Week 2 HW: DNA Read, Write & Edit
Professor Jacobson:
The error rate of DNA polymerase differs in organisms and also because there are different types of polymerase enzymes. Usually this accuracy is 1 in 100,000 bp before proof reading and error correction. (I used an AI prompt to confirm, the prompt: What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?).
The length of the human genome is around 3.2 billion bp. To compare, without error correction, this would cause around 32,000 errors if the entire genome is replicated .To correct for this, biology employs DNA repair mechanisms and proof reading that massively reduce the error rate.An average protein in humans is around 375 aa long (Brocchieri, 2005). Each of those aa can be 1 of 20 types. Each of those Amino acids are coded by 3 codons on average. Codons are 3 nucleotide sequences that code for an amino acid and there are 64 of them. So, for an average protein 20375 different aa chains of length 375 . The number of different DNA encodings are 3375.
In practice most codons don’t code due to lack of specific tRNAs in the cell. These tRNAs match specific codons to specific amino acids before sending them to the ribosome for assembly. As a result if tRNAs do not recognise the specific codon, it stalls protein synthesis. (AI prompt: what are some of the reasons that all of these different codes don’t work to code for the protein of interest?).
Proteins also follow strict structural rules and if the amino acids change, the structure become unstable leading to protein destruction.
Dr LeProust:
Solid phase phosphoramidite synthesis.
As the of an oligonucleotide increase, the coupling efficiency affects the base pair added, even with a 99.9% coupling efficiency after 200nt the probability of the correct bp being added is around 37% and this decreases further as the length increases. At longer lengths, purification becomes difficult. (Gene Synthesis: Methods and Applications, 2011) (AI prompt : what is coupling efficiency).
As mentioned due to coupling efficiency, long lengths of 2000bp would have a lot of errors and may even prevent the stabilisation of the nucleotide.
Prof. George Church:
- The 10 essential amino acids are called essential cause they are usually synthesised from external sources like food. They are not made in the body and are pretty necessary for normal functioning. They are phenylalanine, valine, tryptophan, threonine, isoleucine, methionine, histidine, leucine, lysine and arginine. (Lopez and Mohiuddin, 2024).
In my view, Lysine Contingency worked as a great concept in Jurassic park initially, but like in the movies, this did not seem to work. Lysine is already an essential amino acid, so it does not make sense to add a lysine contingency, it wouldn’t do much. Even if we ignored that part, Lysine is very abundant in the ecosystem, so unless the animal starves it does not experience lysine deficiency.
Part 1: Benchling & In-silico Gel Art
I made a benchling account and I followed the protocol for the lambda phage. I could not get the accession number. I just copied the entire sequence from the lambda phage fasta file. Lambda NEB.
And pasted it into benchling.Under the project named Test. Then created a new sequence by pasting the bases from the fasta file.
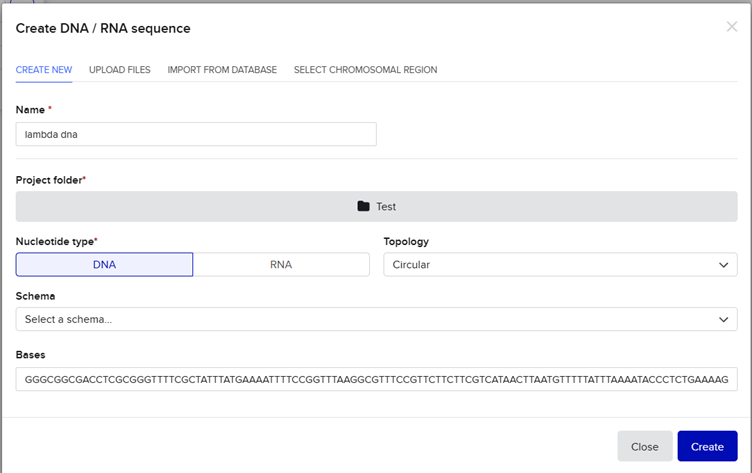
The DNA was then imported and the workspace was split into the entire genome view (on the right) and the individual bases view (on the left).
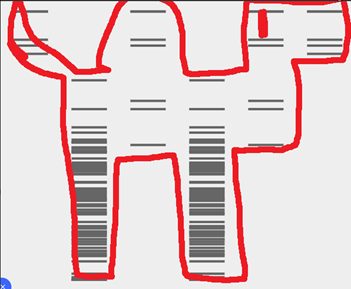
Then I made a list of restriction digestion enzymes that can be used to cut the DNA and named it HTGAA list. Every enzyme showed how many cuts they can produce on this particular genome. Before doing the cuts. I used Ronan’s Automation art website to look for a cool design.
I wanted to initially make the chrome dinosaur art, but that seemed too complex. Then I thought let me make the space invader alien, I failed. I then thought of using the barcoding like pattern to maybe convey a message. In the end I settled for this.


You would need a lot of assumptions for this, but this was the best I could come up with, a sauropod. If there were a lot more horizontal lanes, this would have come better.
The next step was to copy this to Benchling, and the automation art website gave the right enzymes to use. In benchling I made a slightly different verision. (Looks more like a camel)

Part 3: DNA Design Challenge
3.1
I chose Phluorin2. Due to the relevance in my proposal. It shows a higher level of brightness changes and has mammalian codon optimization. It is a modified version of GFP and detects ph changes. Low broghtness low ph, high brightness- high Ph.
This is the AA sequence for the protein MSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLSYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KDDGNILGHK LEYNYNEHLV YIMADKQKNG IKVIFQVHHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LHTQSALSKD PNEKRDHMVF LEFVTAAGIT HGMDELYK
3.2
Using the Twist Biosciences website, I converted the AA sequence to their nucleotide sequence using the e coli codon usage table
ATGTCAAAGGGGGAGGAACTTTTTACCGGTGTAGTACCAATTCTGGTTGAGCTCGATGGCGATGT TAATGGCCATAAGTTCTCTGTGTCTGGCGAAGGCGAAGGCGATGCCACGTATGGAAAATTAACCC TGAAATTTATATGCACGACTGGGAAATTGCCTGTGCCATGGCCGACACTCGTGACCACTCTTAGT TATGGGGTTCAGTGTTTCAGTCGATATCCAGATCACATGAAGCAGCATGACTTCTTCAAGAGCGC CATGCCAGAAGGCTATGTCCAGGAGCGTACTATCTTTTTCAAAGATGATGGAAATTACAAAACTC GTGCGGAGGTGAAATTCGAAGGCGATACTCTTGTTAATCGGATCGAGTTGAAAGGAATTGACTTC AAGGATGACGGTAACATTTTAGGGCATAAACTGGAATATAATTACAACGAACATCTGGTCTATAT TATGGCTGACAAACAGAAAAACGGCATTAAAGTAATTTTCCAGGTACATCATAATATTGAAGATG GCAGCGTTCAACTGGCAGATCATTATCAACAGAATACGCCTATAGGCGACGGTCCGGTCCTGTTA CCAGATAACCATTACCTGCACACTCAATCAGCGCTTTCGAAGGATCCGAATGAAAAGAGAGACCA CATGGTATTTCTGGAGTTCGTCACAGCGGCGGGGATCACCCATGGCATGGATGAGCTGTATAAAT AA This is with the stop codon at the end.
3.3
I used the host organism as e coli since that is what I will probably use to test this gene. Now we use codon optimisation because of anticodon availability. Now, there are around 3 ways to code for a single amino acid but organisms prefer only of those 3 ways because the anticodons that detect that sequence is available in the organism. Hence for efficient translation and minimal errors, it is best to use codon sequences that are optimal for that organism.
3.4
For cell dependent systems, I would first insert the nucleotide sequence into a cloning vector. This will be then inserted into an e-coli cell, which will hopefully produce more copies and into more cells. This is then observed in the colonies, just like you would a regular GFP protein.
Part 4: Prepare a Twist DNA Synthesis Order
4.2
I followed the instructions and then annotated the inserted sequences.
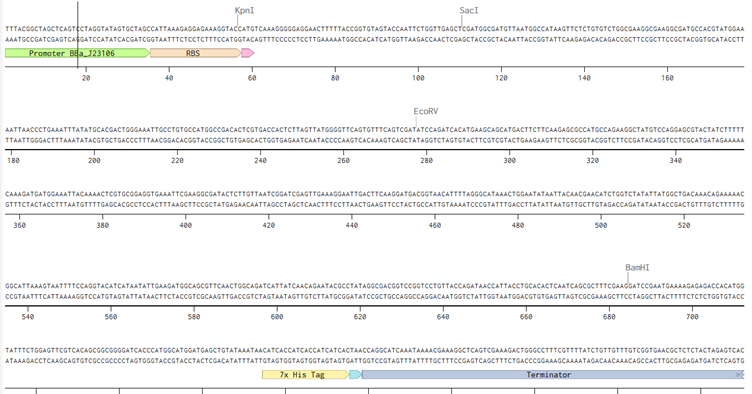
Now since my sequence is different, idk if this would change because through mere observation I noticed that the sequence I have inserted already had a stop codon, the sequence also had the promoter already built in. This is the link Link
4.6
After importing my sequence to Twist and using the pTwist high copy number as my cloning vector. I imported the entire genbank file to Benchling.
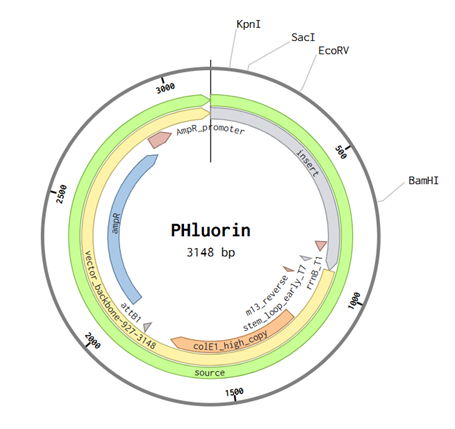
Part 5: DNA Read/Write/Edit
5.1 DNA Read
i.
I would like to sequence a lung cell. Two types, one affected by cancer and the other a normal cell. This is to check for points of differences between the cells that could be used as biomarkers. Which can then be converted to a signal input for phluorin, maybe.
ii.
I would use Whole Genome Sequencing (WGS). Second Generation NGS.
The input would be DNA extracted from the lung cell and a cancerous lung cell.
Steps: Preparing the library: DNA Extraction Fragmentation End Repair & A-Tailing Adapter Ligation PCR Amplification Size Selection & Cleanup
How are the bases decoded? Step 1: Cluster Generation (Bridge Amplification) • Library fragments bind to complementary oligos on the flow cell. • DNA bends over and binds to nearby oligos. • PCR amplification forms clusters (many identical copies of each fragment). This increases signal strength.
Step 2: Sequencing by Synthesis (SBS) Illumina uses: • Fluorescently labeled nucleotides • Reversible terminator chemistry Process:
- Add all 4 labeled nucleotides (A, T, C, G).
- Only one nucleotide is incorporated per cycle.
- Laser excites fluorophore.
- Camera records emitted fluorescence.
- Terminator and dye are cleaved.
- Cycle repeats. Each cycle identifies one base.
Base Calling • Each cluster emits fluorescence. • The color corresponds to A, T, C, or G. • Software converts fluorescence signals into nucleotide sequences. • This produces short DNA reads.
Output Illumina produces: • Millions to billions of short reads • Typically 100–150 bp paired-end reads • Stored in FASTQ format, containing: o DNA sequence o Quality score (Phred score)
The output would be the FASTQ file
5.2 DNA write
5.3 DNA Edit
i.
The genome of a chicken to make a chicken-saurus. There are genes that are inactive in a chicken that were active in their ancestors (dinosaurs). By activating those genes we can potentially make them express and look more like a dinosaur. I would do this because Dinosaurs are cool and it would be harder to synthesize an entire Trex genome than to modify a chicken’s existing genome. Other reasons could be to understand better the evolution and the bifurcation of birds and reptiles.
ii.
I would use CRISPR to edit the DNA cause it is the easiest and most cost effective for these kind of things. The steps include making a guide RNA that detects the sequence in a chicken cell, usually in this case something that targets the repressors and/or promoters /enhancers. So the basic outline would be to identify the repressor that prevents the elongation of tail length for example and then make a guide RNA that detects that repressor and use CRISPR to cut out that part therby making it active
First would be to do a comparative analysis between the chicken and other closely related ancestors (like crocodiles) to identify Conserved developmental genes Regulatory DNA differences, Suppressed ancestral pathways.
Then you would make a genetic map identifying the relevant pathways and the promoters, enhancers and repressors associated with that pathway.
Next we either insert promoters in case of activating dormant genes or removing repressors using CRISPR and the guide RNA. This would be the input.Limitations: Polygenic Traits, many of the pathways are controlled by many repressors/promoters and thereby editing one gene is not enough, this adds complexity.
Modifying one gene could disrupt other genes downstream potentially reducing viability.
Mosaicism only some cells could be modified and then other repair mechanisms may override the genetic modifications stopping the experiment prematurely.
References:
Brocchieri, L. (2005). Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Research, 33(10), pp.3390–3400. doi:https://doi.org/10.1093/nar/gki615.
Gene Synthesis: Methods and Applications. (2011). Methods in Enzymology, [online] 498, pp.277–309. doi:https://doi.org/10.1016/B978-0-12-385120-8.00012-7.
Lopez, M.J. and Mohiuddin, S.S. (2024). Biochemistry, Essential Amino Acids. [online] PubMed. Available at: https://www.ncbi.nlm.nih.gov/books/NBK557845/.
AI used: OpenAI’s ChatGPT 4.1