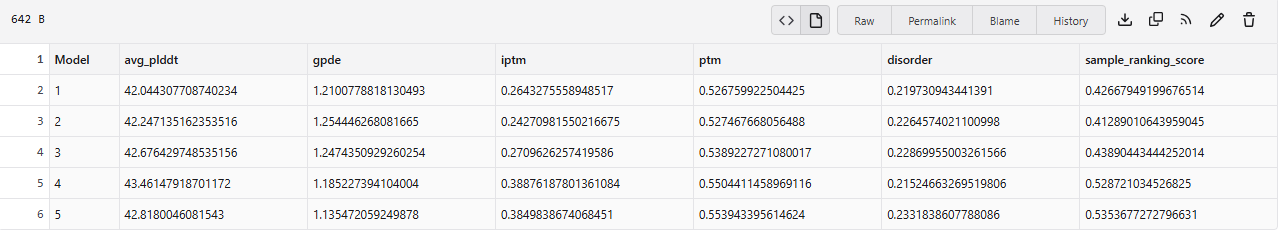
Group Final Project
Bacteriophage Engineering
GROUP MEMBERS: Deep Dalvi,Fabrizio Flores Huamán, Ganapathy Nayagam, Sheila Ramani
Hypothesis: Engineering Lysis Protein Stability via Targeted Mutations and Chaperone-Independent Folding
We hypothesize that stability can be enhanced by:
(i) introducing mutations that promote independent folding or co-folding with the chaperone DnaJ, (ii) leveraging evolutionary conservation and generative protein design to generate variants with improved thermodynamic stability and host compatibility.
Specific Aims and Validation Pipeline:
Mutation Design via Conservation and Predictive Modeling
Perform Clustal Omega alignment of homologous lysis proteins to identify conserved residues, followed by mutational scanning (e.g., via deep mutational scanning) and ESM2 embedding predictions to nominate stabilizing mutations. Predicted single and combinatorial mutants will be modeled for folding changes using ESM2, with validation of fold accuracy via AlphaFold-Multimer (AF2-Multimer) to assess independent folding propensity.

Clustal omega showing multiple alignments
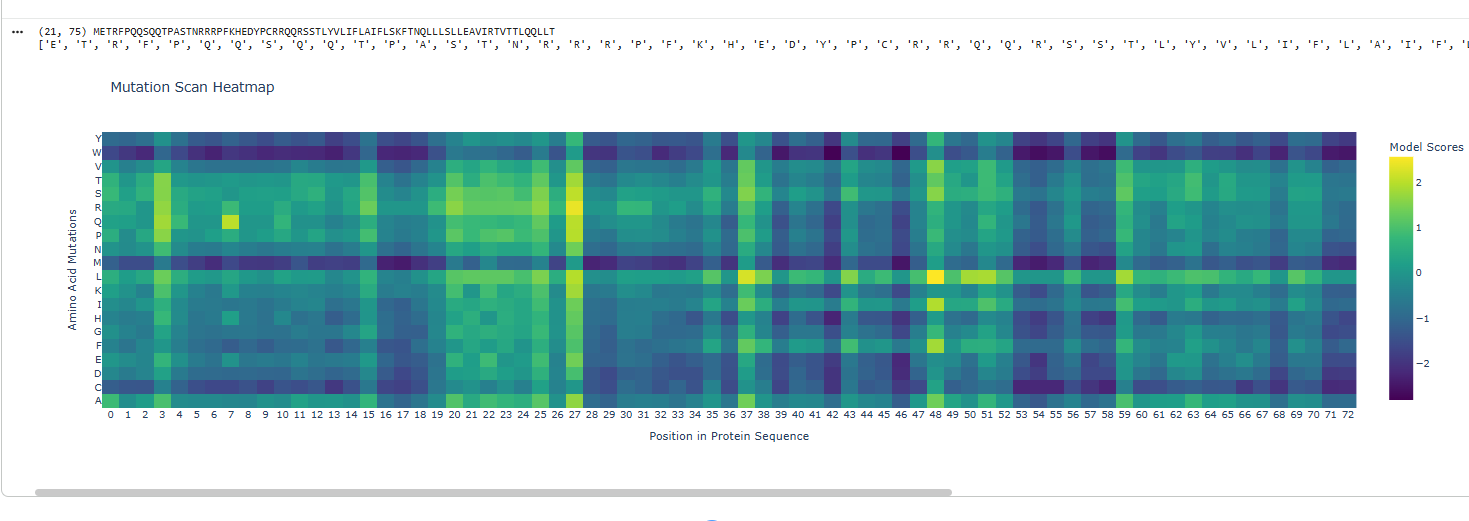
Mutational maps gives an idea of which areas are prone to disastrous effects on mutation.
Generative Design for DnaJ Co-Folding
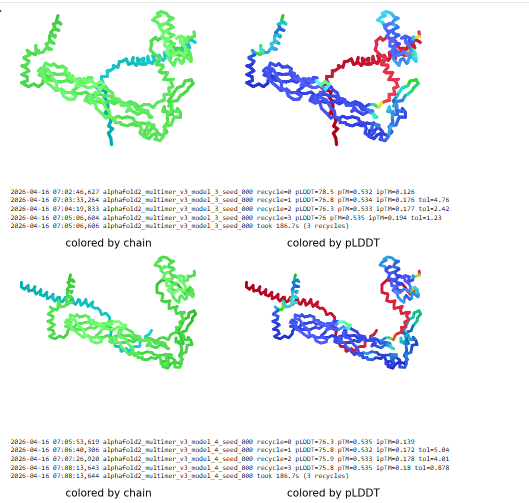
Employ protein generative models (e.g., RFdiffusion or ProteinMPNN) to design lysis protein variant optimized for co-folding with DnaJ. Co-complex structures will be predicted and ranked by AF2-Multimer confidence metrics (pLDDT > 80), prioritizing designs with buried interfaces and reduced aggregation risk.
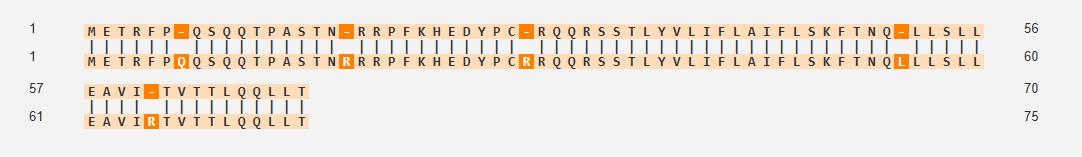
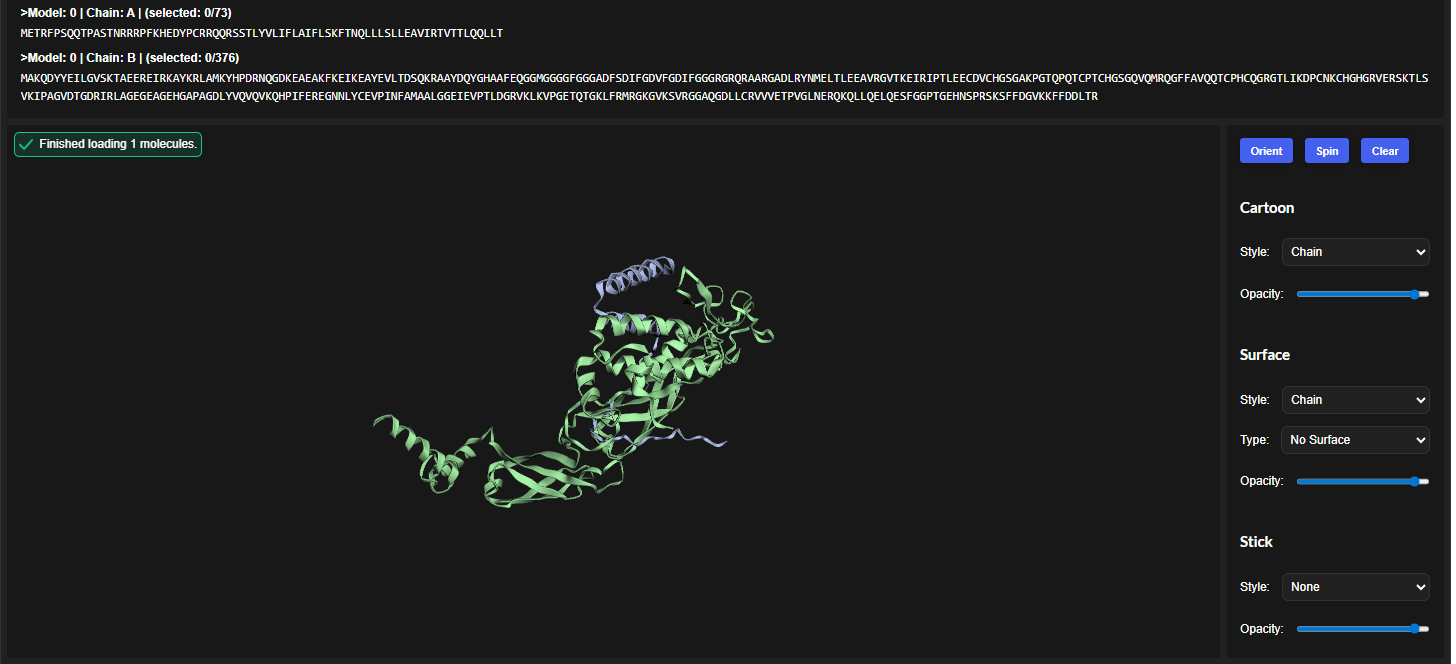
Based on the Clustal omega results mutation was done on the below sites and the protein co folded with DnaJ.
Courtesy: Neurosnap and tamarindBio and VectorBuilder
Mutational sites for creating the mutant lysis protein
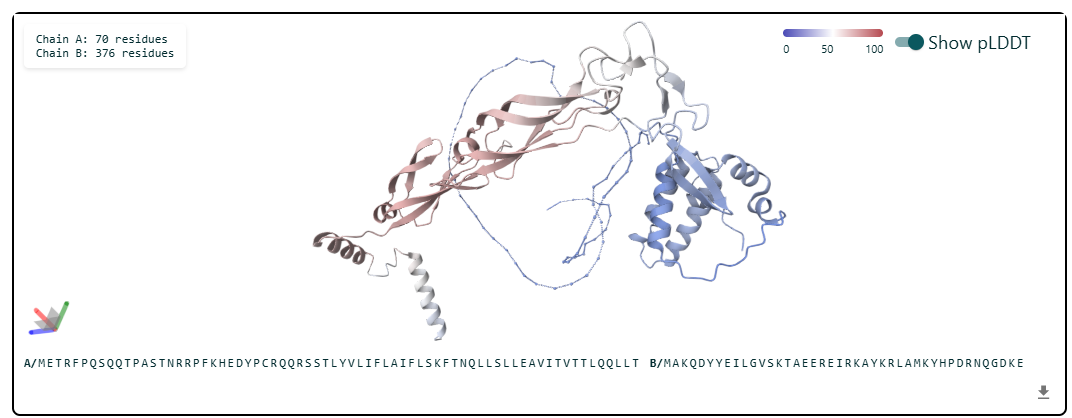
The cofolded structure predictions after mutation
Evolutionary Analysis and Chaperone Generalization
Use pBLAST to survey lysis protein orthologs across bacteria, reconstructing evolutionary trajectories of stability determinants and chaperone interactions. Top candidates will be redesigned for co-folding with alternative chaperones (e.g., DnaK or GroEL), with AF2-Multimer analysis of mature protein folds and in silico stability assays (e.g., ΔΔG predictions) against E. coli host factors to minimize proteotoxicity.

Courtesty: pBLAST tool.
Tools and Goals
The main engineering goal is to prevent or reduce the biologically important interaction between DnaJ and the lysis protein, because host-mediated adaptation of this pathway may reduce lysis efficiency and contribute to resistance-associated escape. To address this, the proposed workflow pursues three solution classes: mutation-driven DnaJ independence, engineered co-folding with DnaJ under controlled structural constraints, and folding rescue by alternative or orthogonal chaperones.
The computational tools consist of sequence alignment for conservation analysis, mutational scanning to identify sensitive and permissive residues, structure prediction to evaluate folding outcomes, generative design to explore new sequence-structure solutions, and evolutionary comparison to detect transferable mechanisms across lysis protein families.
Together, these tools support a design-build-test framework for generating lysis modules that are more stable and less vulnerable to host-dependent failure modes.
Mutations at sites 7 and 8 removed QQ and left a gap in the sequence
Mutations at site 7 8 and 9 removed QQS and left a gap
Deletions that remove the entire N‑terminal basic region (and therefore also remove 7–9) eliminate DnaJ binding and DnaJ requirement, and actually accelerate lysis, because the N‑terminal domain is no longer there to interfere with target binding when not chaperoned by DnaJ.(Jennifer et. al)
Mutation at 61 site E reoved and a gap is left
Position 61 falls within this TM domain that drives membrane insertion, oligomerization into higher‑order complexes, and the formation of large lesions spanning the outer membrane, peptidoglycan, and inner membrane.Chosen as one mutation was to be done in the C-domain of the protein.(Mezhyrova J et.al)
Mutation at site 3 with I instead of T
Position 3 is located in the N-terminal soluble domain of the L-protein.May alter the kinetics, efficiency, or interaction with host proteins (like DnaJ) due to their location in the regulatory N-terminal domain. ( Chamakura et al.)
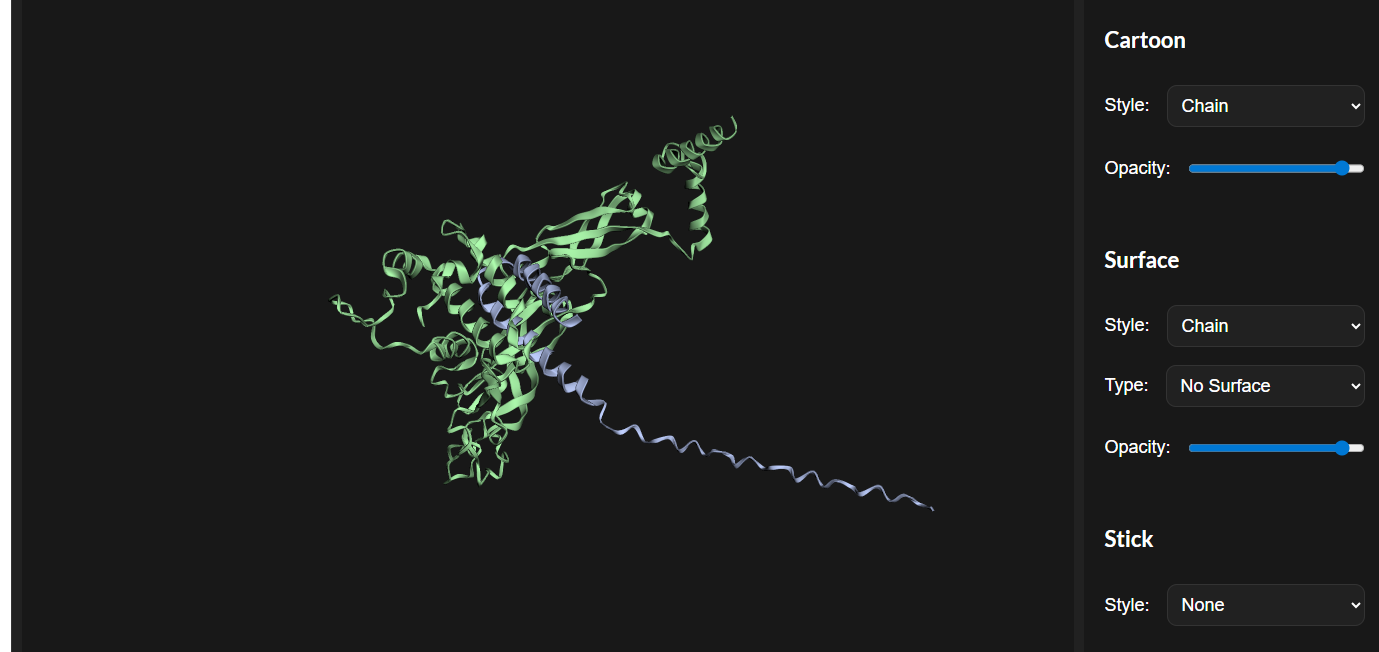
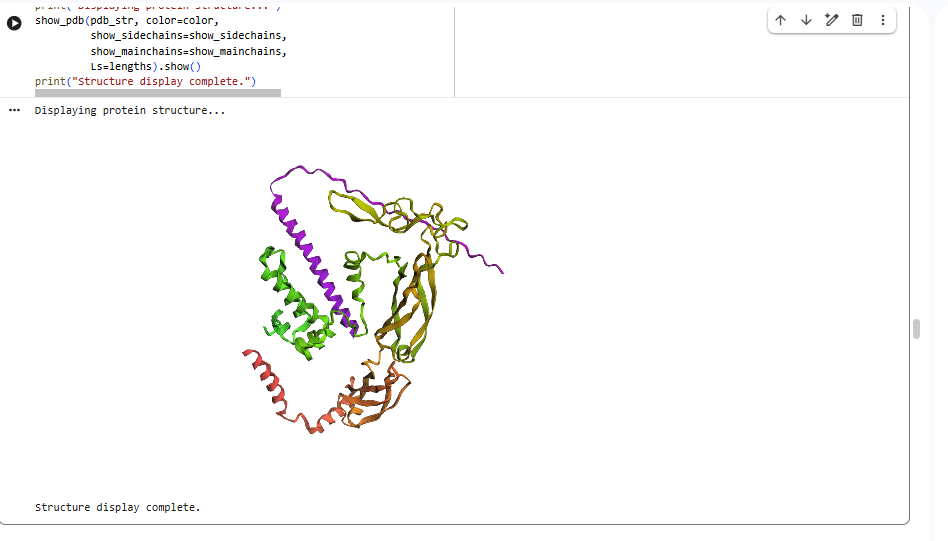
esmfold predicted structure
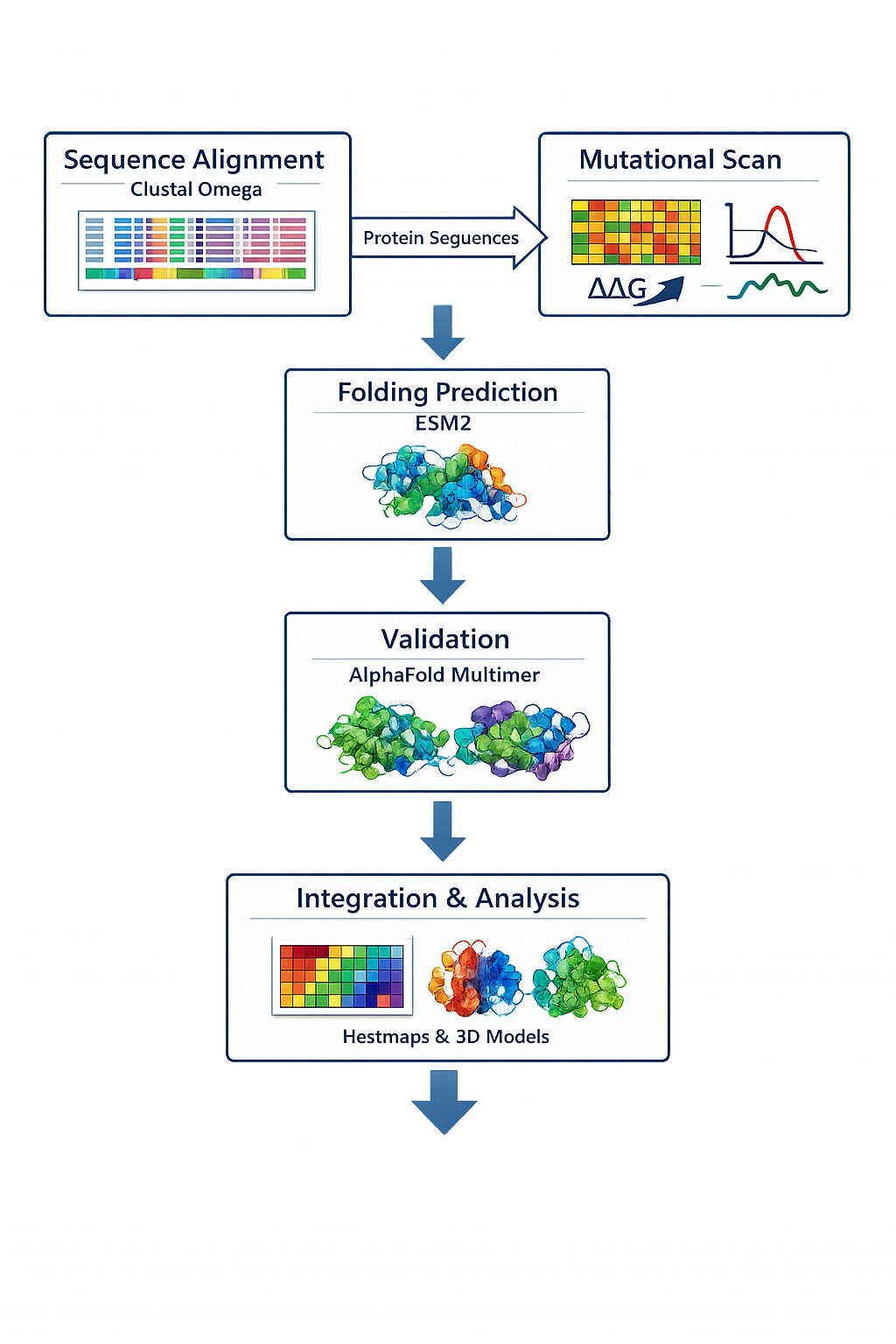
Validation Pipeline
The schematic pipeline begins with multiple sequence alignment and mutational interpretation, then proceeds to structure-guided mutant design, co-folding assessment with DnaJ, and final structural evaluation of the redesigned lysis protein. Structural predictions from AlphaFold-Multimer and ESM-based folding tools will be used to prioritize candidates before downstream experimental validation.
Courtesy: Copilot
Potential Pitfalls
A major risk is that mutation of residues involved in chaperone recognition may also perturb the native fold, membrane interaction properties, or lytic activity of the protein, leading to partial or complete loss of function. This is especially relevant for compact lysis proteins in which small sequence changes can have disproportionately large structural effects.
A second risk is that co-folding with alternative chaperones may produce unexpected host interactions, altered trafficking, or proteostasis stress in E. coli, particularly if the engineered complex is not part of the native folding network. Therefore, structural confidence must be paired with functional testing, because even high-confidence predicted folds may not guarantee productive biological activity.
Expected Outcome
If the hypothesis is correct, the resulting lysis protein variants should show improved predicted structural stability, decreased dependence on native DnaJ interaction, and retention of a mature fold compatible with lytic activity in E. coli. More broadly, this strategy could establish a generalizable framework for reengineering host-dependent phage lysis proteins into more robust synthetic biology modules for antimicrobial and biosensing applications.
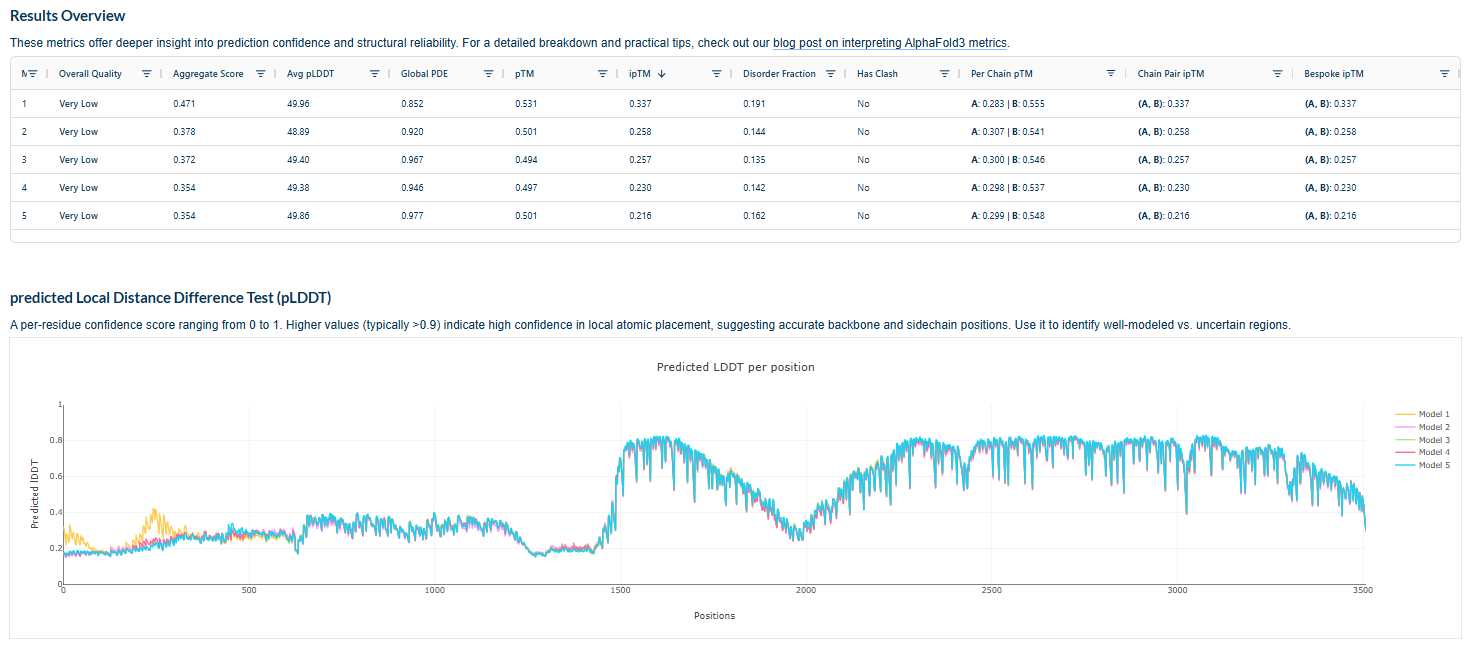
Courtesy: Alphafold3 colab notebook


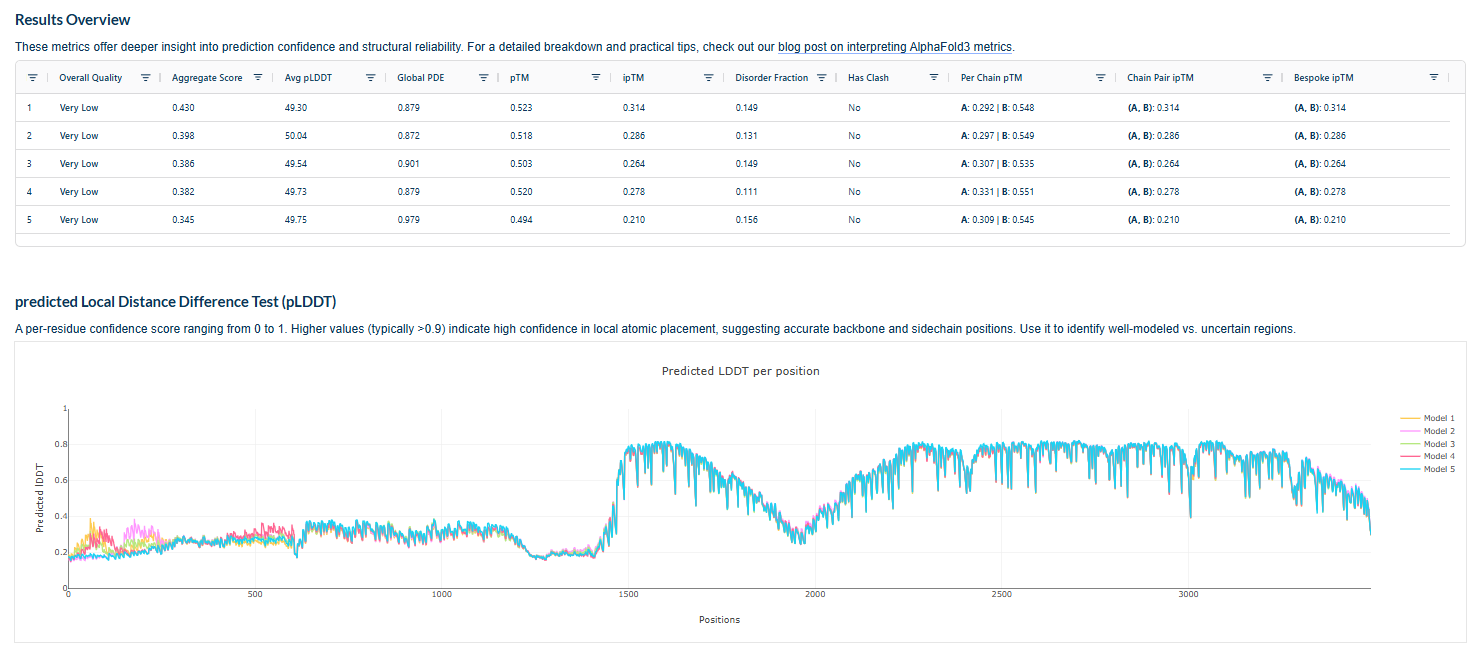
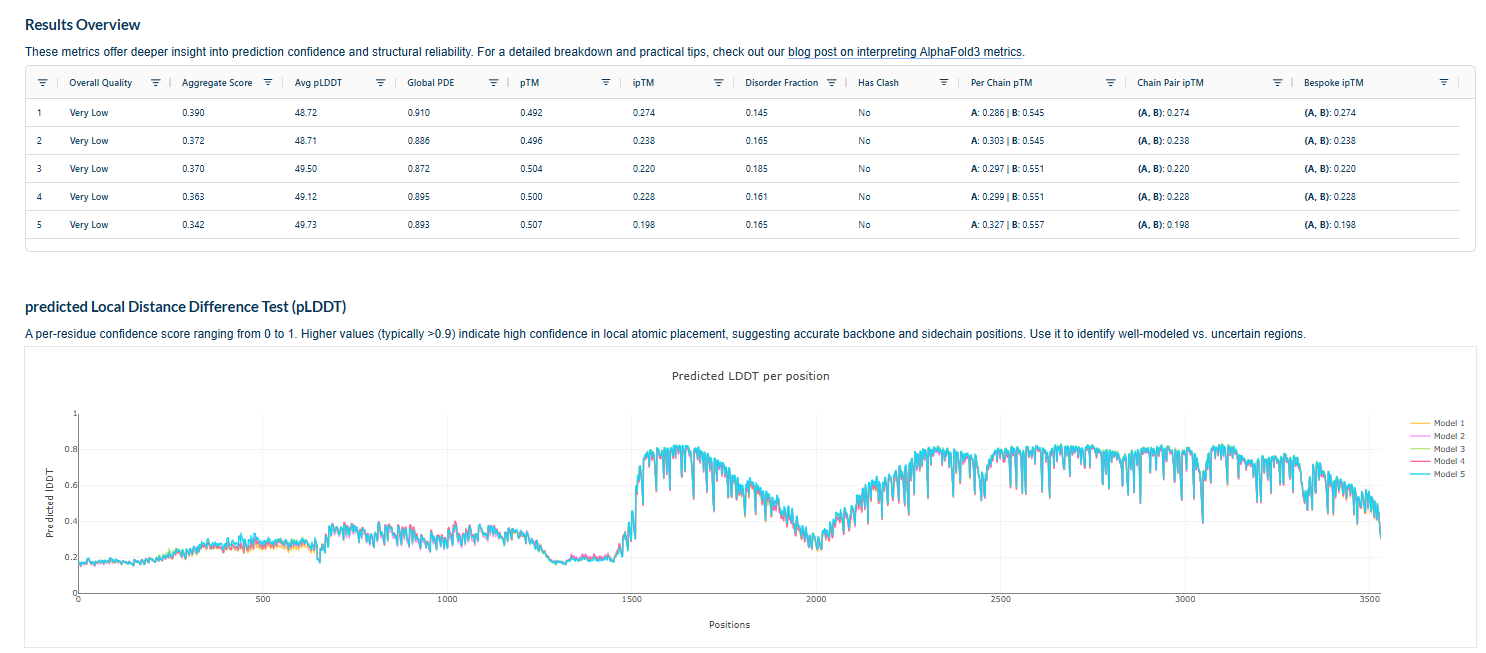
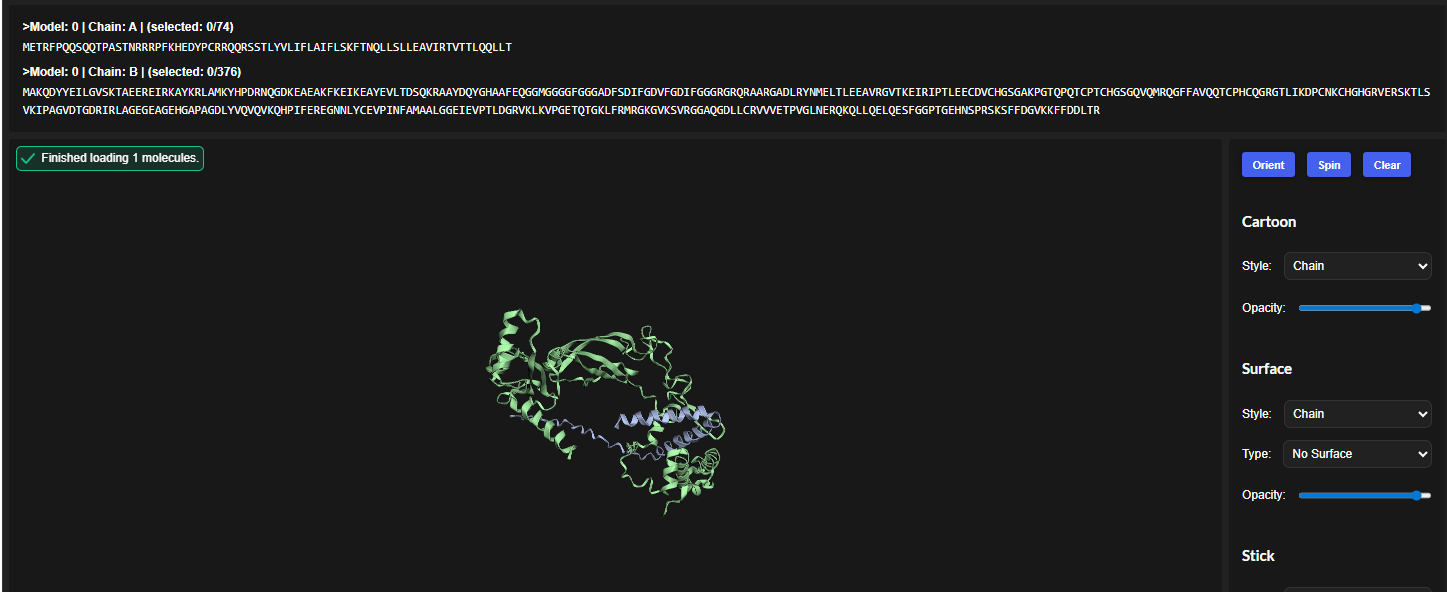
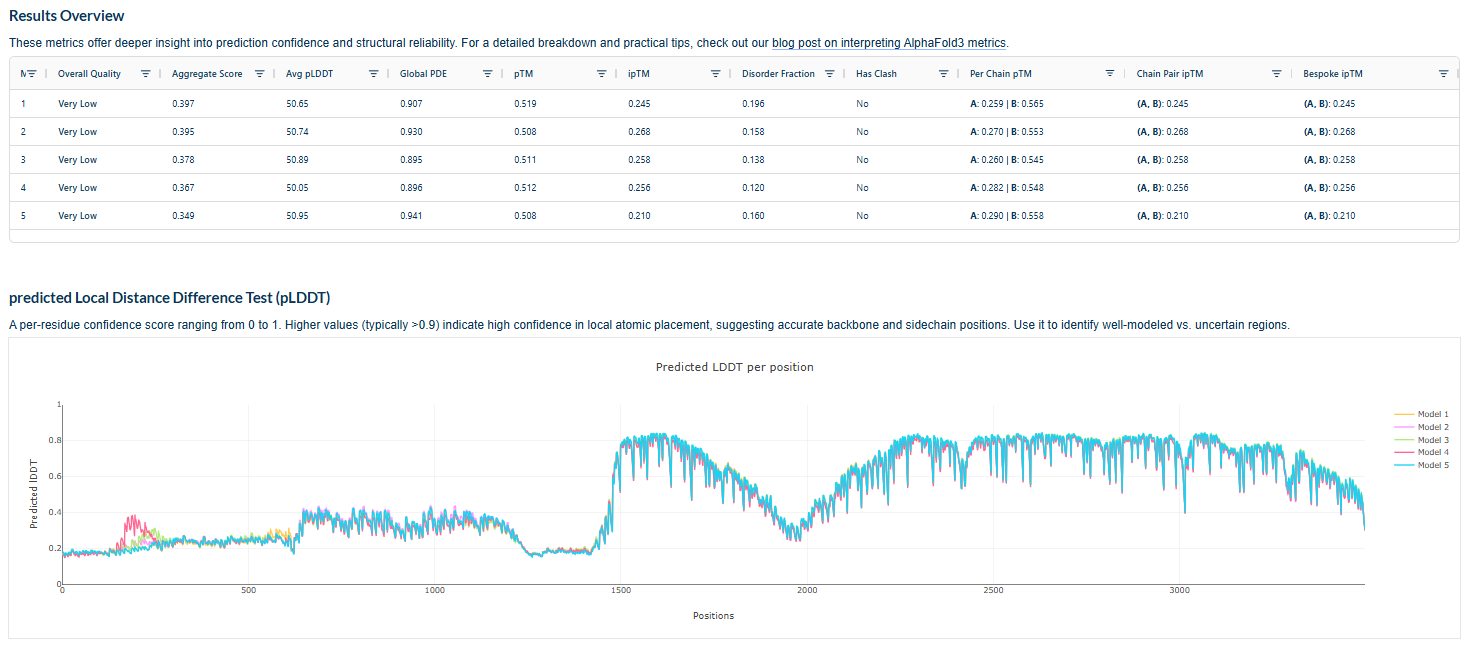
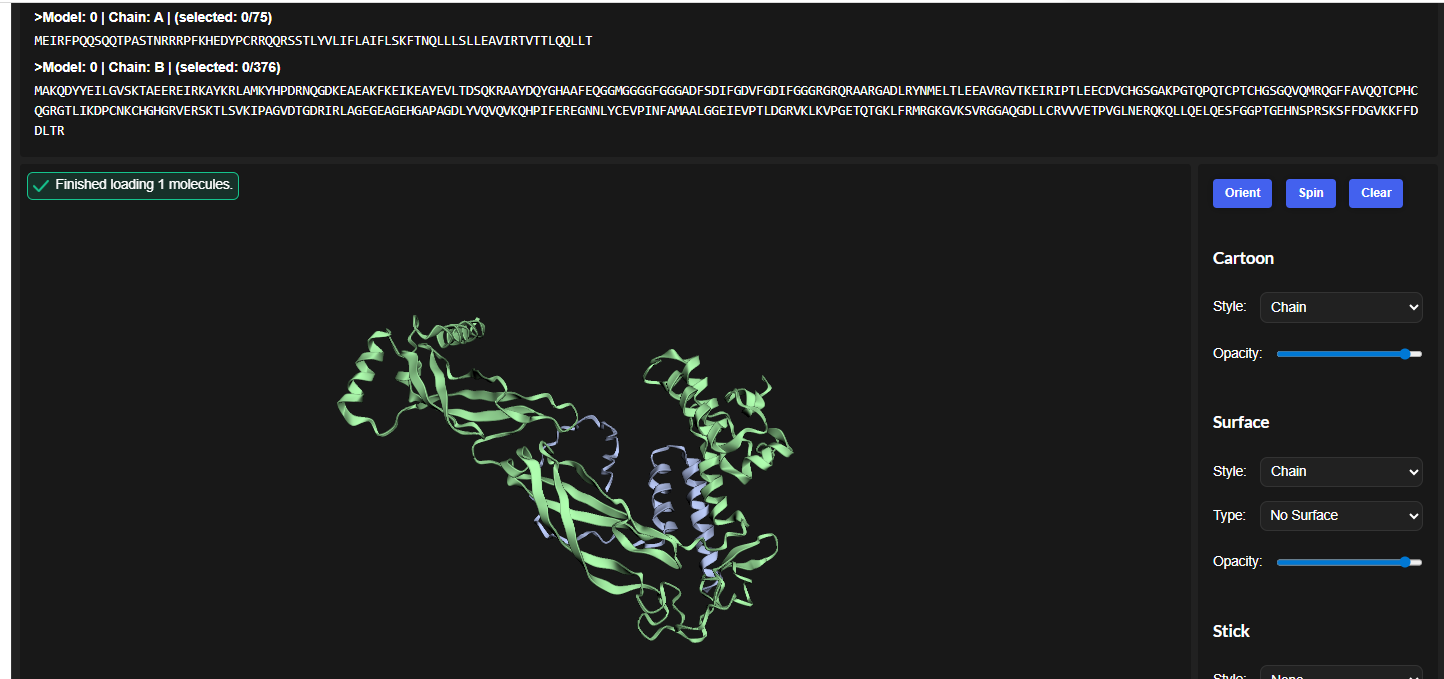
Plots for the models
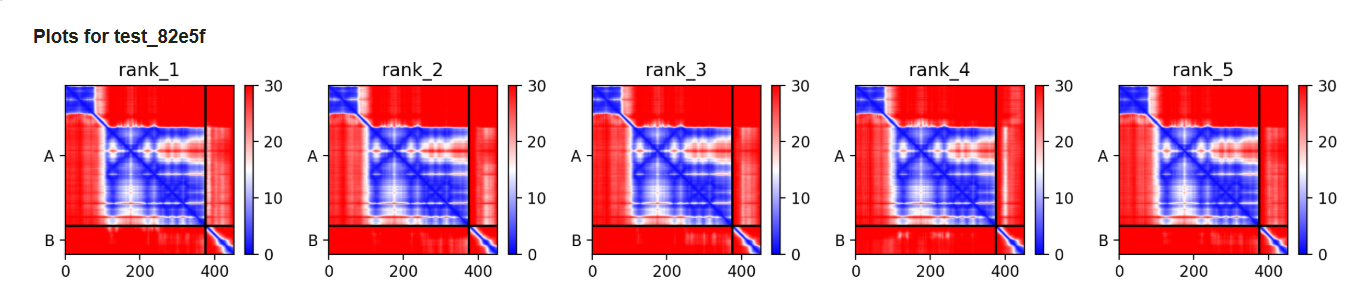
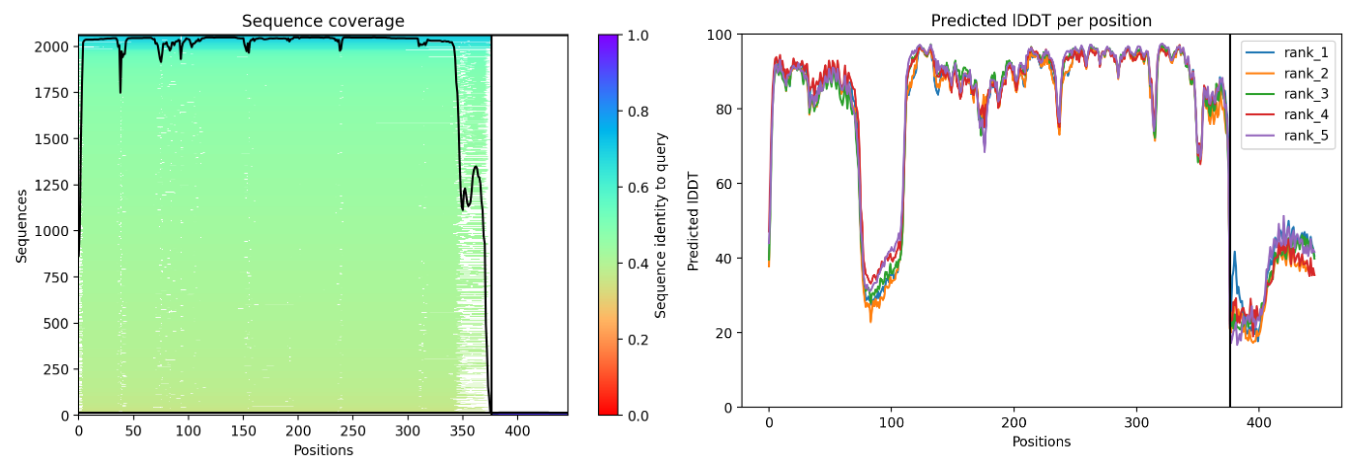
The predicted model using Alphafold3 for the mutant.

Prediction Interpretation The image by itself does not strongly support a specific co-folded structure.possibly, but this plot alone does not show evidence that it already co-folds well.A better test is to run a complex prediction with the relevant partner and inspect whether the interface residues are consistently high confidence and whether the relative placement is stable.
References:
Chamakura KR, Edwards GB, Young R. Mutational analysis of the MS2 lysis protein L. Microbiology (Reading). 2017 Jul;163(7):961-969. doi: 10.1099/mic.0.000485. Epub 2017 Jul 21. PMID: 28691656; PMCID: PMC5775895.
Chamakura KRTran JS, Young R2017.MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ. J Bacteriol199:10.1128/jb.00058-17.https://doi.org/10.1128/jb.00058-17
Mezhyrova J, Martin J, Börnsen C, Dötsch V, Frangakis AS, Morgner N, Bernhard F. In vitro characterization of the phage lysis protein MS2-L. Microbiome Res Rep. 2023 Jul 20;2(4):28. doi: 10.20517/mrr.2023.28. PMID: 38045926; PMCID: PMC10688784.
4.Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). https://doi.org/10.1038/s41586-024-07487-w
5.Zeming Lin et al. ,Evolutionary-scale prediction of atomic-level protein structure with a language model.Science379,1123-1130(2023).DOI:10.1126/science.ade2574
6.Abramson, J., Jumper, J., Evans, R. et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 455–466.
“Microsoft Copilot.” Microsoft, 2026, copilot.microsoft.com.
Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J. (1990) “Basic local alignment search tool.” J. Mol. Biol. 215:403-410.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539. DOI: 10.1038/msb.2011.75
Sequence Alignment tool available on VectorBuilder (https://vectorbuilder.com)