Week-05-hw-protein-design-part-2
Homework: Protein Design II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
1.Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2.Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
3.Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.

4.To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.

5.Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Part 2: Evaluate Binders with AlphaFold3
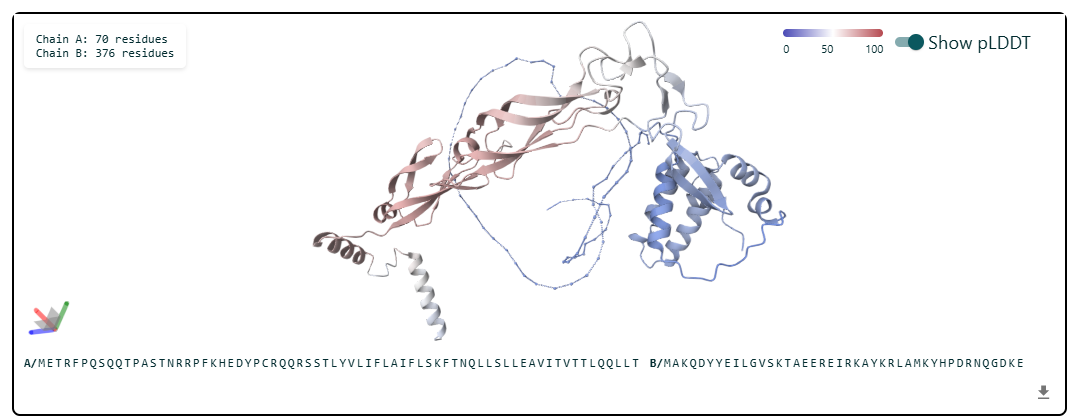
1.For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
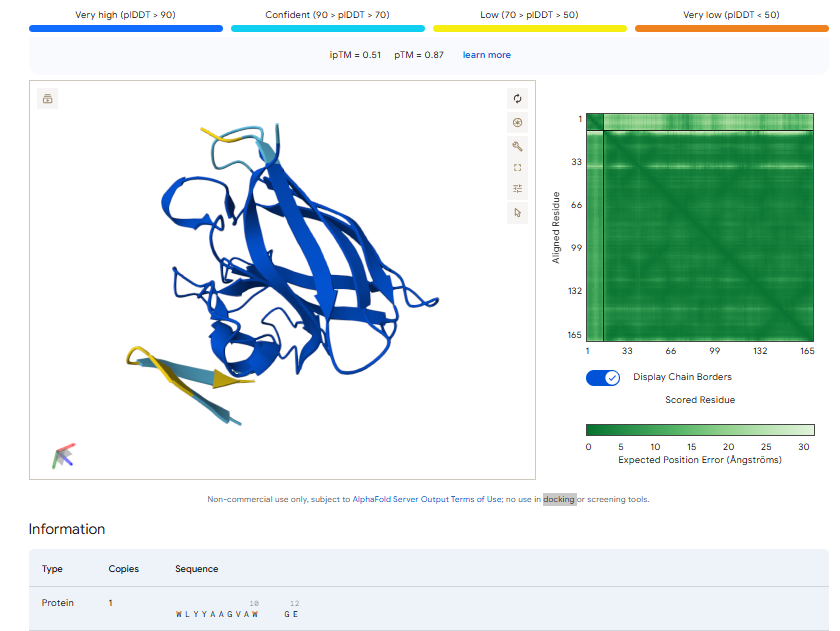
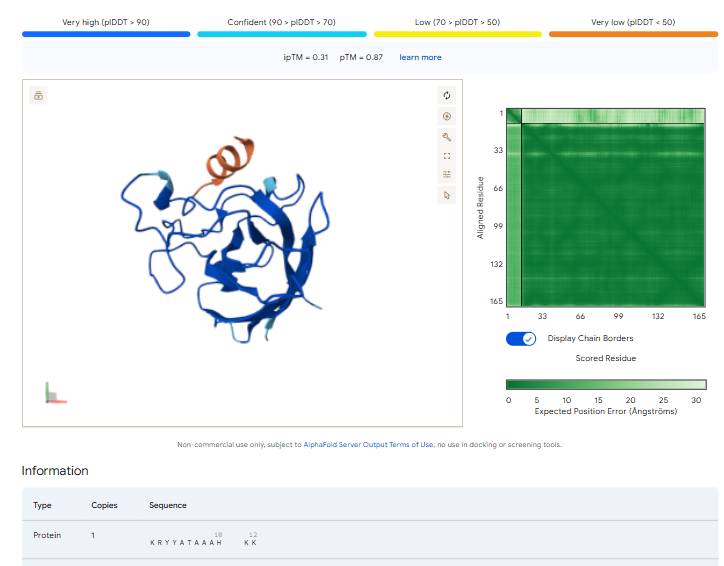
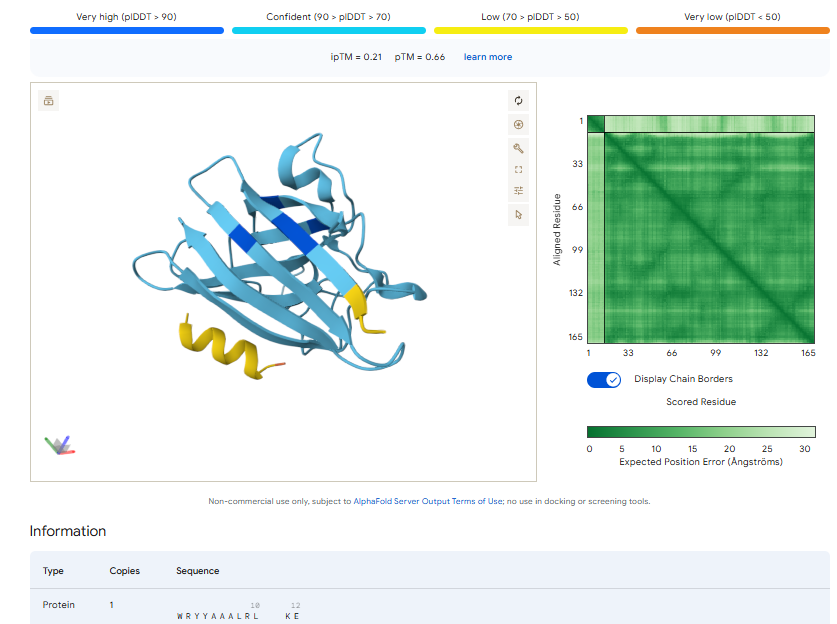
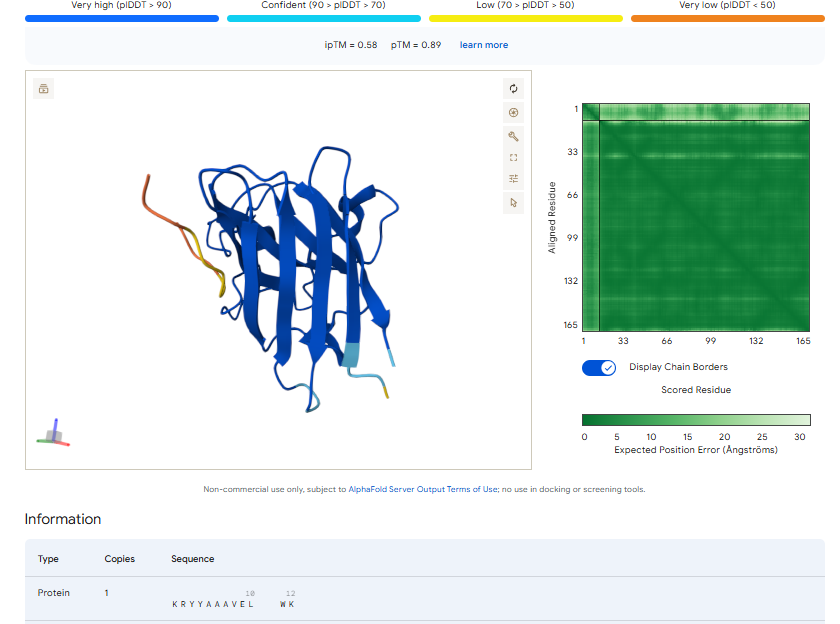
3.Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In all the figures the peptide appears to bind to the β-barrel region or approach the dimer interface. It appears surface bound.
4.In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
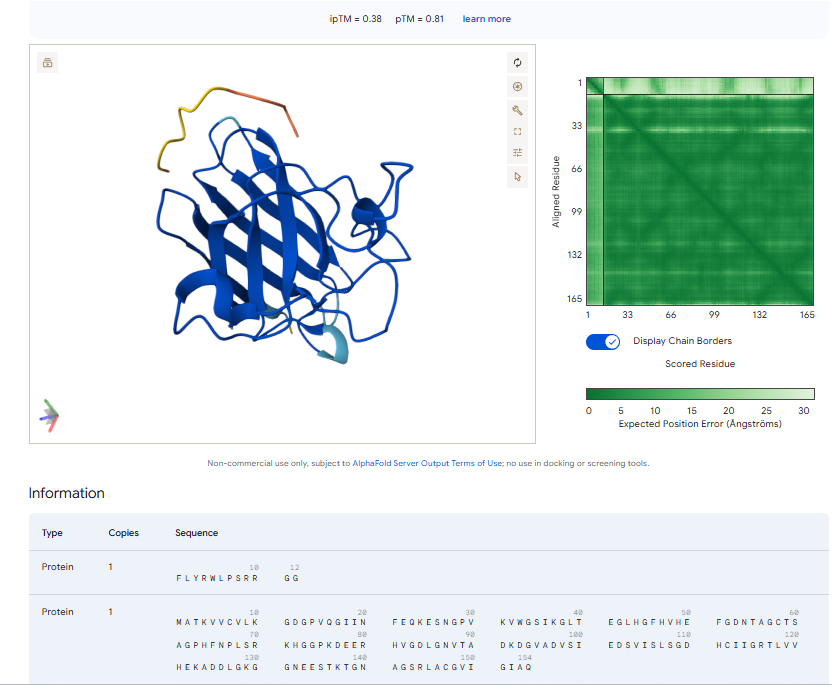
The above image of known binder shows ipTM value of 0.38 and pTm of 0.81.
The ipTm values observed for other binders only one comes close to ipTM of 0.31 and pTm of 0.87.The rest are all binders have higher ipTM in the range of 051-0.60.The (KRYYAAAVELWK) due to its balanced high ipTM, affinity, solubility, and specificity for SOD1 A4V—ideal for therapeutic prototyping in cell-free systems or biosensor integration may be a good hit. None deeply penetrate pockets, typical for PepMLM 12-mers on structured targets like SOD1.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
1.Paste the peptide sequence.
2.Paste the A4V mutant SOD1 sequence in the target field.
3.Check the boxes
1,Predicted binding affinity
2.Solubility
3.Hemolysis probability
4.Net charge (pH 7)
5.Molecular weight
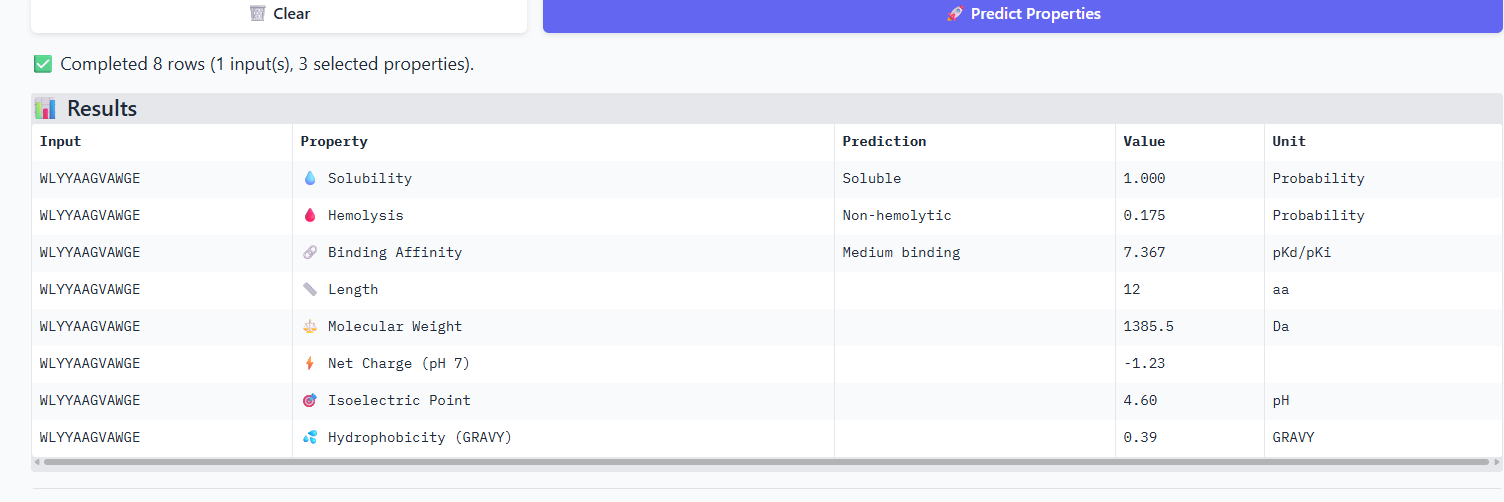
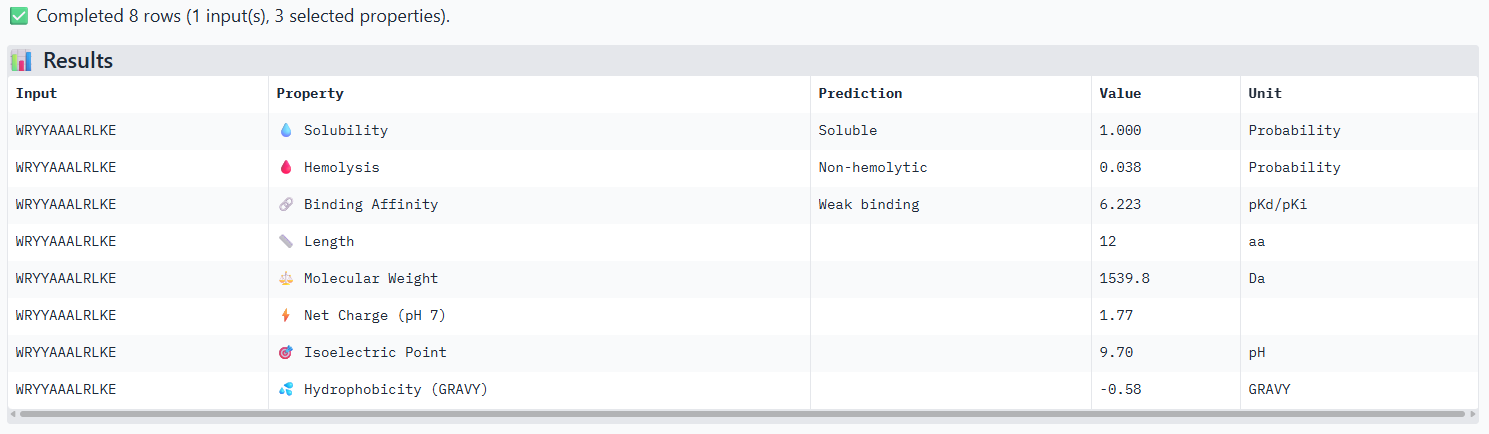
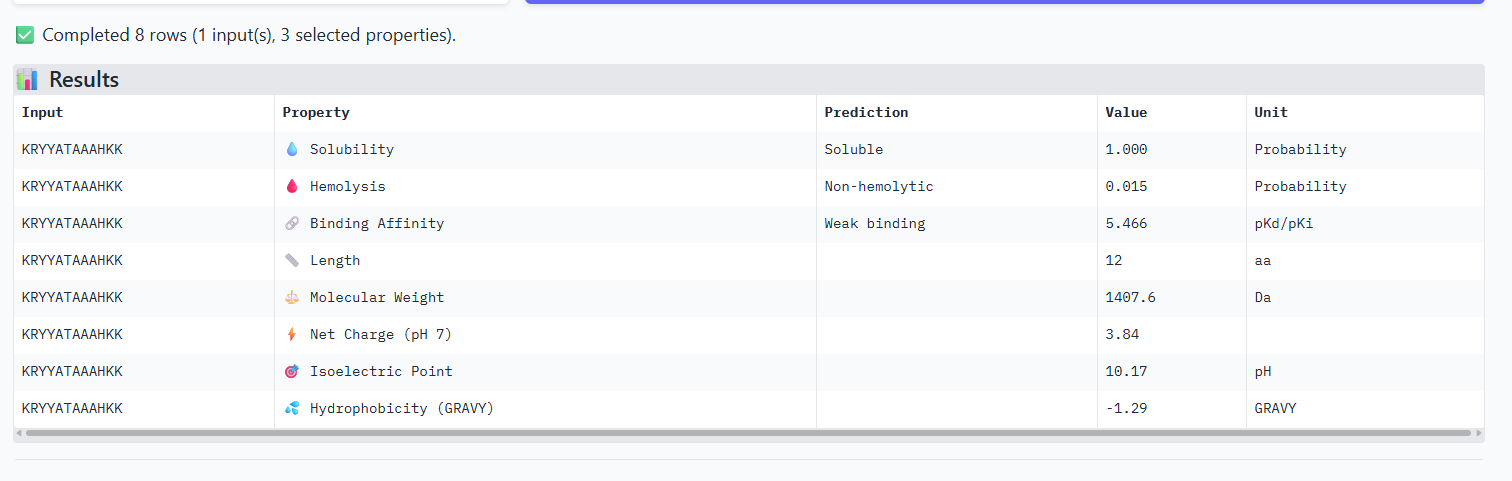
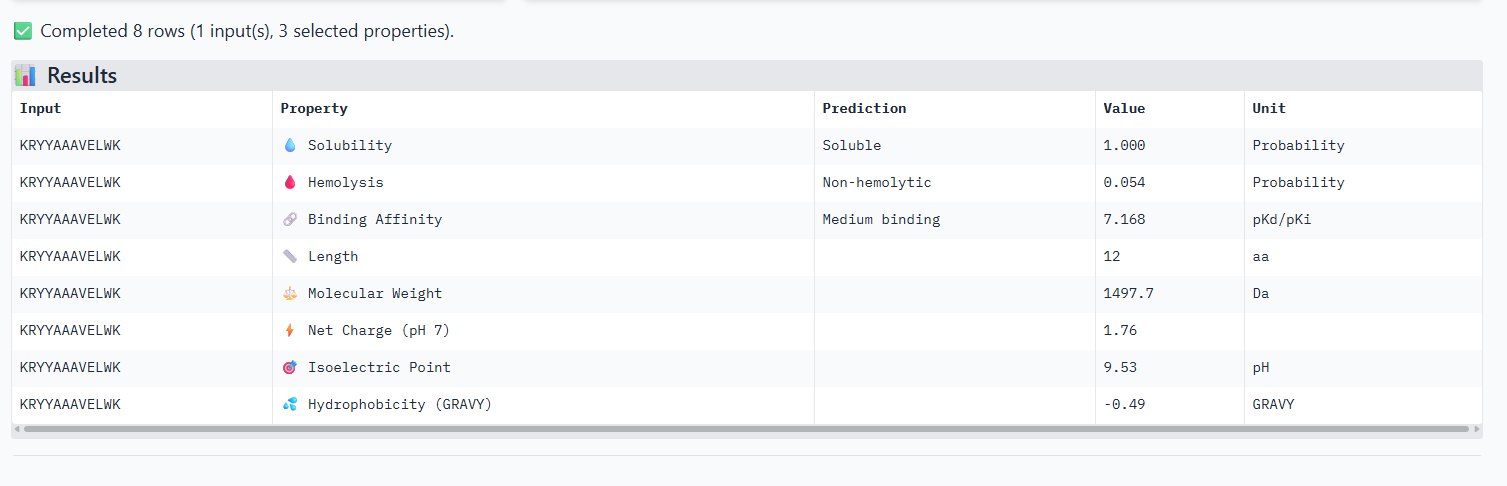
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
The iPTM scores and binding affinity are related, the highter the score the higher the bonding affinity.I could not find any strong binder only found medium binding binders. All of them were soluble and non-haemolytic.
Choose one peptide you would advance and justify your decision briefly.
I would like to go with KRYYAAAVELWK binder as it has 7.168 binding affinity, higher than all four. Has high specificity for target SOD1 mutant protein. It has high iPTM score of 0.58.
Part 4: Generate Optimized Peptides with moPPIt
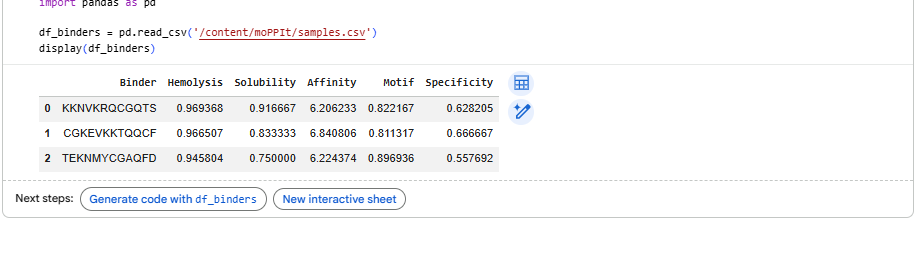
4.After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
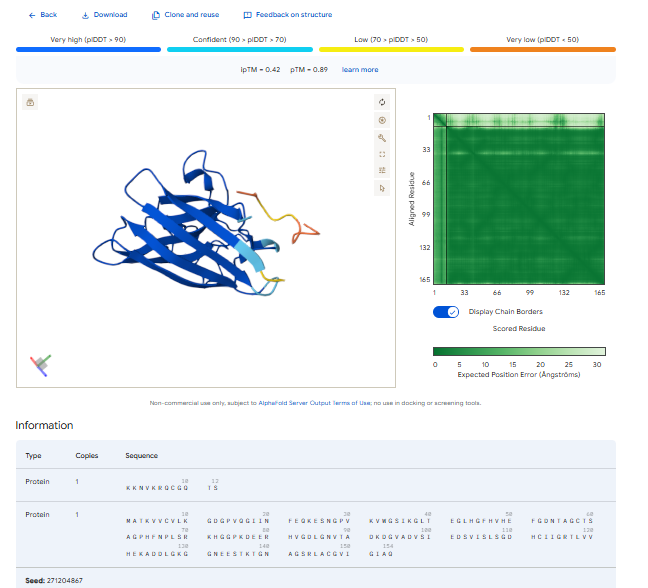
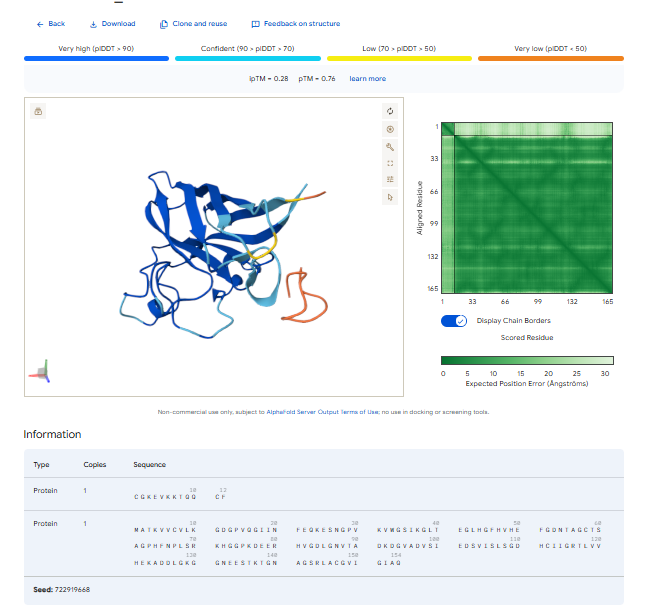
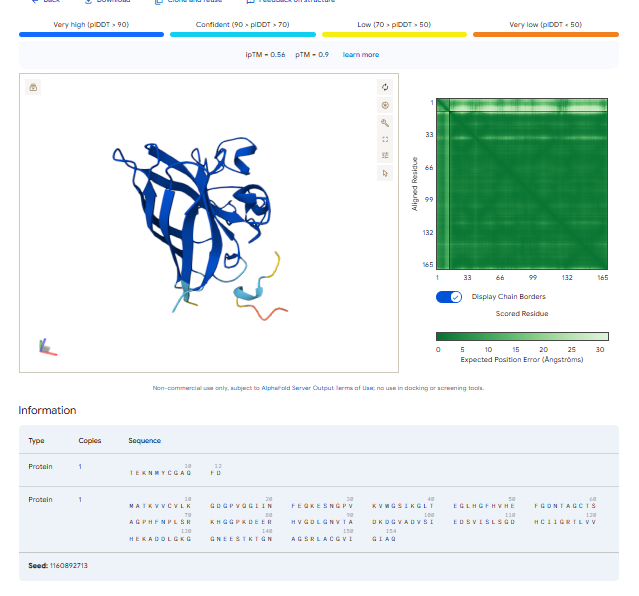
Higher values (closer to 1) indicate better performance; affinity measures binding likelihood to the target motif, the specificty of binder 1 is best compared to the other two.
Evaluating peptides like KKKRGGST and TEKVQAGCF from moPPIt for clinical advancement requires a rigorous, multi-stage preclinical pipeline to confirm computational predictions and ensure safety/efficacy.Sequences can be refined using ProteinMPNN for improved affinity/specificity.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
Part 0: Sign-up to Boltz Lab
Part 1: Structural Predictions in the Sandbox
Running Your Three Predictions Navigate to the Boltz Sandbox at lab.boltz.bio and log in to your account.
Go to Sandbox → New Prediction
Name this BRD4 binder JQ1
Select ‘Complex’, add ‘Sequence from RCSB’, and add 3MXF
Continue through Constraints (not needed for this example), and select Jq1 as the Binder for an affinity prediction.
Submit the prediction.
Use the ‘Duplicate Prediction’ in the results review, and remove the small molecule.
Add in the SMILES for the Hit and Lead.
When predictions complete, record your results in the table below
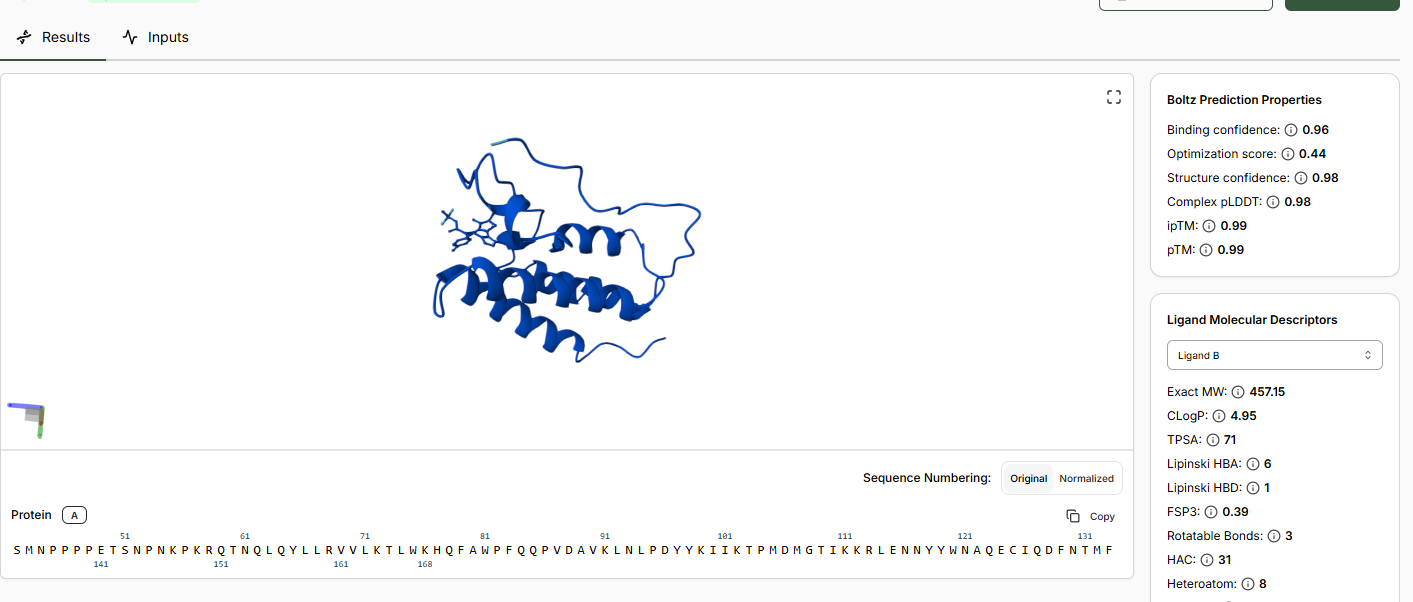
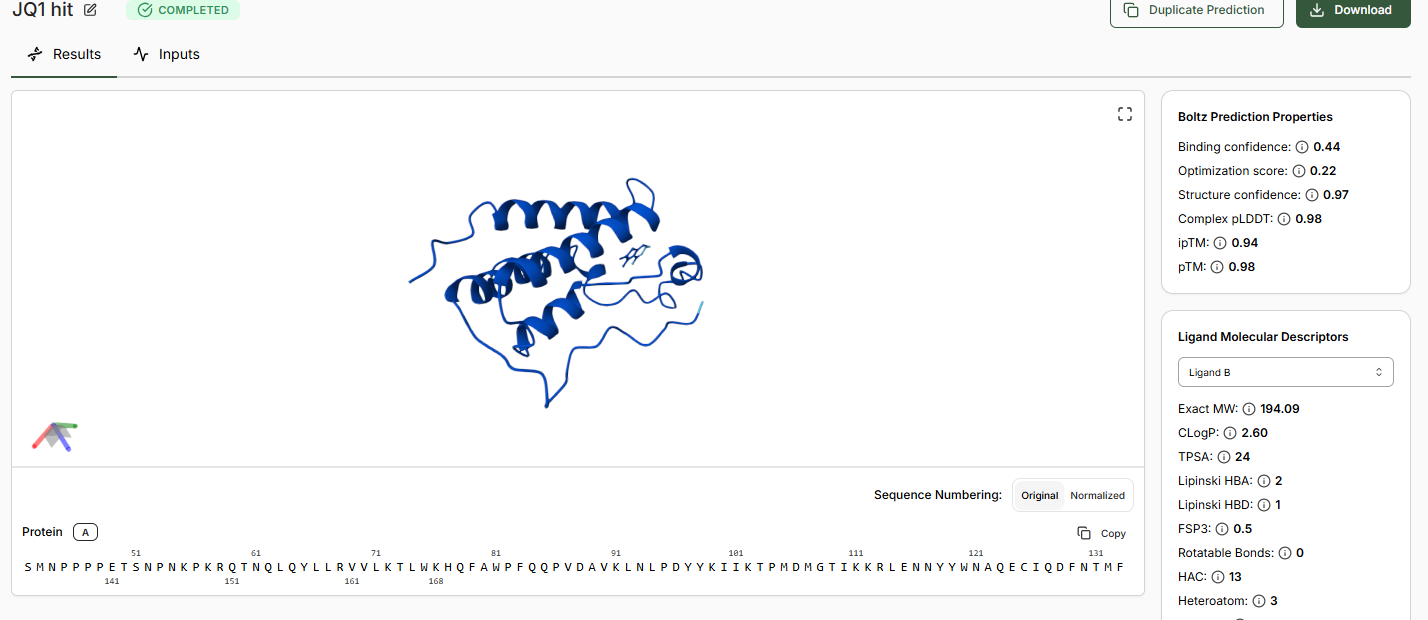
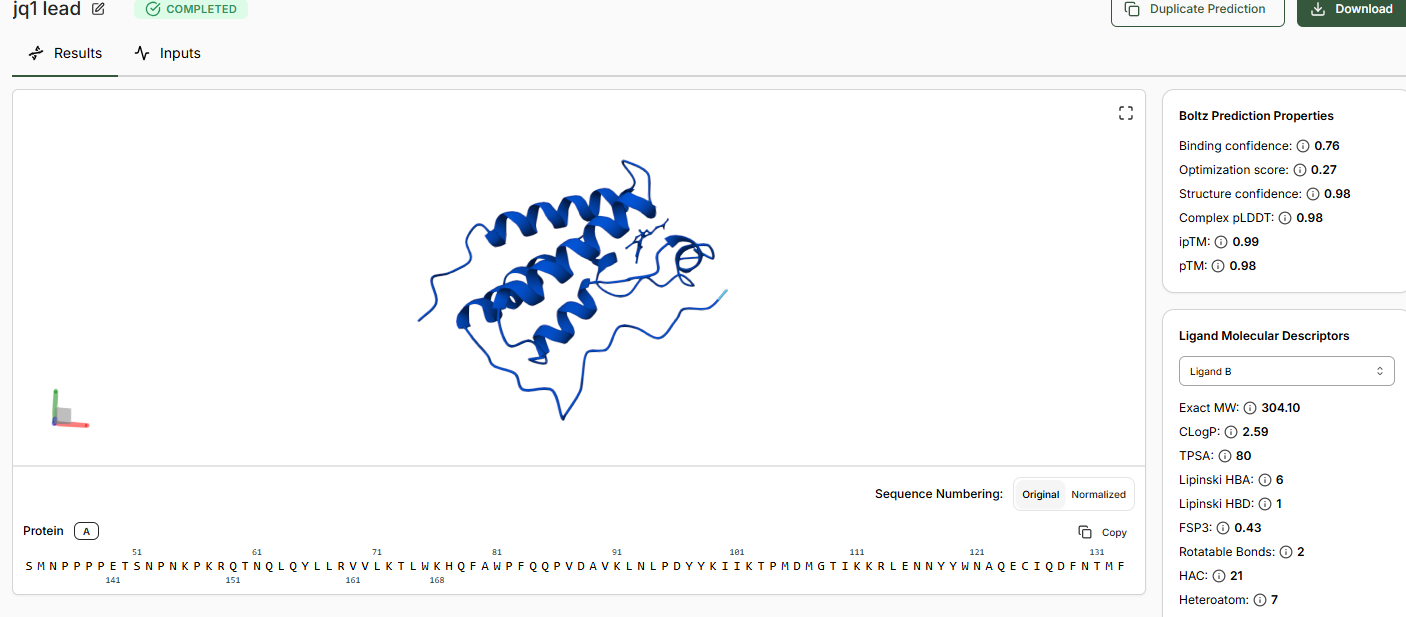
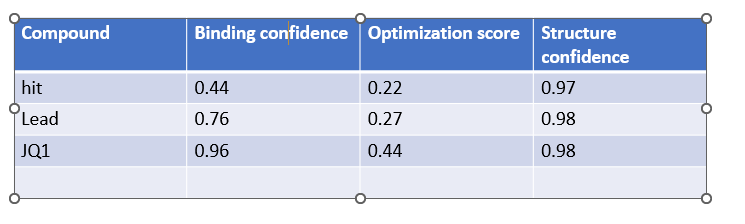
Does Binding Confidence increase as you move from hit to clinical candidate? What would you expect, and why might it deviate?
Yes the binding confidence increases from hit to clinical candidate. The goal of drug discovery is to incraese the binding affinity and selectivity of the drug without comprimising on efficacy,potency, pharmocological properties, its a balance of all these properties.
It might deviate due to the in vitro environment assay not matching with the complex environment in body and due to the balancing of all drug properties like binding strength, potency etc.
•Inspect the predicted binding pose for JQ1. Can you identify potential key binding interactions.
The approach is there towards the helices of the BRD4 domain.
•Compare the Optimization Scores. How do the scores compare for JQ1 vs the Lead.
The optimization scores are 0.27 for lead and 0.44 for JQ1. The difference in the values tell us that the lead has initial activity but compared to JQ1 requires more modifications for binding affinity, selectivity and pharmocological properties.
Part 2: Setting Up a BRD4 Design Project
Now you will create a small molecule Design Project - the Boltz Lab workflow for virtual screening and lead optimisation. We will set up BRD4 as a target using the clinical candidate as our structural reference. 2.1 Creating the Target
- From the dashboard, create a Design Projects via ‘New Project’
- Name your project: ‘BRD4 Workshop '
- Select ‘Small Molecule’
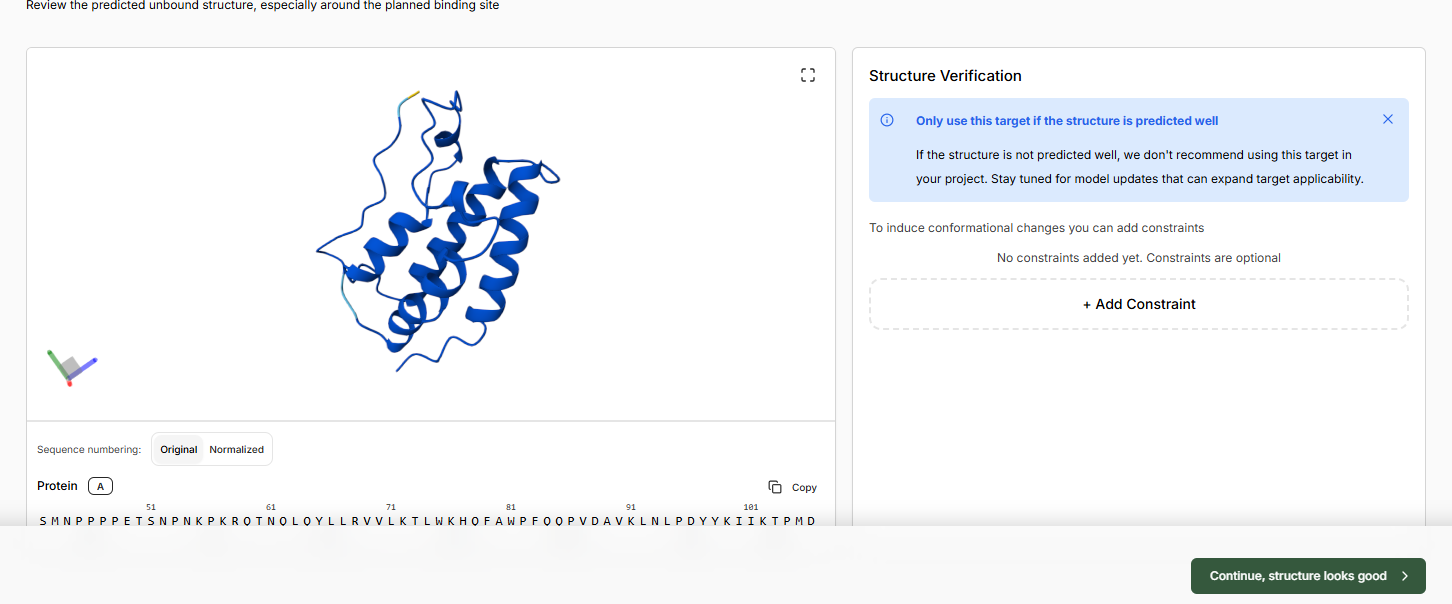
- Click Add Target and add the protein structure as in the Sandbox using PDB code 3MXF
- Continue and let the apo structure complete. Continue if the structure looks good.
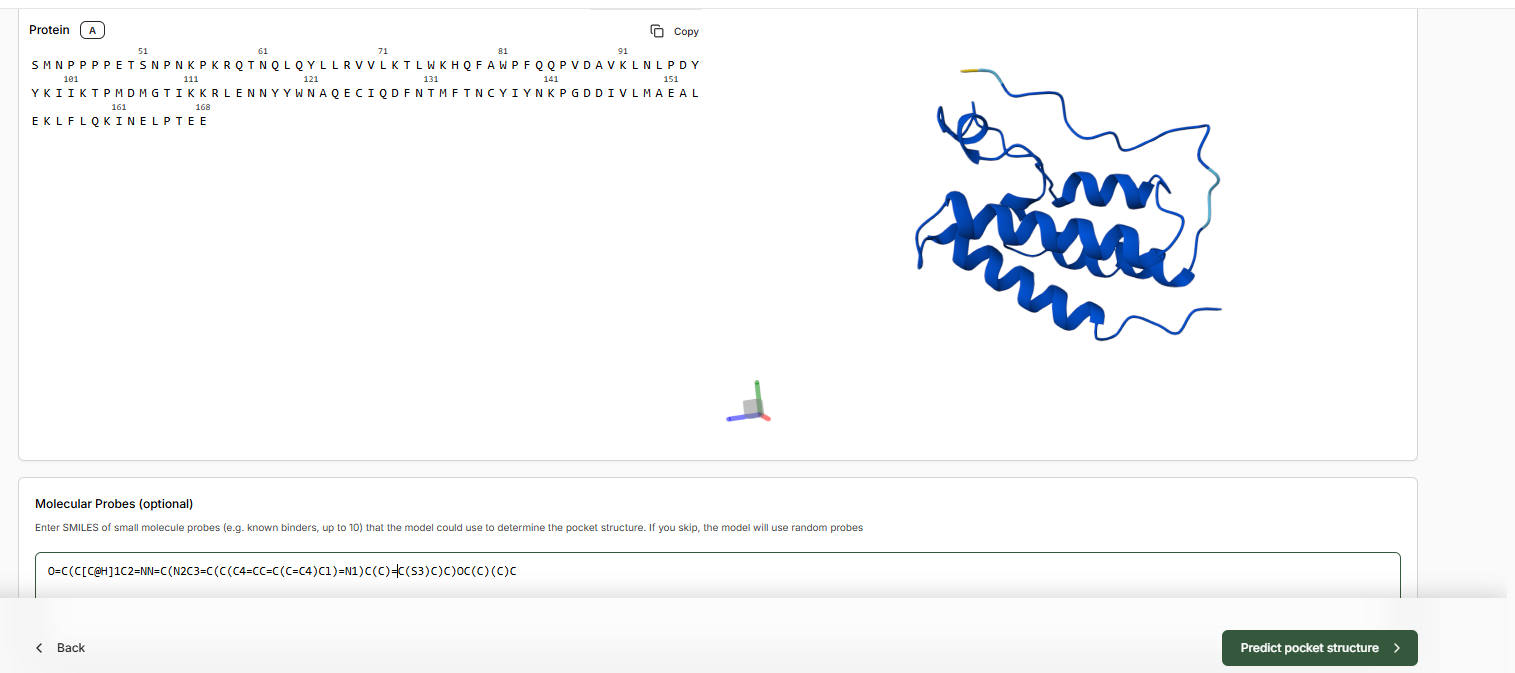
- Leave binding residue selection blank, the platform will auto-detect the pocket
- In the Molecular Probe field, paste the JQ1 SMILES.
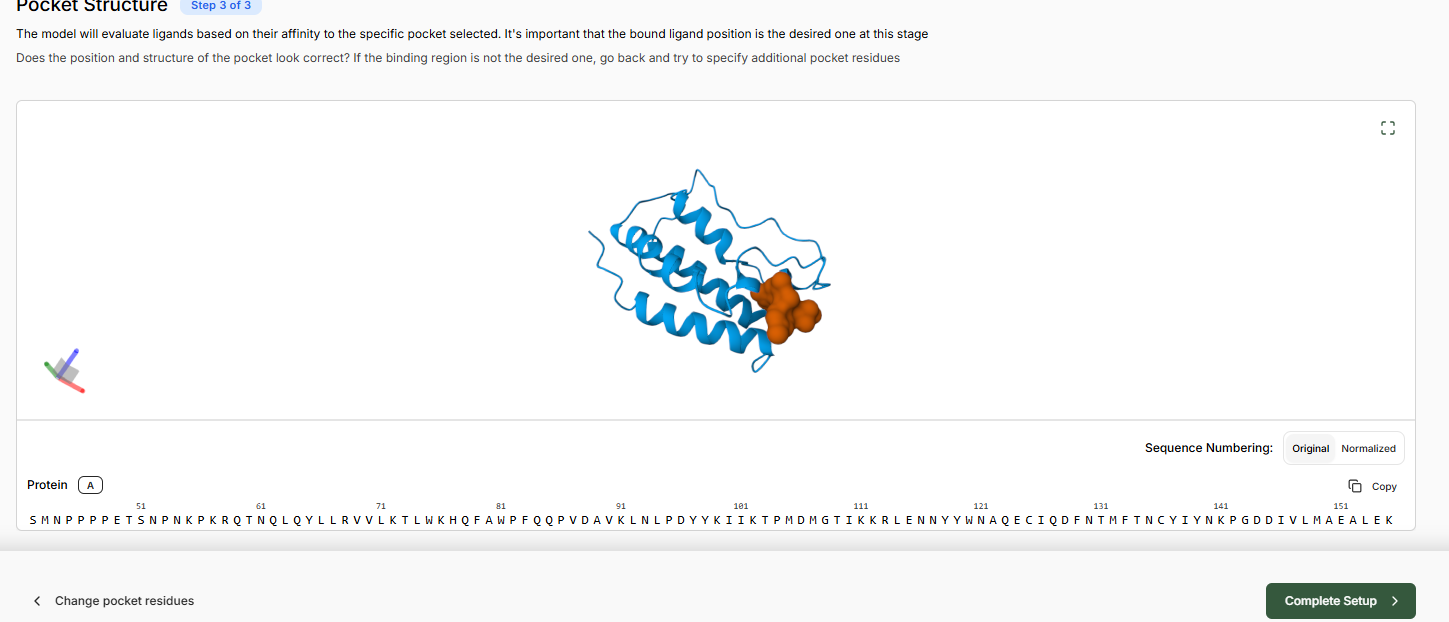
- Predict Pocket Structure and complete the Target Set-Up
Part 3: Running Your Virtual Screen
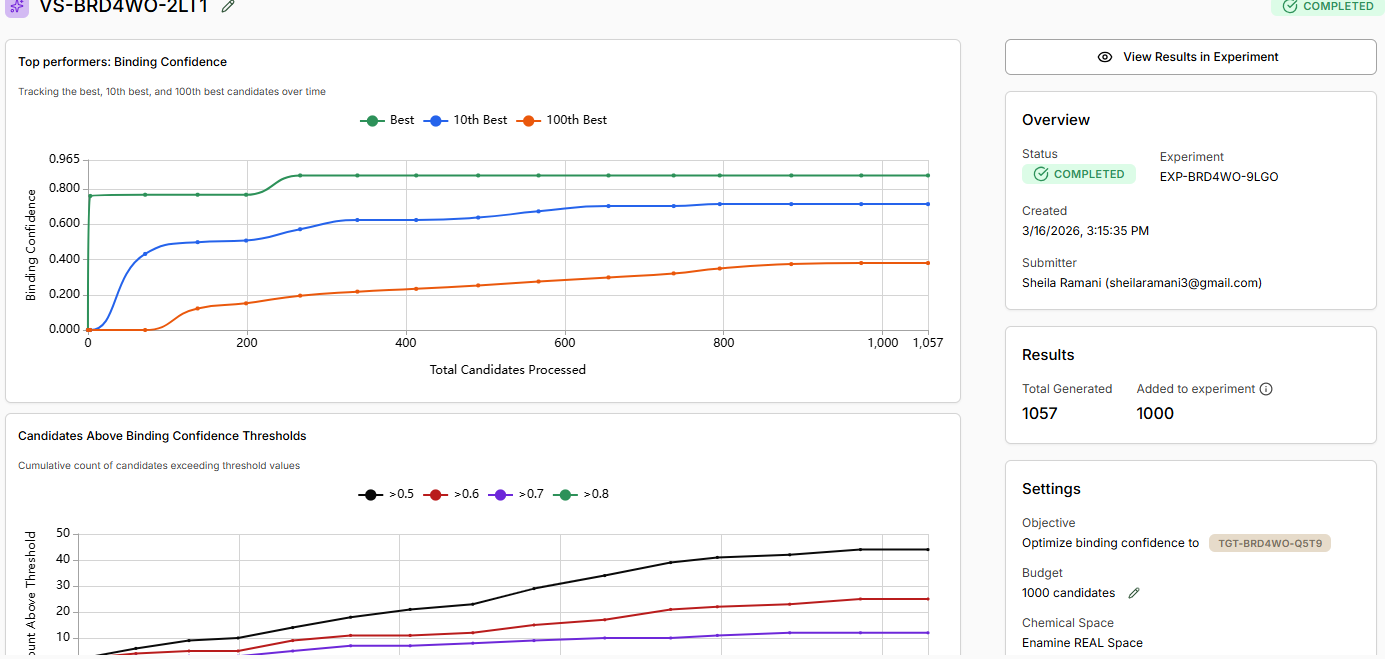
Run a Generative Design Campaign We will utilize the Boltz Lab small-molecule generative workflow. This generates novel molecules optimised for BRD4 binding using Boltz-2 as the scoring function.
- After creating the design project, Boltz Lab will prompt you to Generate binders with AI.
- Name your experiment, provide a relevant hypothesis, and Create the Experiment.
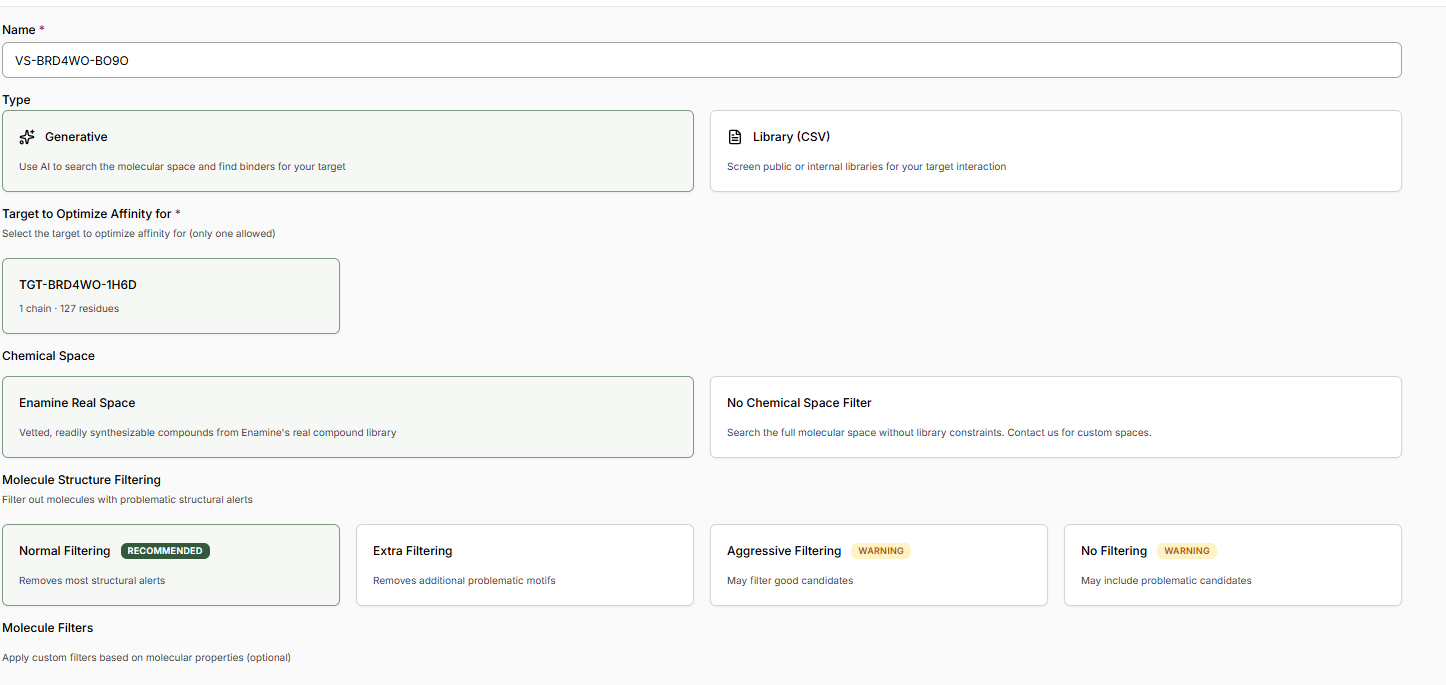
- The New Virtual Screen will be pre-configured with a Generative screen using the Enamine REAL space.
- Keep ‘Normal Filtering’ selected. This will ensure we only generate molecules acceptable to a medicinal chemist.
- Decide if you would like to apply any Molecule Filters. We recommend the ‘Drug-Like’ Preset.
- Select a custom number of Binders and enter 1K.
- Start the Virtual Screen.
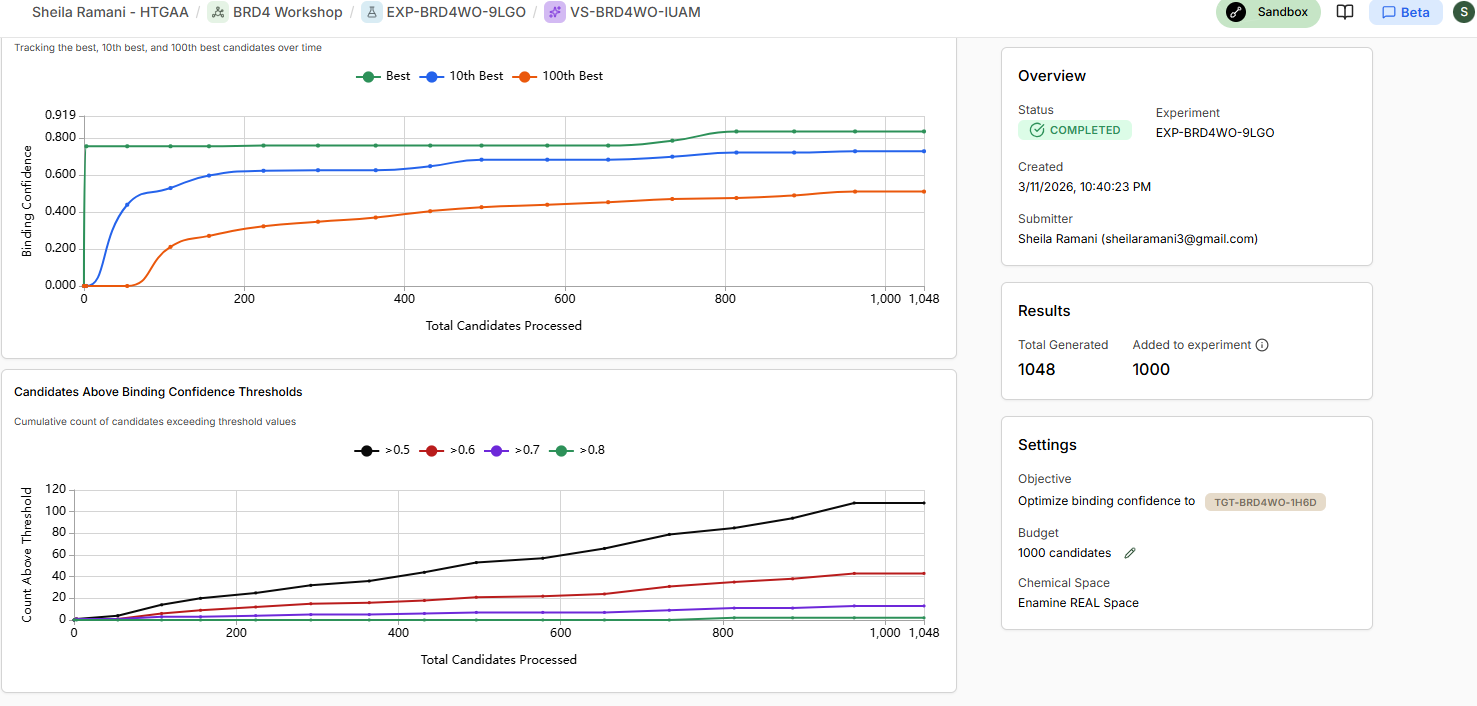
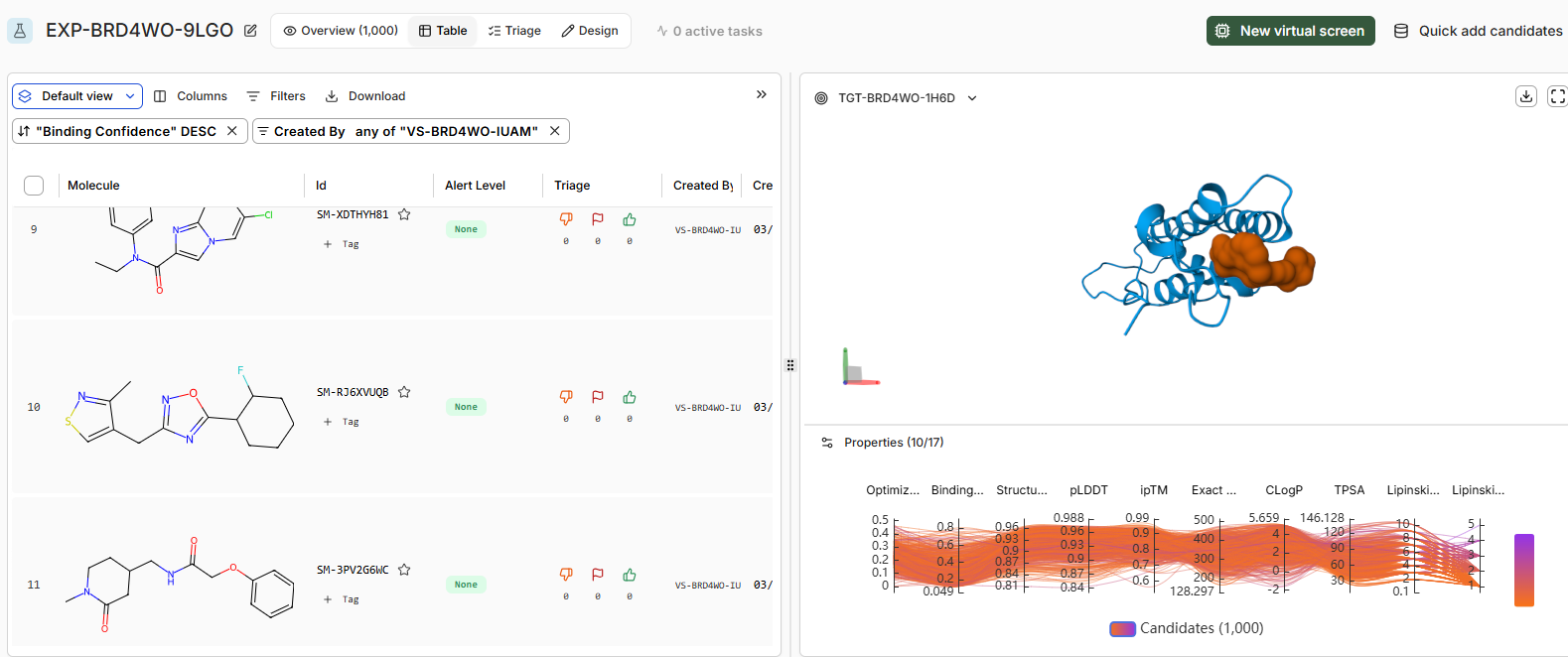
- Allow binders to be generated, and View Results in Experiment
Part 4: Analysis and Discussion
As your experiment completes, use the ‘Quick Add Candidates’ on the experiment screen to add JQ1 as a benchmark for generated designs.
Interpreting Your Results
As your experiment completes, use the ‘Quick Add Candidates’ on the experiment screen to add JQ1 as a benchmark for generated designs. From your screen output, identify three categories of molecules:
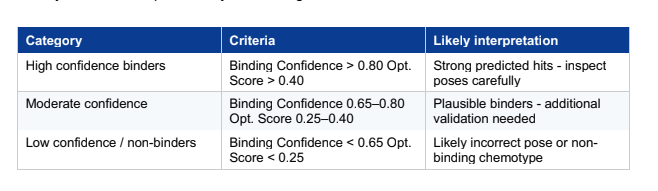
There is only 1 candiadate as high confidence binder SM-Z2EJ9VV7
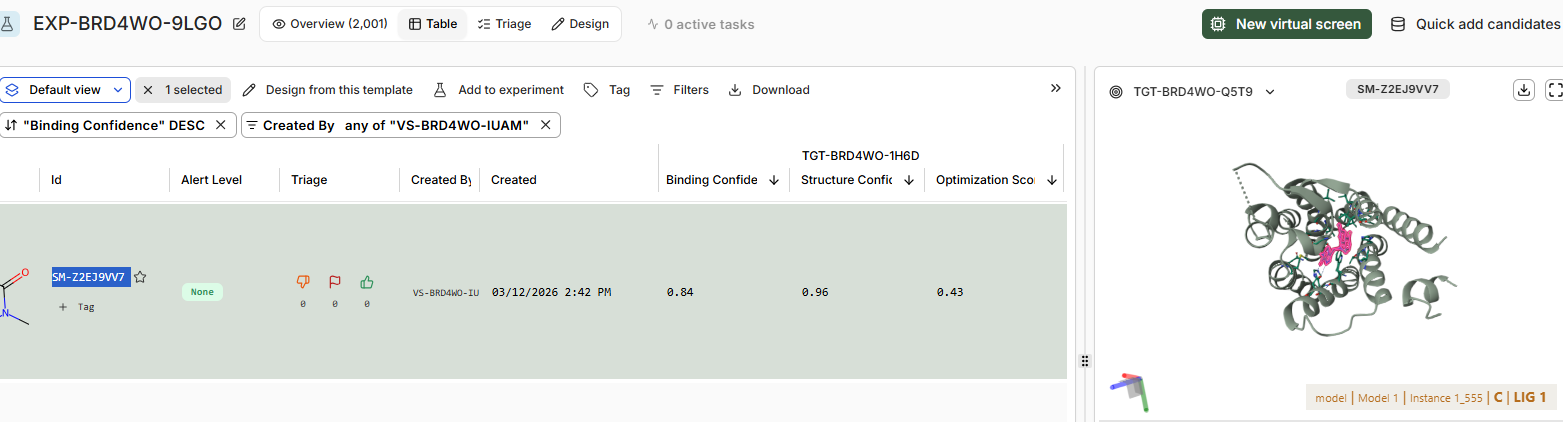
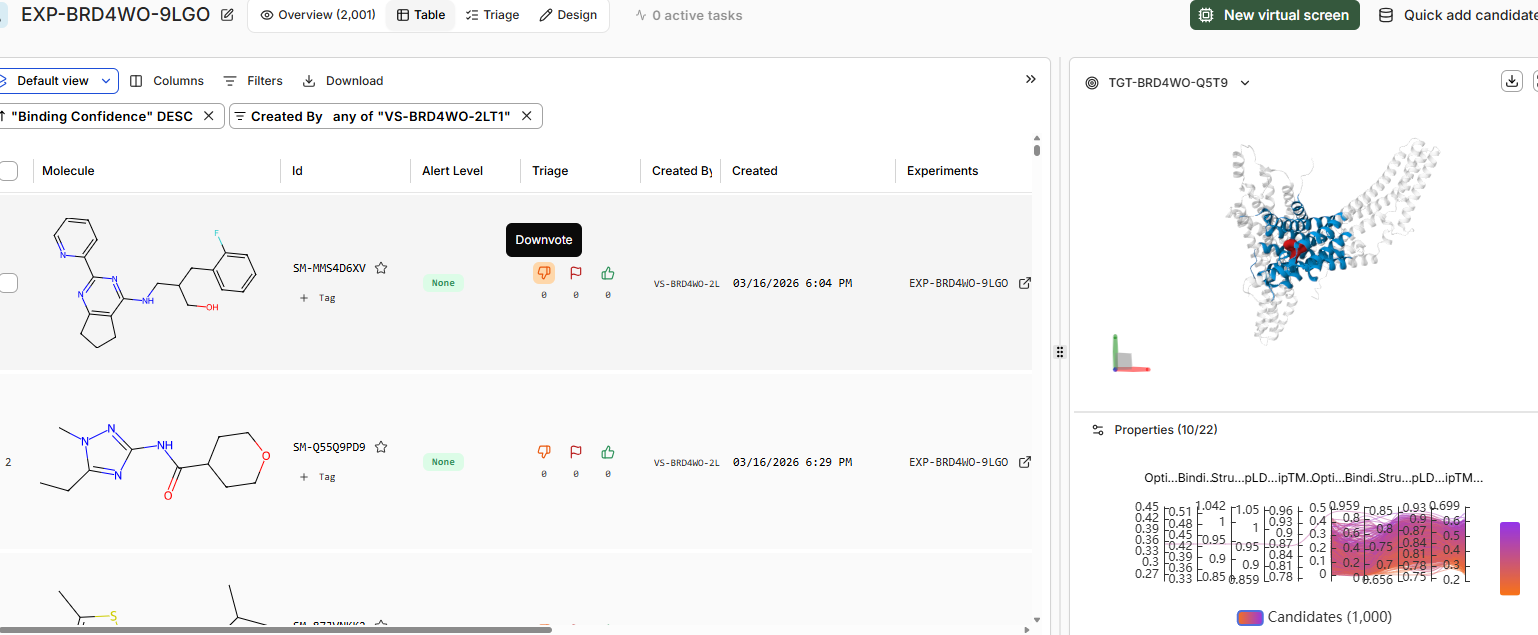
22 of them are in the moderate confidence binder category, and rest in low confidence / no binders category.
Discussion:
As the virtual screen completes, assess the following:
• How does JQ1 in the Design Project screen alongside the library. Does it score as the top compound?
Binding confidence is 0.96 as compared to 0.84 of the candidate compound.
Yes it scores as the top compound.
• How do the top scoring binders compare in binding pose to JQ1?
The binding pose of the top binder is similar to JQ1.Key differences include denser packing and fewer disordered loops (less yellow) in the high candidate, suggesting improved prediction reliability. Perceptual image similarity is moderate, confirming related but distinct poses.
• Try adding a second target to your project via the dropdown in the structure viewer, for example, BRD2 (PDB: 5UEN). Re-run the top scoring binders against BRD2 and compare which compounds score highly for BRD4 but not BRD2. This is a selectivity analysis - a key part of real BET inhibitor programs.
The top binder for BRD4 binds with binding confidence of 0.30 to the BRD2 domain.
SM-MMS4D6XV is the top binder for BRD2 domain but binding confidence of same is 0.43 with the BRD4 domain.
Part C: Final Project: L-Protein Mutants

Evolutionary Analysis and Chaperone Generalization
Use pBLAST to survey lysis protein orthologs across bacteria, reconstructing evolutionary trajectories of stability determinants and chaperone interactions. Top candidates will be redesigned for co-folding with alternative chaperones (e.g., DnaK or GroEL), with AF2-Multimer analysis of mature protein folds and in silico stability assays (e.g., ΔΔG predictions) against E. coli host factors to minimize proteotoxicity.

Courtesty: pBLAST tool.
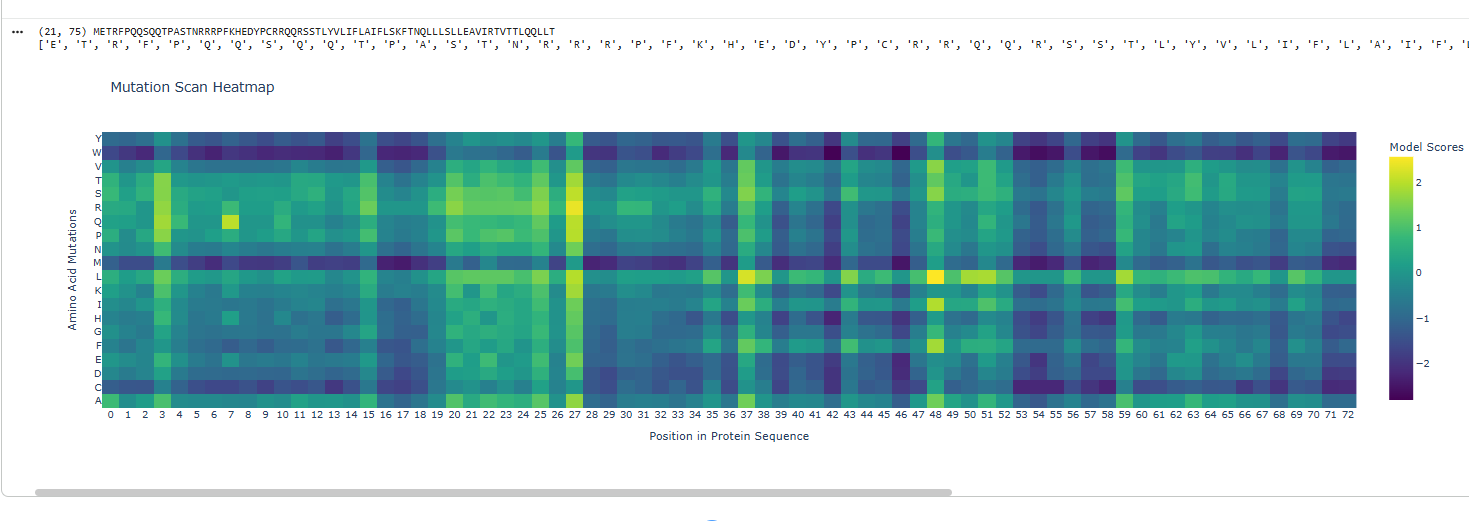
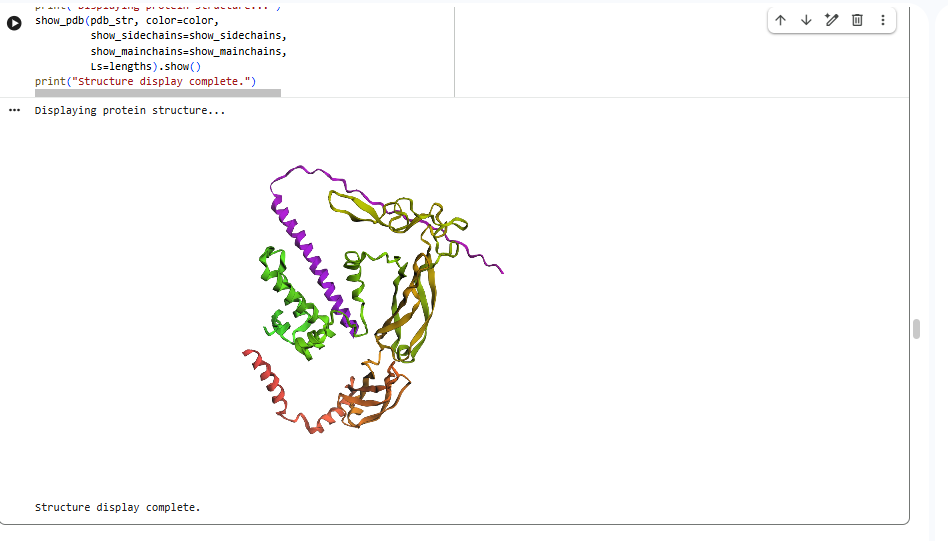
Based on the Clustal omega results mutation was done on the below sites and the protein co folded with DnaJ.
Coutesy: Neurosnap and tamarindBio
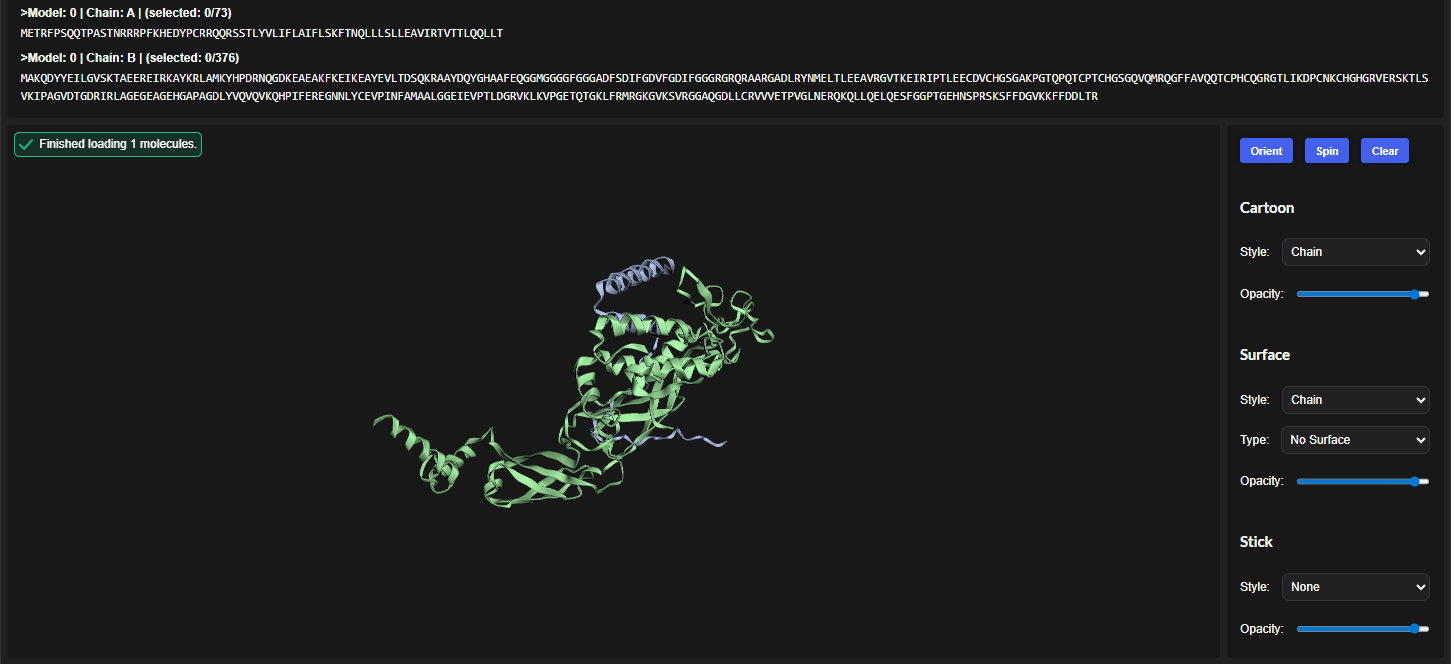
Mutations at site 7 8 and 9 removed QQS and left a gap
Deletions that remove the entire N‑terminal basic region (and therefore also remove 7–9) eliminate DnaJ binding and DnaJ requirement, and actually accelerate lysis, because the N‑terminal domain is no longer there to interfere with target binding when not chaperoned by DnaJ.(Jennifer et. al)
Mutation at 61 site E reoved and a gap is left
Position 61 falls within this TM domain that drives membrane insertion, oligomerization into higher‑order complexes, and the formation of large lesions spanning the outer membrane, peptidoglycan, and inner membrane.Chosen as one mutation was to be done in the C-domain of the protein.(Mezhyrova J et.al)
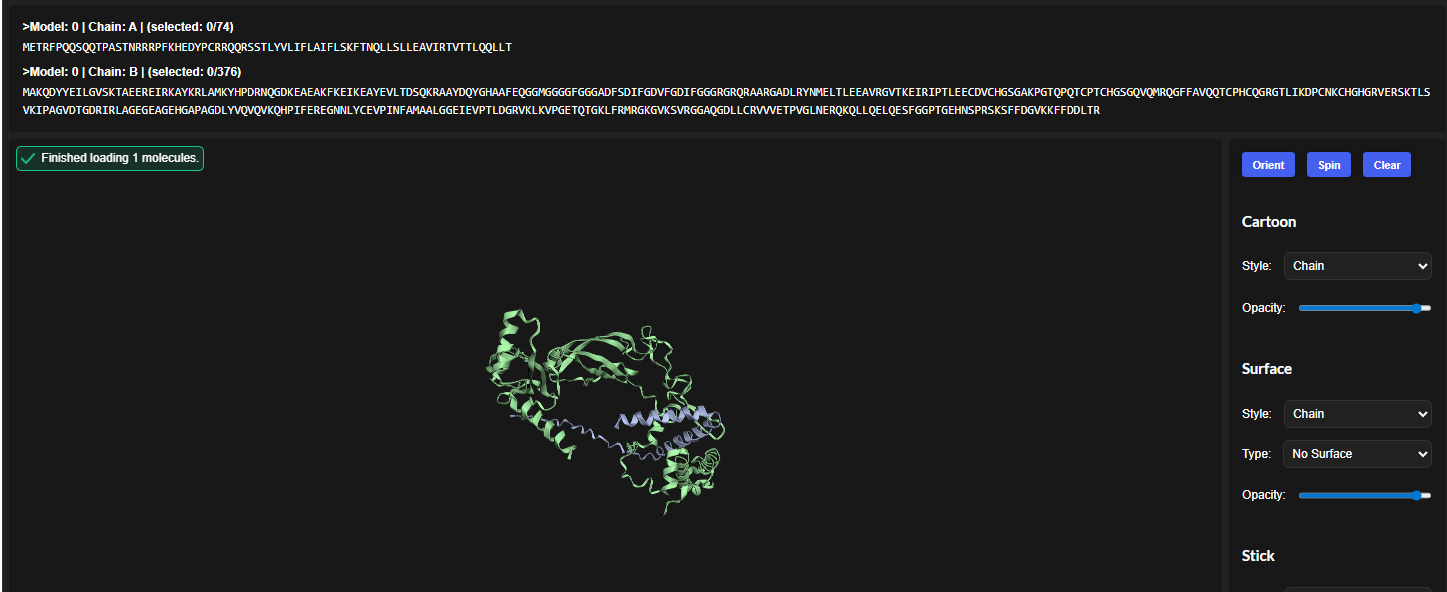
Mutation at site 3 with I instead of T
Position 3 is located in the N-terminal soluble domain of the L-protein.May alter the kinetics, efficiency, or interaction with host proteins (like DnaJ) due to their location in the regulatory N-terminal domain. ( Chamakura et al.)
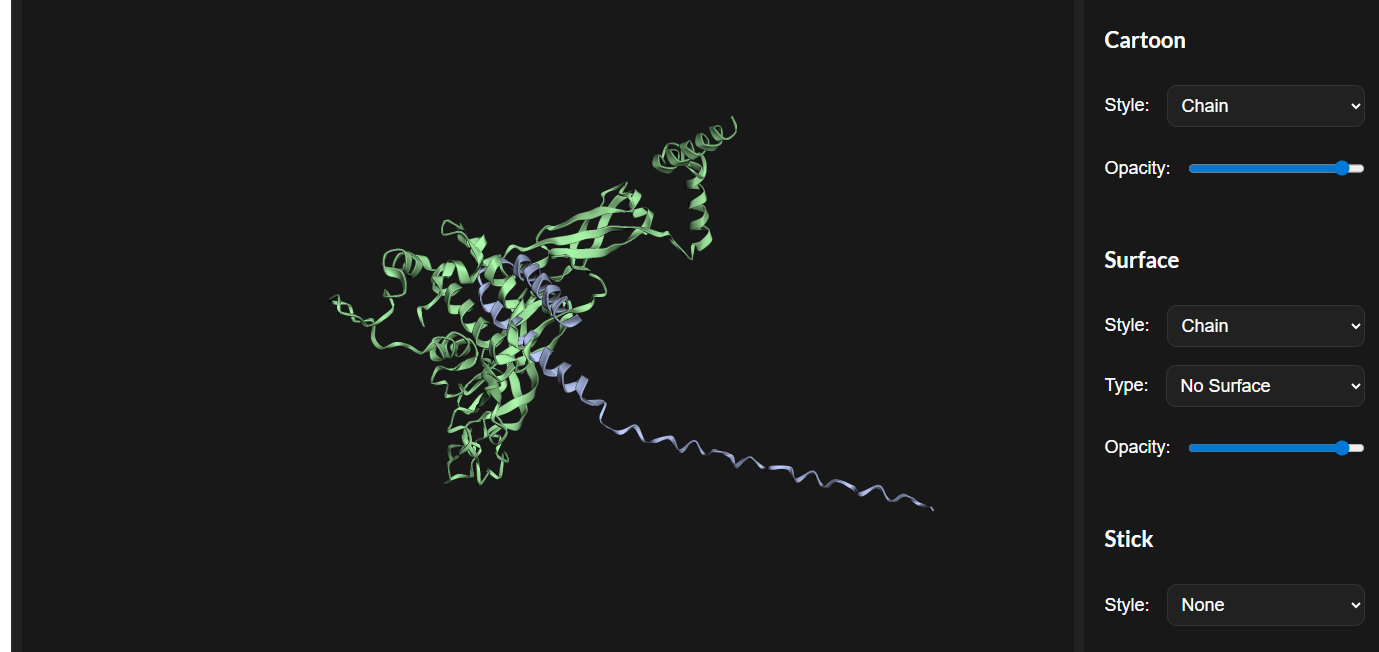
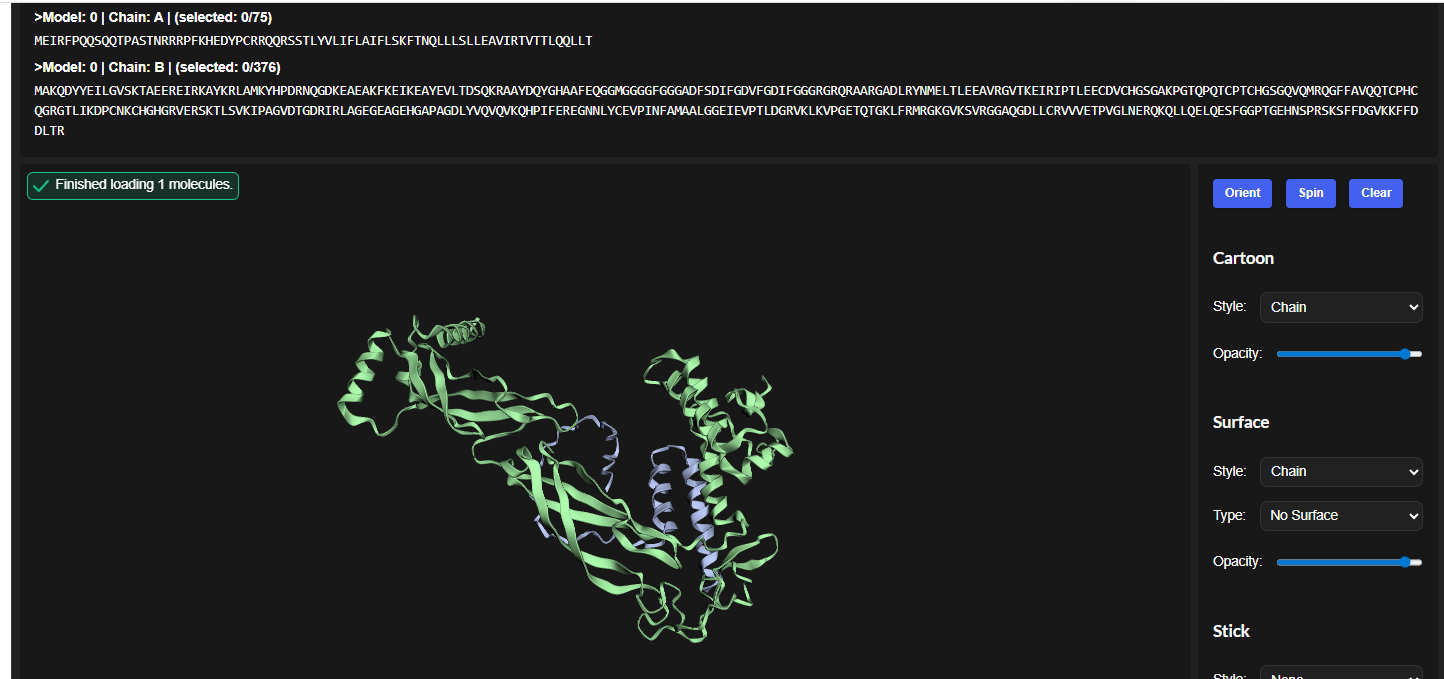
This shows image with all mutations.
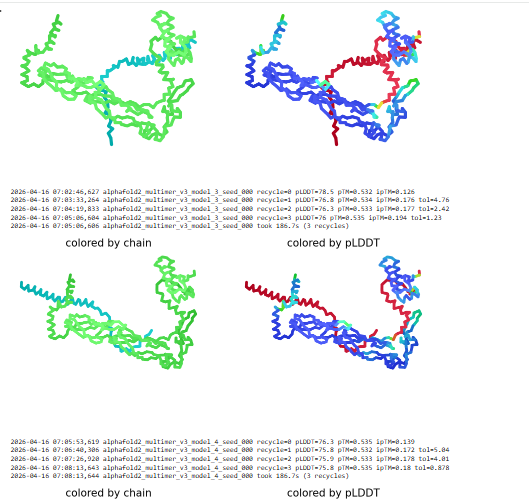
Courtesy: ESM fold
Expected Outcome
If the hypothesis is correct, the resulting lysis protein variants should show improved predicted structural stability, decreased dependence on native DnaJ interaction, and retention of a mature fold compatible with lytic activity in E. coli. More broadly, this strategy could establish a generalizable framework for reengineering host-dependent phage lysis proteins into more robust synthetic biology modules for antimicrobial and biosensing applications.


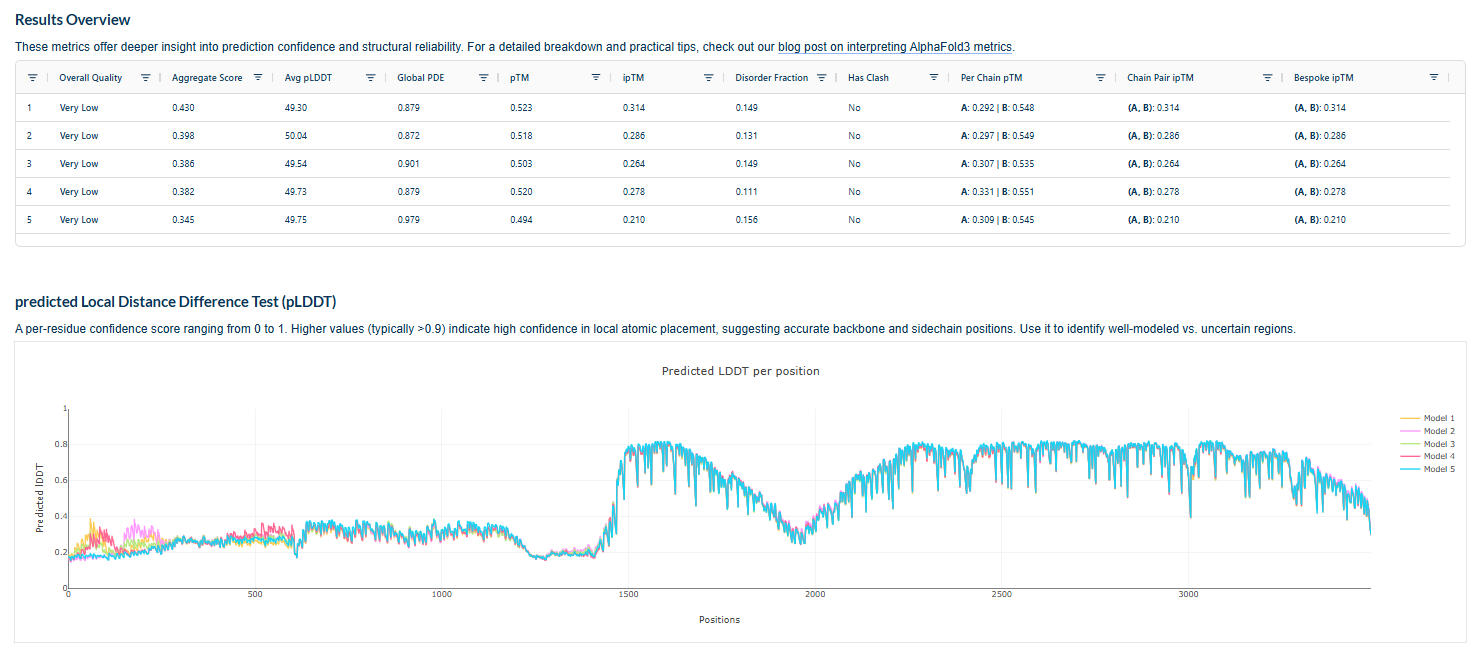
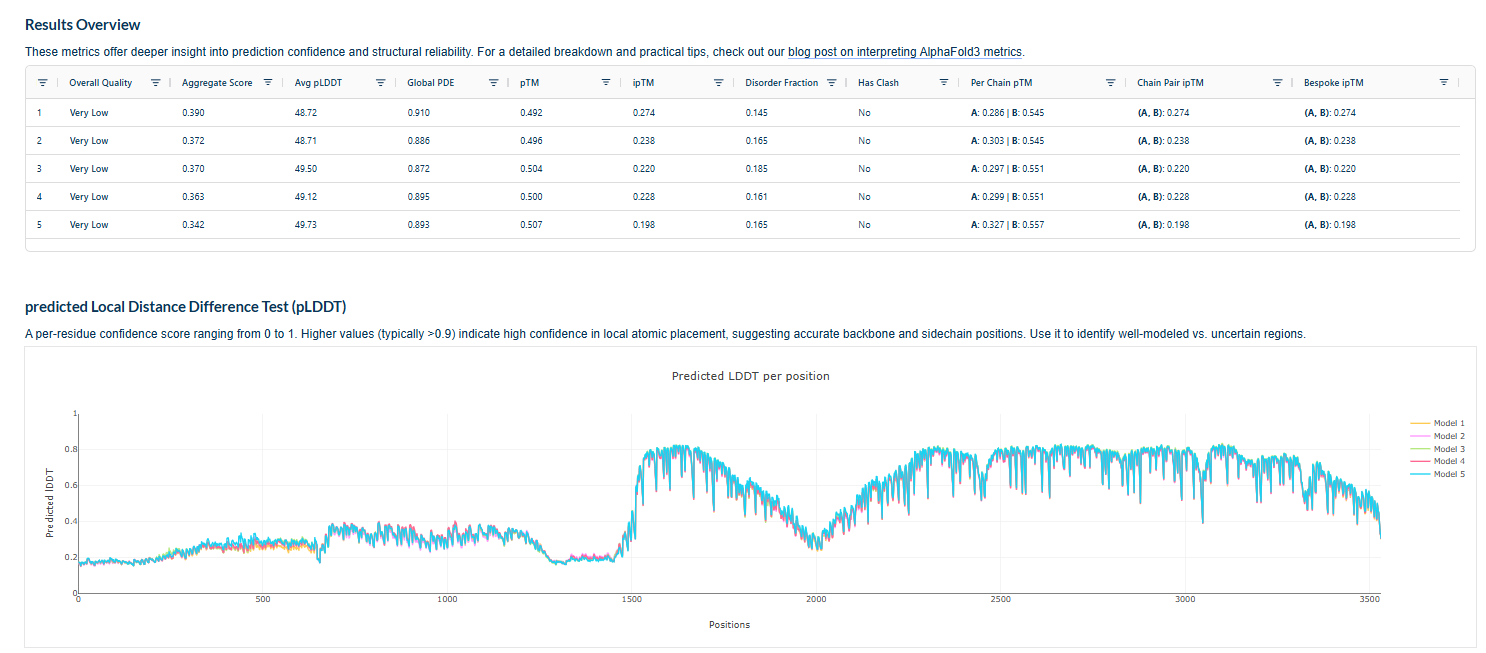
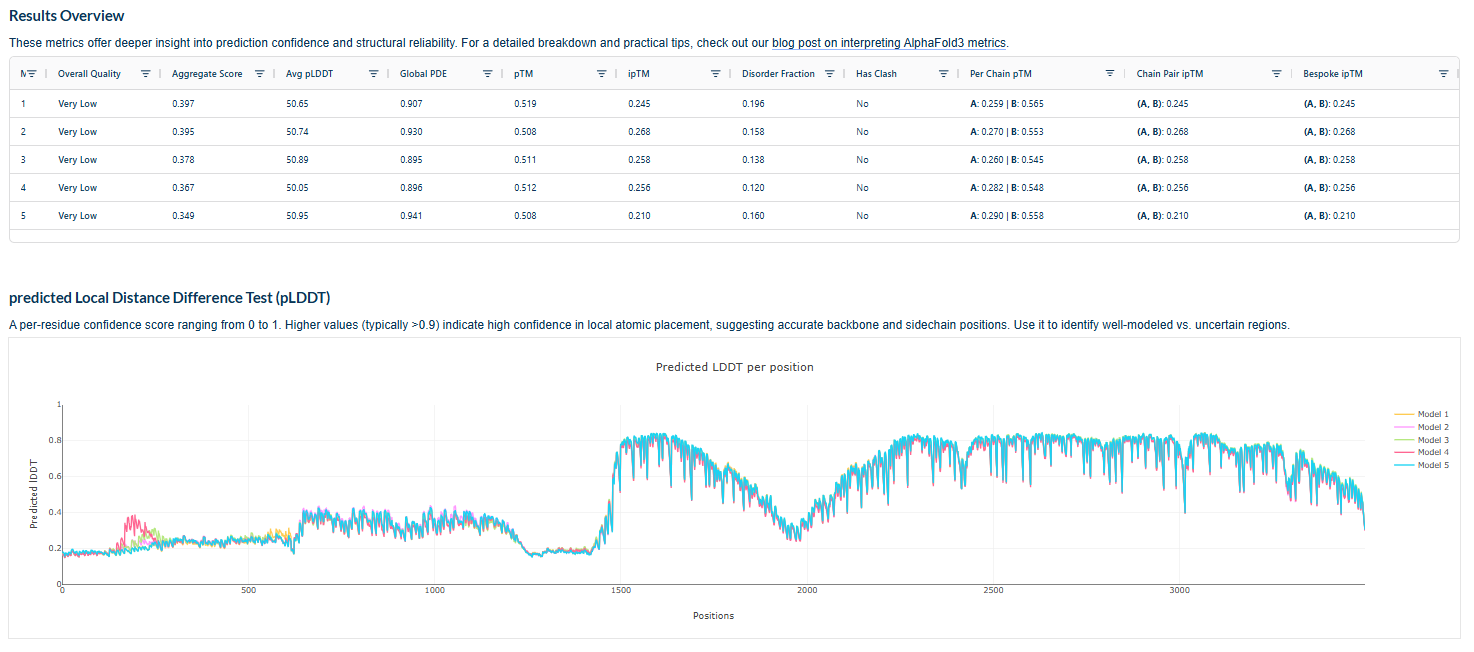
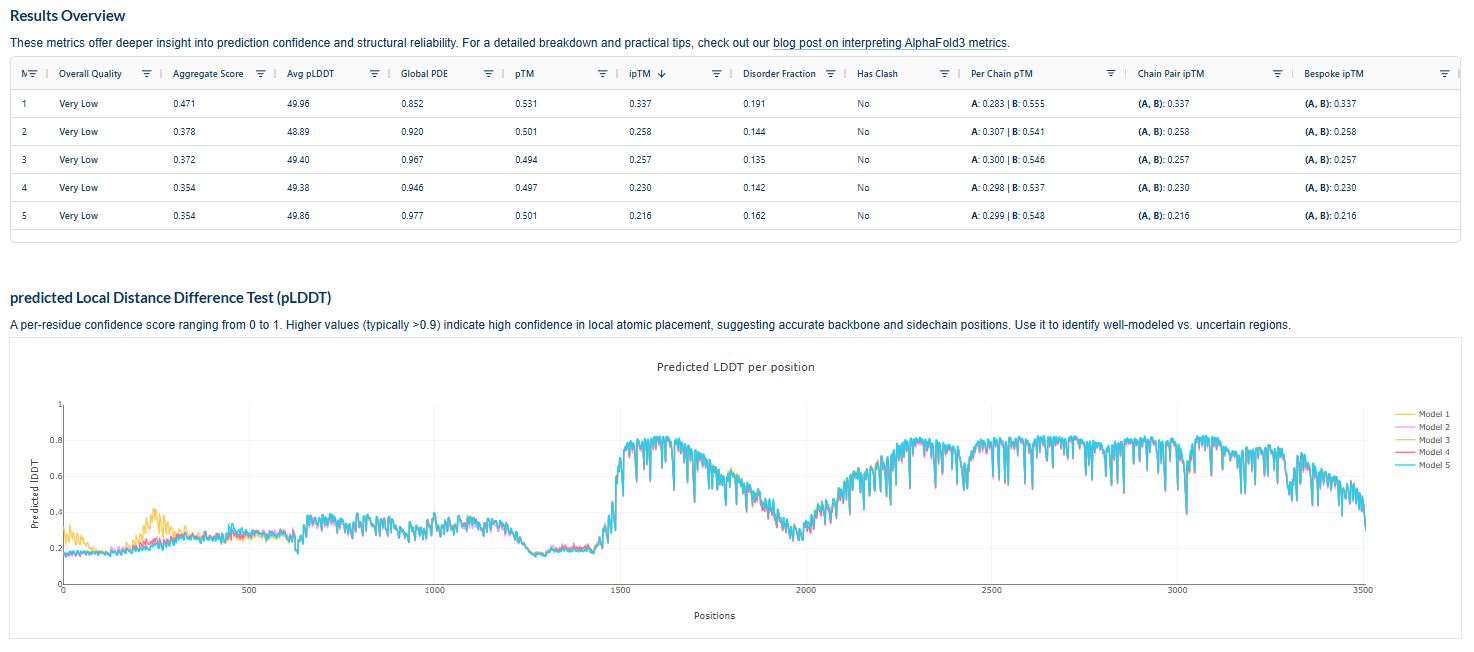
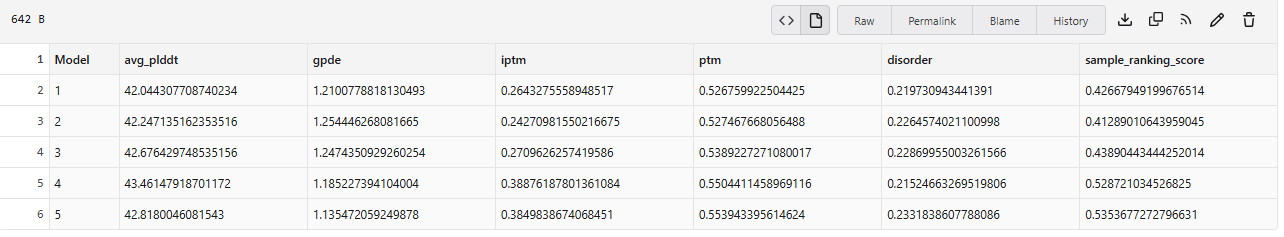
Plots and graphs for models
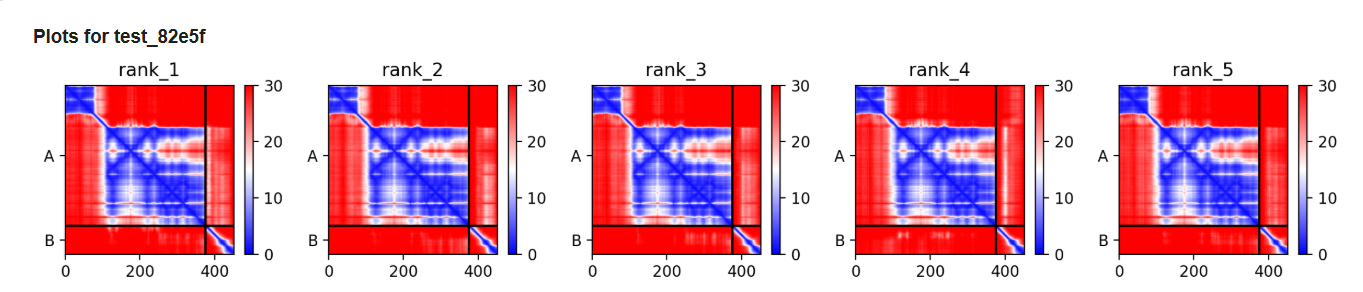
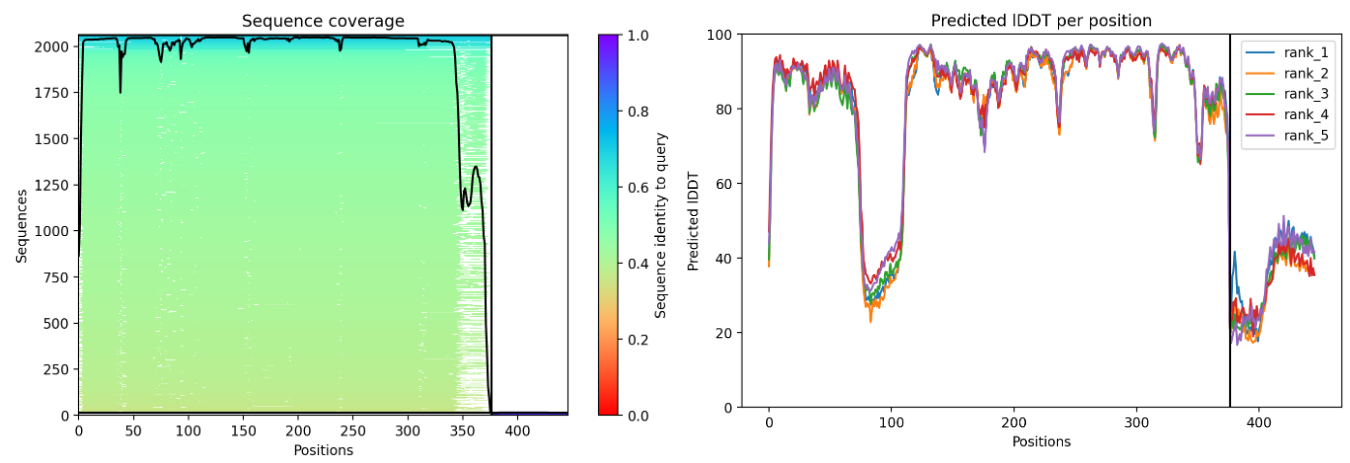
The predicted model using Alphafold3 for the mutant.

Prediction Interpretation
The image by itself does not strongly support a specific co-folded structure.possibly, but this plot alone does not show evidence that it already co-folds well.A better test is to run a complex prediction with the relevant partner and inspect whether the interface residues are consistently high confidence and whether the relative placement is stable.
References:
1.Chamakura KR, Edwards GB, Young R. Mutational analysis of the MS2 lysis protein L. Microbiology (Reading). 2017 Jul;163(7):961-969. doi: 10.1099/mic.0.000485. Epub 2017 Jul 21. PMID: 28691656; PMCID: PMC5775895.
2.Chamakura KRTran JS, Young R2017.MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ. J Bacteriol199:10.1128/jb.00058-17.https://doi.org/10.1128/jb.00058-17
3.Mezhyrova J, Martin J, Börnsen C, Dötsch V, Frangakis AS, Morgner N, Bernhard F. In vitro characterization of the phage lysis protein MS2-L. Microbiome Res Rep. 2023 Jul 20;2(4):28. doi: 10.20517/mrr.2023.28. PMID: 38045926; PMCID: PMC10688784.
4.Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). https://doi.org/10.1038/s41586-024-07487-w
5.Zeming Lin et al. ,Evolutionary-scale prediction of atomic-level protein structure with a language model.Science379,1123-1130(2023).DOI:10.1126/science.ade2574
PepMLM Colab linked from the HuggingFace PepMLM-650M model card- generating peptide binders
Abramson, J et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (2024).alphafoldserver.com to evaluate structure of binder
PeptiVerse-Zhang, Y., Tang, S., Chen, T., Mahood, E., & Chatterjee, P. (2026). PeptiVerse: A unified platform for therapeutic peptide property prediction. arXiv. doi.org
moPPit Colab linked from the HuggingFace moPPIt model card- to generate optimised peptides
Passaro, S., et al. (2025). Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. bioRxiv, 2025.06.14.659707. https://doi.org/10.1101/2025.06.14.659707- for drug discovery
Wohlwend, J., et al. (2024). Boltz-1: Democratizing Biomolecular Interaction Modeling. bioRxiv. https://doi.org/10.1101/2024.11.19.624167
“Microsoft Copilot.” Microsoft, 2026, copilot.microsoft.com.
13.Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J. (1990) “Basic local alignment search tool.” J. Mol. Biol. 215:403-410.
14.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539. DOI: 10.1038/msb.2011.75
15.Sequence Alignment tool available on VectorBuilder (https://vectorbuilder.com)
16.Lysis protein mutated structure predictions were generated using OpenFold3, an open-source reproduction of the AlphaFold 3 architecture (Abramson et al., 2024; OpenFold Consortium, 2024), implemented on the Neurosnap platform (Neurosnap Inc., 2024).
17.Lytic protein complete 5 point mutated structures and complex interactions were modeled using the OpenFold3 architecture (Abramson et al., 2024; OpenFold Consortium, 2024), deployed via the cloud-based Tamarind Bio interface (Tamarind Bio, Inc., 2024).
18.Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. doi.orgCamacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10(1), 421. doi.org