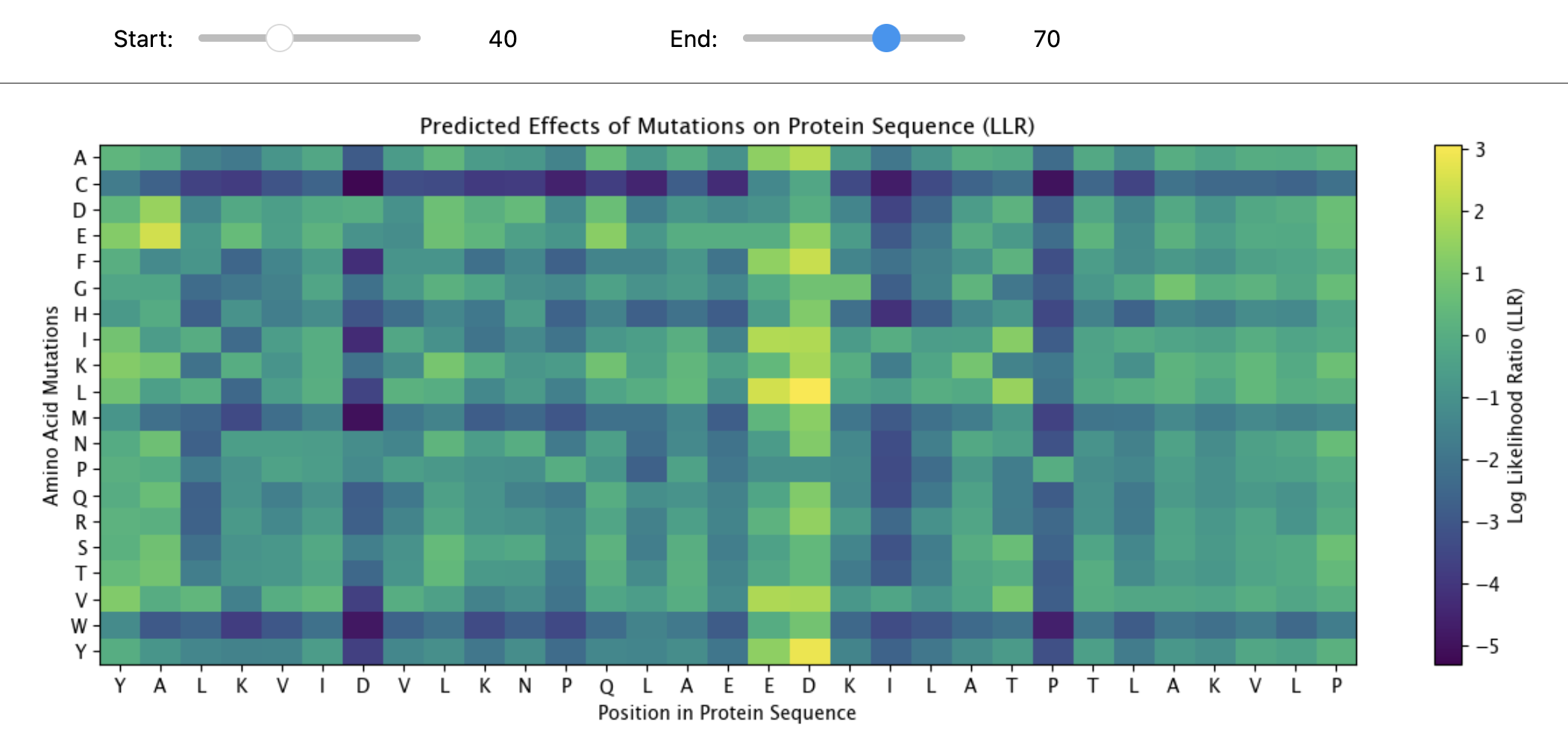
c.(Bonus)Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
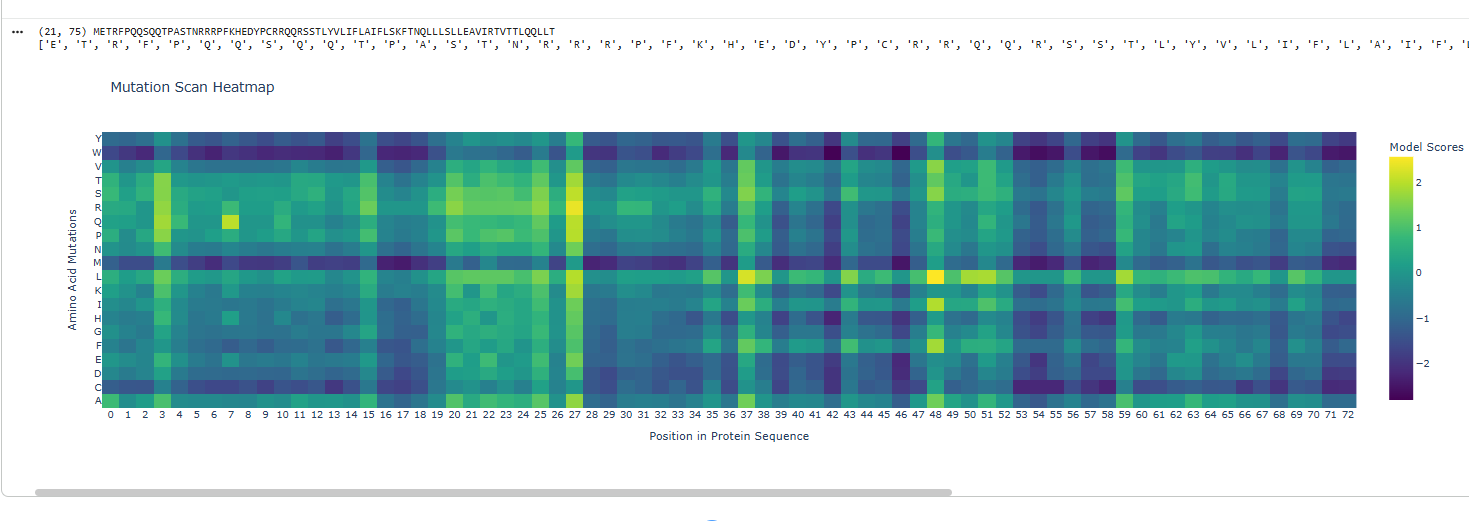
“MAPLRKTYVLKLYVAGNTPNSVRALKTLNNILEKEFKGVYALKVIDVLKNPQLAEEDKILATPTLAKVLPPPVRRIIGDLSNREKVLIGLDLLYEEIGDQAEDDLGLE”
The UniProt Consortium UniProt: the Universal Protein Knowledgebase in 2025 Nucleic Acids Res. 53:D609–D617 (2025)
The protein sequence provided corresponds to the Circadian clock oscillator protein KaiB in cyanobacteria. It is frequently used as a test sequence for analyzing protein structure, stability, and mutations in AI-driven protein language models.
Yes, the two images are structurally similar in that they are both heatmaps designed to visualize the effects of amino acid mutations on a protein sequence.
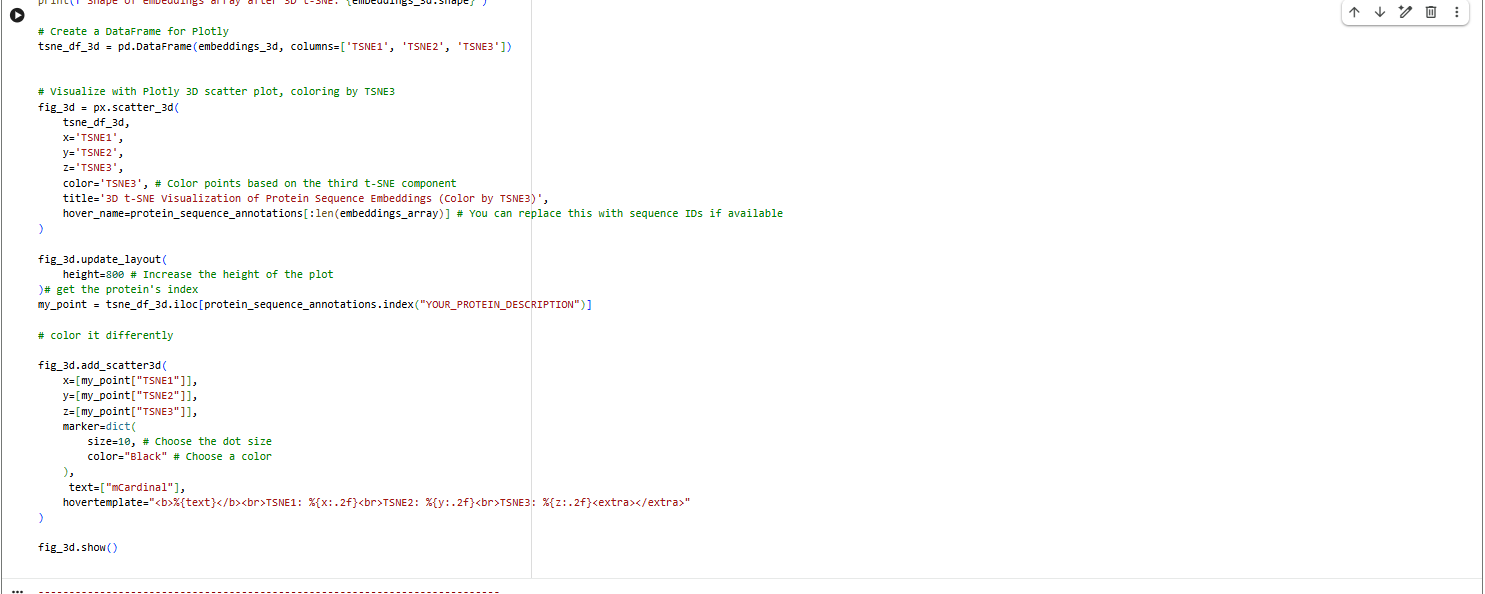
Latent Space Analysis
a.Use the provided sequence dataset to embed proteins in reduced dimensionality.
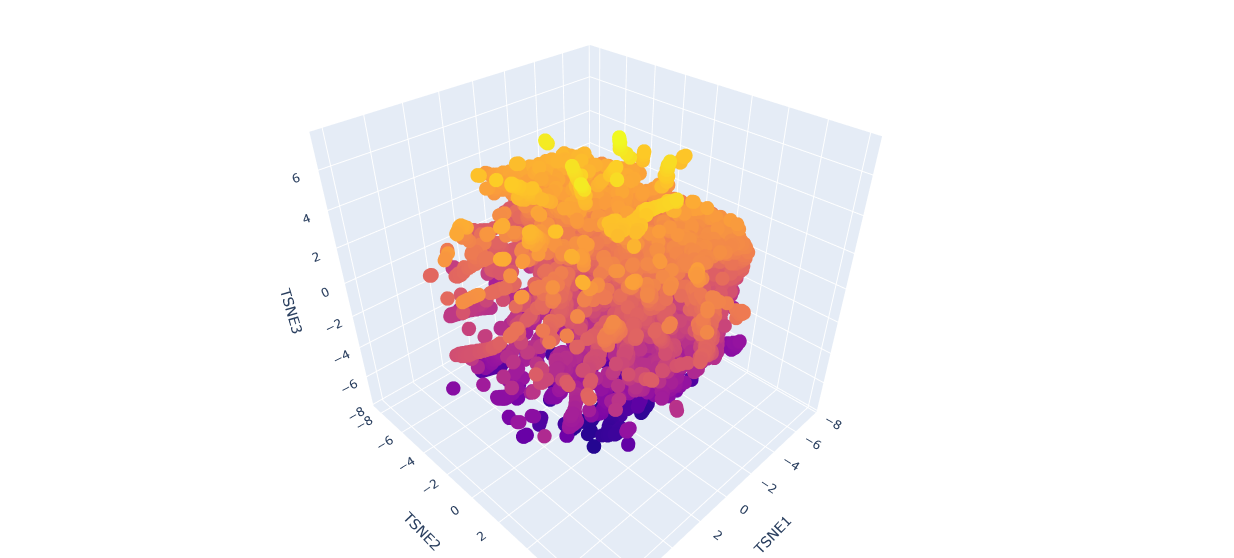
b.Analyze the different formed neighborhoods: do they approximate similar proteins?
The proteins in the neighbourhood approximate similar proteins.
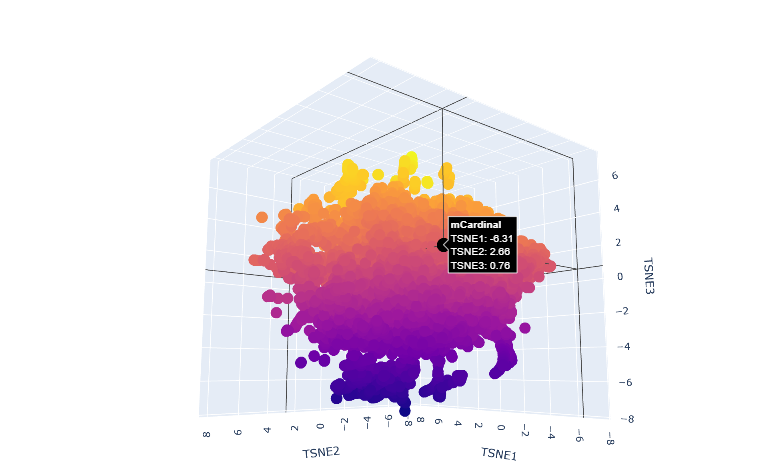
c.Place your protein in the resulting map and explain its position and similarity to its neighbors.
The orange-to-purple gradient likely shows point density (denser orange/yellow central cloud for common mCardinal-like sequences like Vibrio cholorae. The main orange-yellow hexagonal group (tSNE1 ≈ -1 to 3, tSNE2 ≈ -2 to 2) likely includes related far-red/red emitters like mNeptune, eqFP578, mKate2, or TagRFP, derived from similar Anthozoa.
C2 Protein Folding
Folding a protein
1.Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
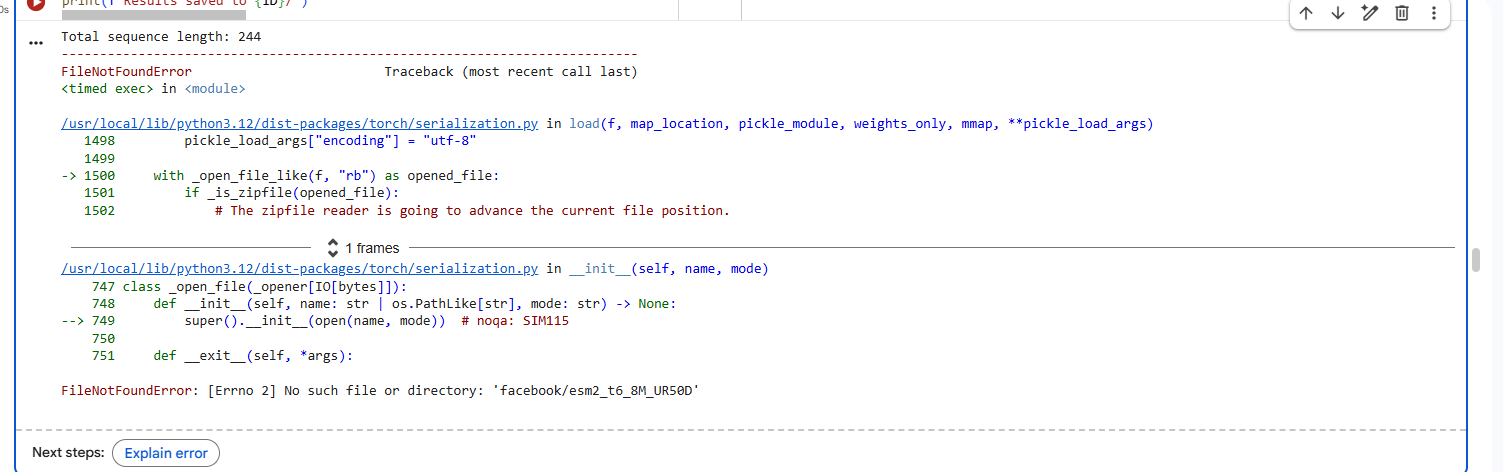
I faced repeated errors while folding the protein.
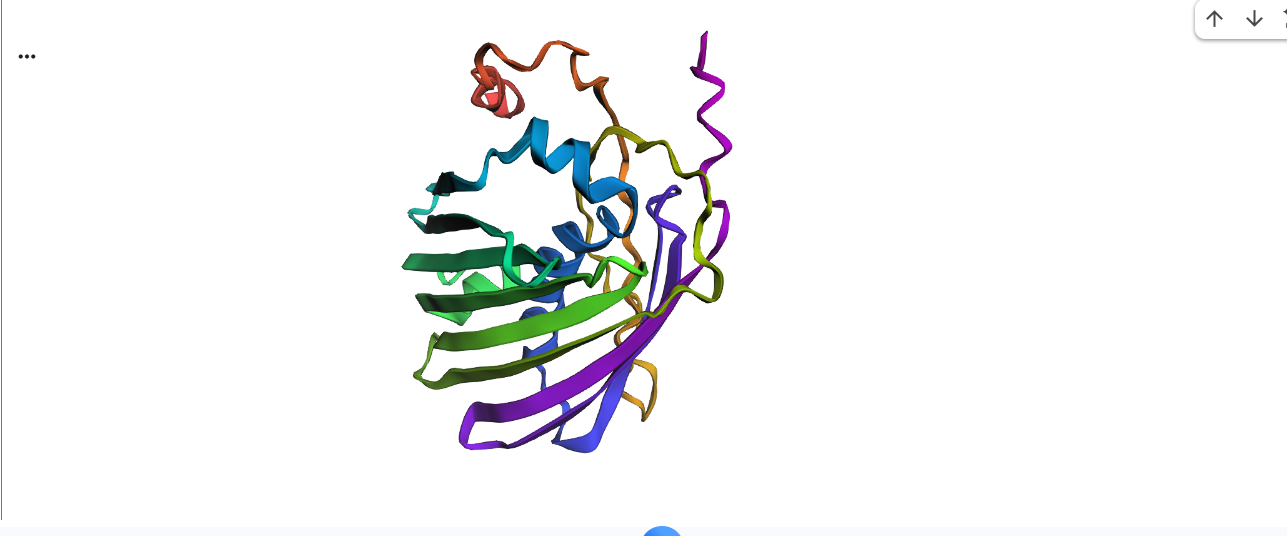
Yes the predicted co-ordinates match the original structure.
2.Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I deleted 4 residues GEEL from the beginning of sequence and did a mutational scan.
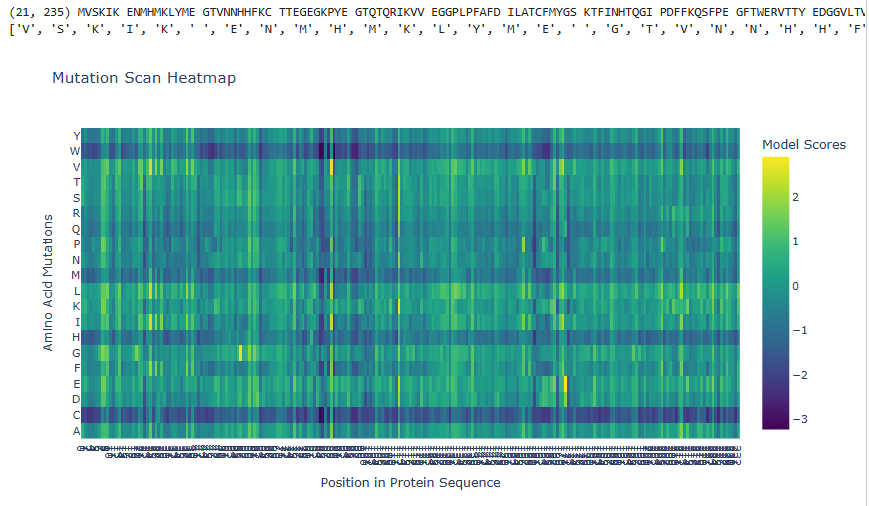
Yes the proteins still folds back into its native structure after a segment of deletion in sequences.It is resilient to mutations.
C3 Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1.Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
2.Input this sequence into ESMFold and compare the predicted structure to your original.
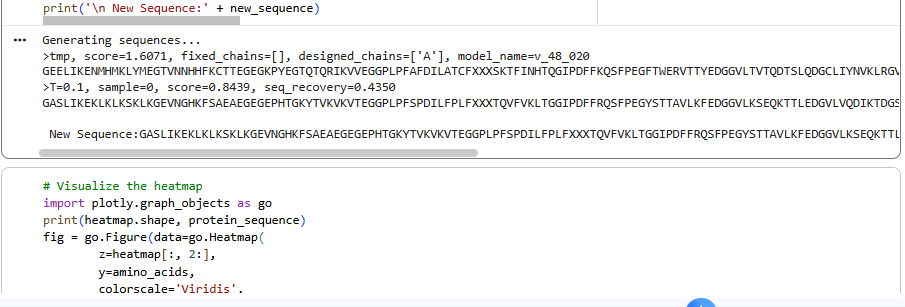

ESMFold 3D Structure: The predicted 3D structure had a ptm score of 0.785 and an average pLDDT of 73.443, indicating a generally good confidence in the protein’s fold and supporting that the designed sequence folds into a stable, well-defined structure.
Visual inspection of the 3D structure provided qualitative assessment of its compactness, secondary structure elements, and overall tertiary fold, confirming the design folds as expected.It is sinmilar to original structure.
Part D. Group Brainstorm on Bacteriophage Engineering
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
The group plans to stabilize the protein by making it fold without DnaJ chaperone protein of host.
Write a 1-page proposal (bullet points or short paragraphs) describing: Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
This can be done by predicting mutations using mutational scans and protein ESM2 fold to predict the fold after the mutation. This can be validated using Alphafold multimer.
Other generative models can be used to make the protein co-fold with the DnaJ protein.This can be studied using alphafold multimer.
The lysis proteins could be studied and probably a mechanism to apply to co-fold the protein with other chaperones and analyse the fold and stability of mature protein against E.coli host. pBLAST may be used to study different lysis proteins and how it has evolved and can be analysed for using similar mechanisms but folding with a different chaperone.
Why do you think those tools might help solve your chosen sub-problem?
The major goal is to prevent the interaction of DnaJ with lysis protein as E.coli is mutating this mechanism leading to antimicrobial resistance. So the aim is to either make the protein fold independent of DnaJ of host by mutations or co-folding or by adding similar chaperones for it to fold.
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”)
The protein might not fold in a desired way, may lead to loss of function of protein.
Since we are not sure about the interactions of bacteria with proteins co folded with other chaperones.
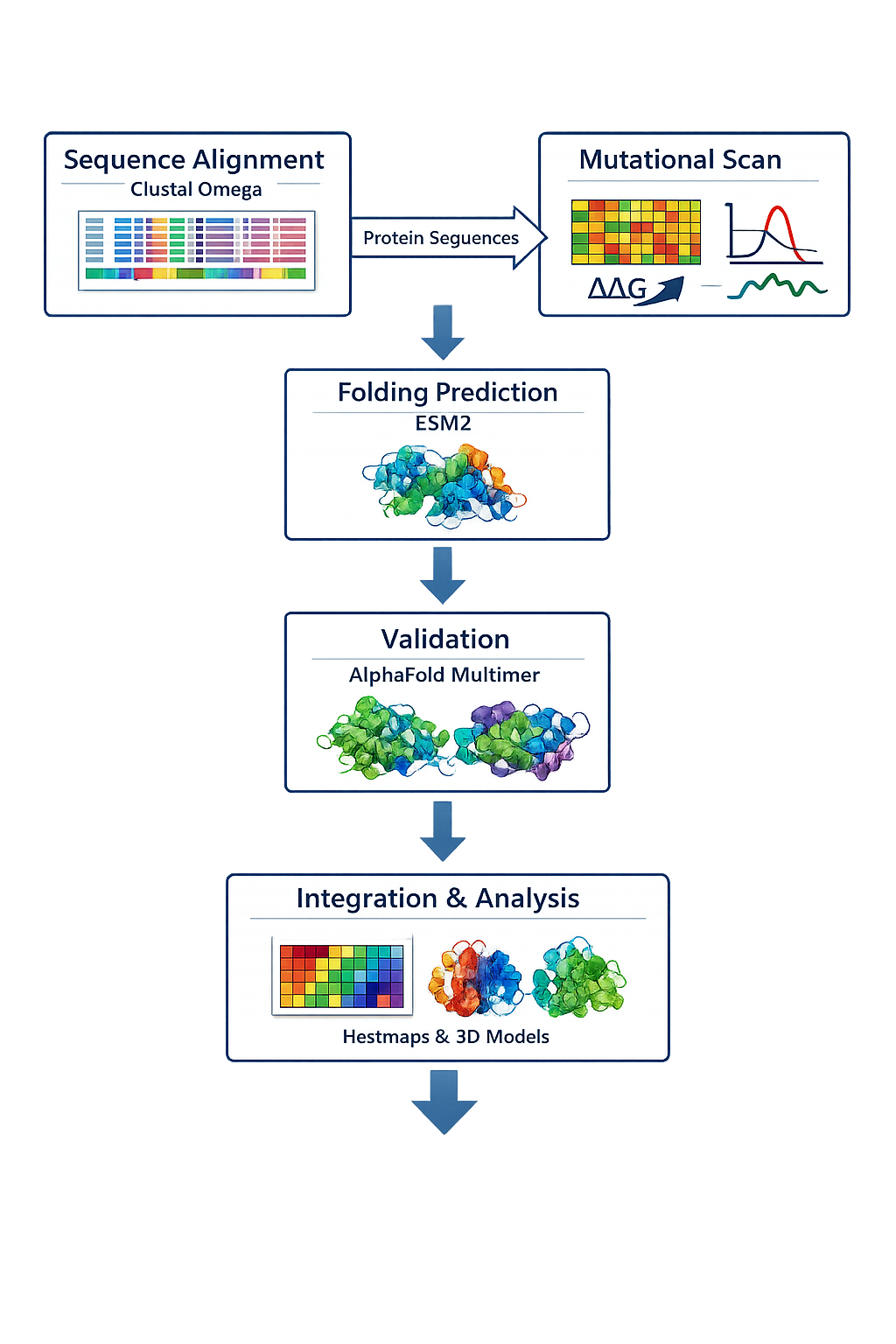
Include a schematic of your pipeline.

5.Each individually put your plan on your HTGAA website Include your group’s short plan for engineering a bacteriophage
I want to mutate the lysis protein at three parts N-terminal, C-terminal and the middle part and see how it co folds with DnaJ protein.I will use esmfold and Alphafold3 to validate the predicted structure.
References
1.Colab notebook to count amino acids.
2.The UniProt Consortium. (2025). UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Research, 53(D1), D609-D617.The UniProt Consortium. (2023). UniProt Tools: BLAST, Align, Peptide search and ID mapping. Current Protocols, 3(3), e697.
Schrödinger, LLC. (2025). The PyMOL Molecular Graphics System (Version 2.6.2). https://www.pymol.org/ generating the protein structure visuals
H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne, The Protein Data Bank (2000) Nucleic Acids Research 28: 235-242 https://doi.org/10.1093/nar/28.1.235. RCSB.org - for mCardinal structure
Lambert, TJ (2019) FPbase: a community-editable fluorescent protein database. Nature Methods. 16, 277–278. doi: 10.1038/s41592-019-0352-8-FP Base-mCArdinal Protein Sequence
HTGAA_ProteinDesign2026.ipynb for esm2 fold, latent space analysis, mutational heat map and inverse folding
https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb -for lysis protein folding
https://www.ebi.ac.uk/jdispatcher/msa/clustalo- alignment tool used to match multiple similar lysis proteins to study conserved areas
“Schematic Validation pipeline” Microsoft Copilot, Microsoft, [21-3-2026], https://copilot.microsoft.com/
“Alpha helices” prompt. Perplexity, Pro version, Perplexity AI, March 2026, www.perplexity.ai.