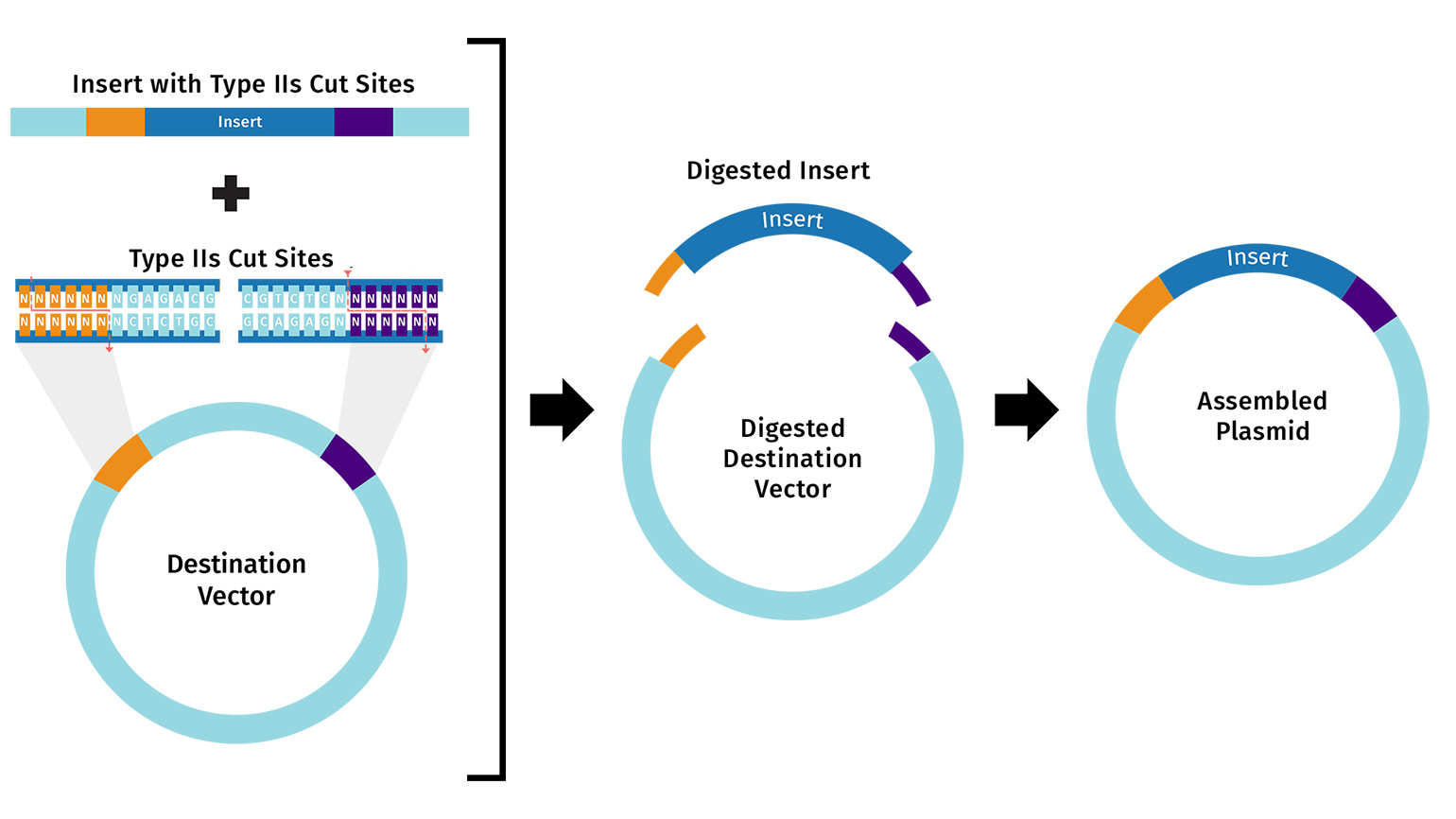Homework
Weekly homework submissions:
Week 01 HW: Principles and Practices
Class Assignment 1. First, describe a biological engineering application or tool you want to develop and why. I want to optimize PETase (polyethylene terephthalate hydrolase). PETase is an enzyme that can break down PET plastics, which are widely used in packaging. By optimizing PETase, we can enhance its efficiency in degrading PET and increase its stability under various conditions. This could lead to more effective recycling processes and help reduce plastic pollution.
Week 02 HW: DNA Read, Write, & Edit
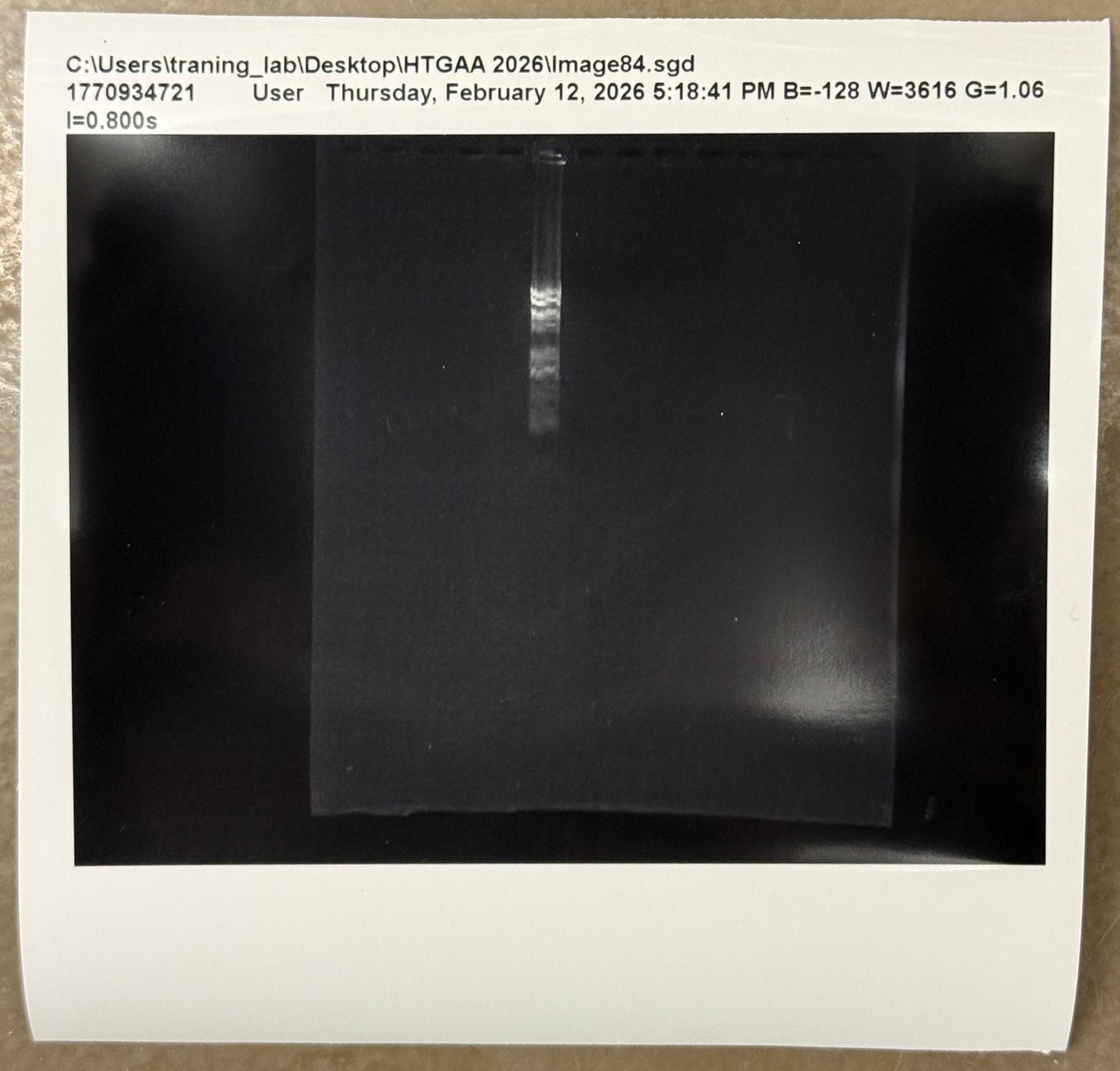
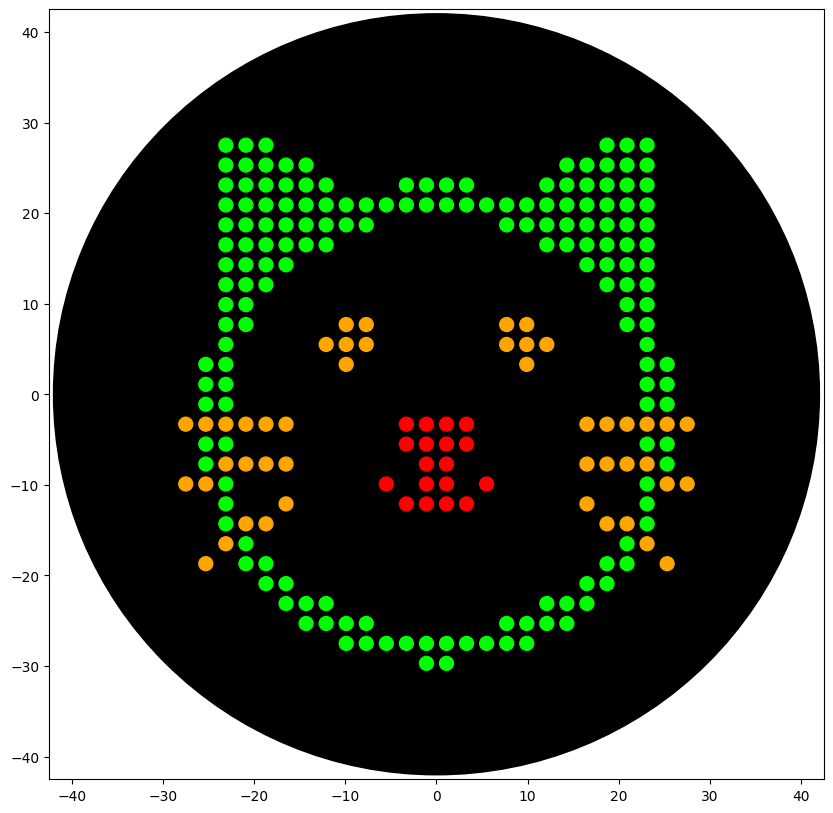
Part 0: Basics of Gel Electrophoresis I have attended the recitation. Part 1: Benchling & In-silico Gel Art I made the gel art below. It is “HT” for “How To grow almost anything”. Part 2: Gel Art - Restriction Digests and Gel Electrophoresis I worked in group with Louisa, Jasmine, and Yutong. We tried to make the cat gel art designed by Louisa, but unfortunately it was not very successful. Photo below:
Python Script for Opentrons Artwork I created a design using opentrons-art.rcdonovan.com Opentrons-Art Website: https://opentrons-art.rcdonovan.com/?id=80fx569l8o4tho4 Google Colab: https://colab.research.google.com/drive/1UPiCmwBP3sIFD_rNVRHeT3YhuiQQ5ZGP#scrollTo=pczDLwsq64mk&line=6&uniqifier=1 The OpentronMock gives the following output: Code:
### YOUR CODE HERE to create your design ### sfgfp_points = [(-3.3, -3.3),(-1.1, -3.3),(1.1, -3.3),(3.3, -3.3),(-3.3, -5.5),(-1.1, -5.5),(1.1, -5.5),(3.3, -5.5),(-1.1, -7.7),(1.1, -7.7),(-5.5, -9.9),(-1.1, -9.9),(1.1, -9.9),(5.5, -9.9),(-3.3, -12.1),(-1.1, -12.1),(1.1, -12.1),(3.3, -12.1)] mrfp1_points = [(-23.1, 27.5),(-20.9, 27.5),(-18.7, 27.5),(18.7, 27.5),(20.9, 27.5),(23.1, 27.5),(-23.1, 25.3),(-20.9, 25.3),(-18.7, 25.3),(-16.5, 25.3),(-14.3, 25.3),(14.3, 25.3),(16.5, 25.3),(18.7, 25.3),(20.9, 25.3),(23.1, 25.3),(-23.1, 23.1),(-20.9, 23.1),(-18.7, 23.1),(-16.5, 23.1),(-14.3, 23.1),(-12.1, 23.1),(-3.3, 23.1),(-1.1, 23.1),(1.1, 23.1),(3.3, 23.1),(12.1, 23.1),(14.3, 23.1),(16.5, 23.1),(18.7, 23.1),(20.9, 23.1),(23.1, 23.1),(-23.1, 20.9),(-20.9, 20.9),(-18.7, 20.9),(-16.5, 20.9),(-14.3, 20.9),(-12.1, 20.9),(-9.9, 20.9),(-7.7, 20.9),(-5.5, 20.9),(-3.3, 20.9),(-1.1, 20.9),(1.1, 20.9),(3.3, 20.9),(5.5, 20.9),(7.7, 20.9),(9.9, 20.9),(12.1, 20.9),(14.3, 20.9),(16.5, 20.9),(18.7, 20.9),(20.9, 20.9),(23.1, 20.9),(-23.1, 18.7),(-20.9, 18.7),(-18.7, 18.7),(-16.5, 18.7),(-14.3, 18.7),(-12.1, 18.7),(-9.9, 18.7),(-7.7, 18.7),(7.7, 18.7),(9.9, 18.7),(12.1, 18.7),(14.3, 18.7),(16.5, 18.7),(18.7, 18.7),(20.9, 18.7),(23.1, 18.7),(-23.1, 16.5),(-20.9, 16.5),(-18.7, 16.5),(-16.5, 16.5),(-14.3, 16.5),(-12.1, 16.5),(12.1, 16.5),(14.3, 16.5),(16.5, 16.5),(18.7, 16.5),(20.9, 16.5),(23.1, 16.5),(-23.1, 14.3),(-20.9, 14.3),(-18.7, 14.3),(-16.5, 14.3),(16.5, 14.3),(18.7, 14.3),(20.9, 14.3),(23.1, 14.3),(-23.1, 12.1),(-20.9, 12.1),(-18.7, 12.1),(18.7, 12.1),(20.9, 12.1),(23.1, 12.1),(-23.1, 9.9),(-20.9, 9.9),(20.9, 9.9),(23.1, 9.9),(-23.1, 7.7),(-20.9, 7.7),(20.9, 7.7),(23.1, 7.7),(-23.1, 5.5),(23.1, 5.5),(-25.3, 3.3),(-23.1, 3.3),(23.1, 3.3),(25.3, 3.3),(-25.3, 1.1),(-23.1, 1.1),(23.1, 1.1),(25.3, 1.1),(-25.3, -1.1),(-23.1, -1.1),(23.1, -1.1),(25.3, -1.1),(-25.3, -5.5),(-23.1, -5.5),(23.1, -5.5),(25.3, -5.5),(-25.3, -7.7),(25.3, -7.7),(-23.1, -9.9),(23.1, -9.9),(-23.1, -12.1),(23.1, -12.1),(-23.1, -14.3),(23.1, -14.3),(-20.9, -16.5),(20.9, -16.5),(-20.9, -18.7),(-18.7, -18.7),(18.7, -18.7),(20.9, -18.7),(-18.7, -20.9),(-16.5, -20.9),(16.5, -20.9),(18.7, -20.9),(-16.5, -23.1),(-14.3, -23.1),(-12.1, -23.1),(12.1, -23.1),(14.3, -23.1),(16.5, -23.1),(-14.3, -25.3),(-12.1, -25.3),(-9.9, -25.3),(-7.7, -25.3),(7.7, -25.3),(9.9, -25.3),(12.1, -25.3),(14.3, -25.3),(-9.9, -27.5),(-7.7, -27.5),(-5.5, -27.5),(-3.3, -27.5),(-1.1, -27.5),(1.1, -27.5),(3.3, -27.5),(5.5, -27.5),(7.7, -27.5),(9.9, -27.5),(-1.1, -29.7),(1.1, -29.7)] azurite_points = [(-9.9, 7.7),(-7.7, 7.7),(7.7, 7.7),(9.9, 7.7),(-12.1, 5.5),(-9.9, 5.5),(-7.7, 5.5),(7.7, 5.5),(9.9, 5.5),(12.1, 5.5),(-9.9, 3.3),(9.9, 3.3)] mwasabi_points = [(-27.5, -3.3),(-25.3, -3.3),(-23.1, -3.3),(-20.9, -3.3),(-18.7, -3.3),(-16.5, -3.3),(16.5, -3.3),(18.7, -3.3),(20.9, -3.3),(23.1, -3.3),(25.3, -3.3),(27.5, -3.3),(-23.1, -7.7),(-20.9, -7.7),(-18.7, -7.7),(-16.5, -7.7),(16.5, -7.7),(18.7, -7.7),(20.9, -7.7),(23.1, -7.7),(-27.5, -9.9),(-25.3, -9.9),(25.3, -9.9),(27.5, -9.9),(-16.5, -12.1),(16.5, -12.1),(-20.9, -14.3),(-18.7, -14.3),(18.7, -14.3),(20.9, -14.3),(-23.1, -16.5),(23.1, -16.5),(-25.3, -18.7),(25.3, -18.7)] scale = 1 def draw_points(points, color="Red"): segments = [] for i in range(0, len(points), 20): segments.append(points[i : i+20]) for seg in segments: pipette_20ul.pick_up_tip() pipette_20ul.aspirate(len(seg), location_of_color(color)) for x, y in seg: adjusted_location = center_location.move(types.Point(x=xscale, y=yscale)) dispense_and_detach(pipette_20ul, 1, adjusted_location) pipette_20ul.drop_tip() draw_points(sfgfp_points, "Red") draw_points(mrfp1_points, "Green") draw_points(azurite_points, "Orange") draw_points(mwasabi_points, "Orange") Result With the help of our TA Ronan, the art was printed with an Opentrons robot. The result is shown below:
Week 04 HW: Protein Design Part I
Part A: Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) 1 Dalton = 1.66 x 10-24 grams. 100 Daltons = 1.66 x 10-22 grams. 1 gram = 6.02 x 1023 molecules. 20% of meat is protein, so 100 grams of proteins in 500 grams of meat. Therefore: 100 x 6.02 x 1023 = 6.02 x 1025 molecules of amino acids.
Week 05 HW: Protein Design Part II
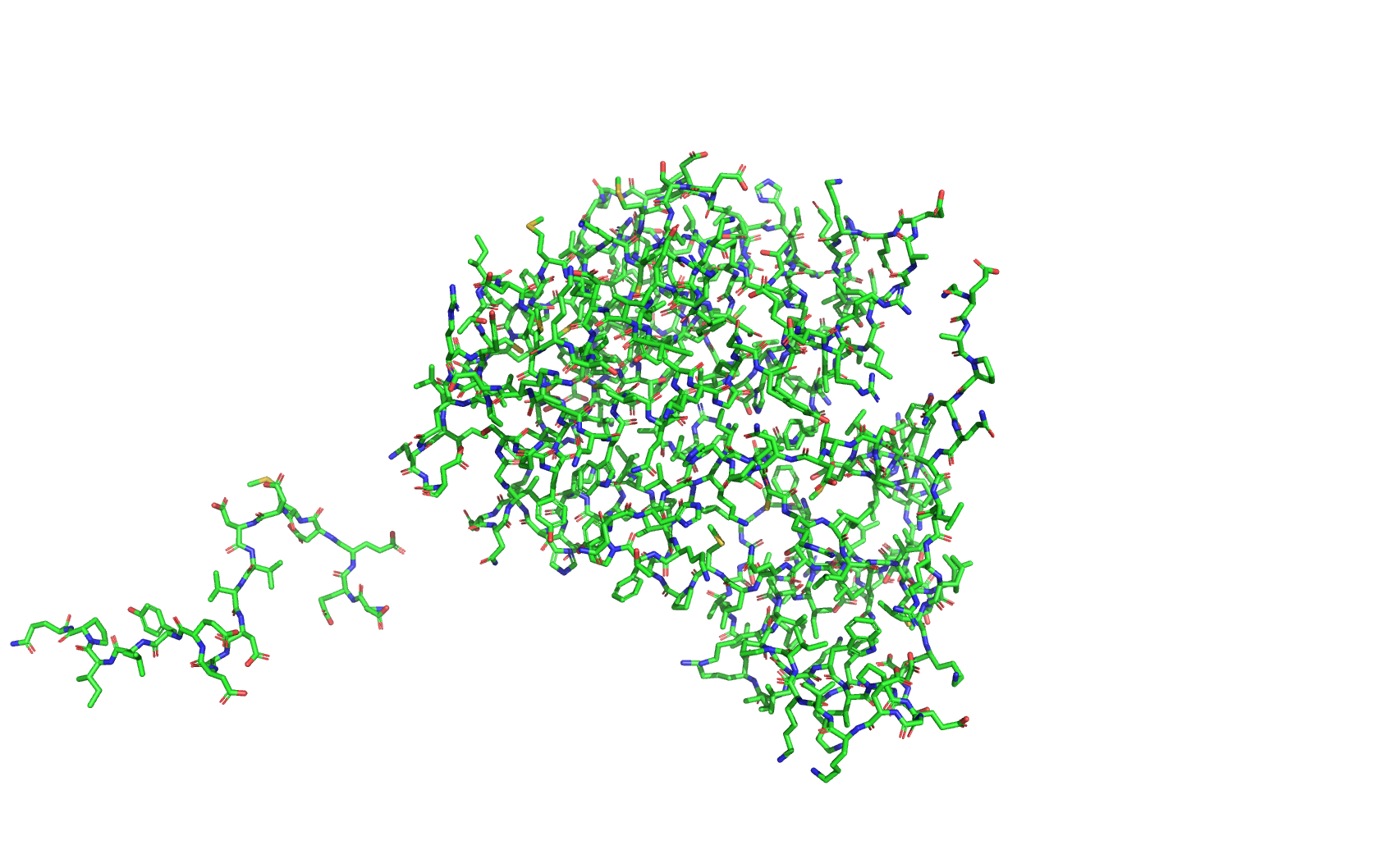
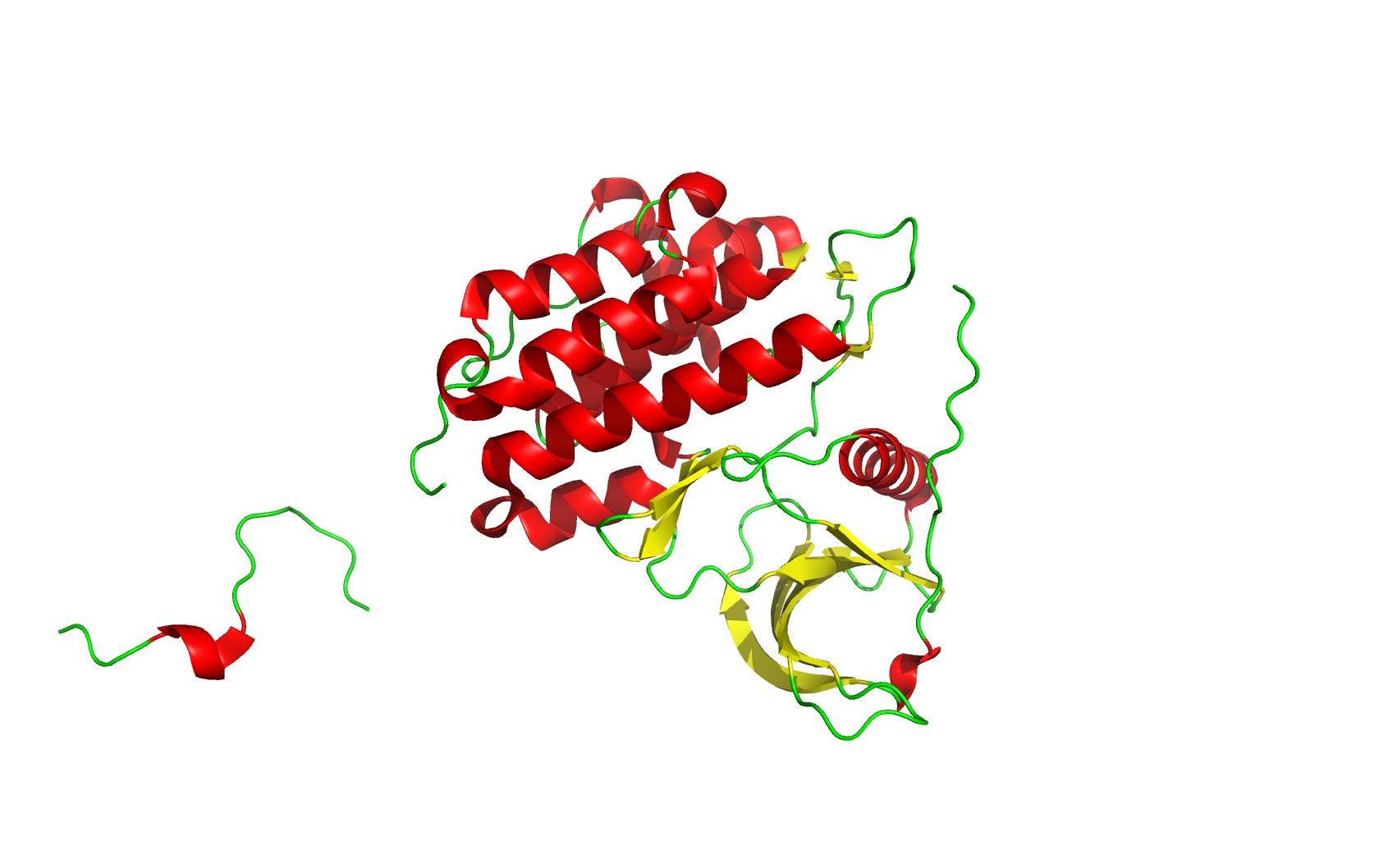
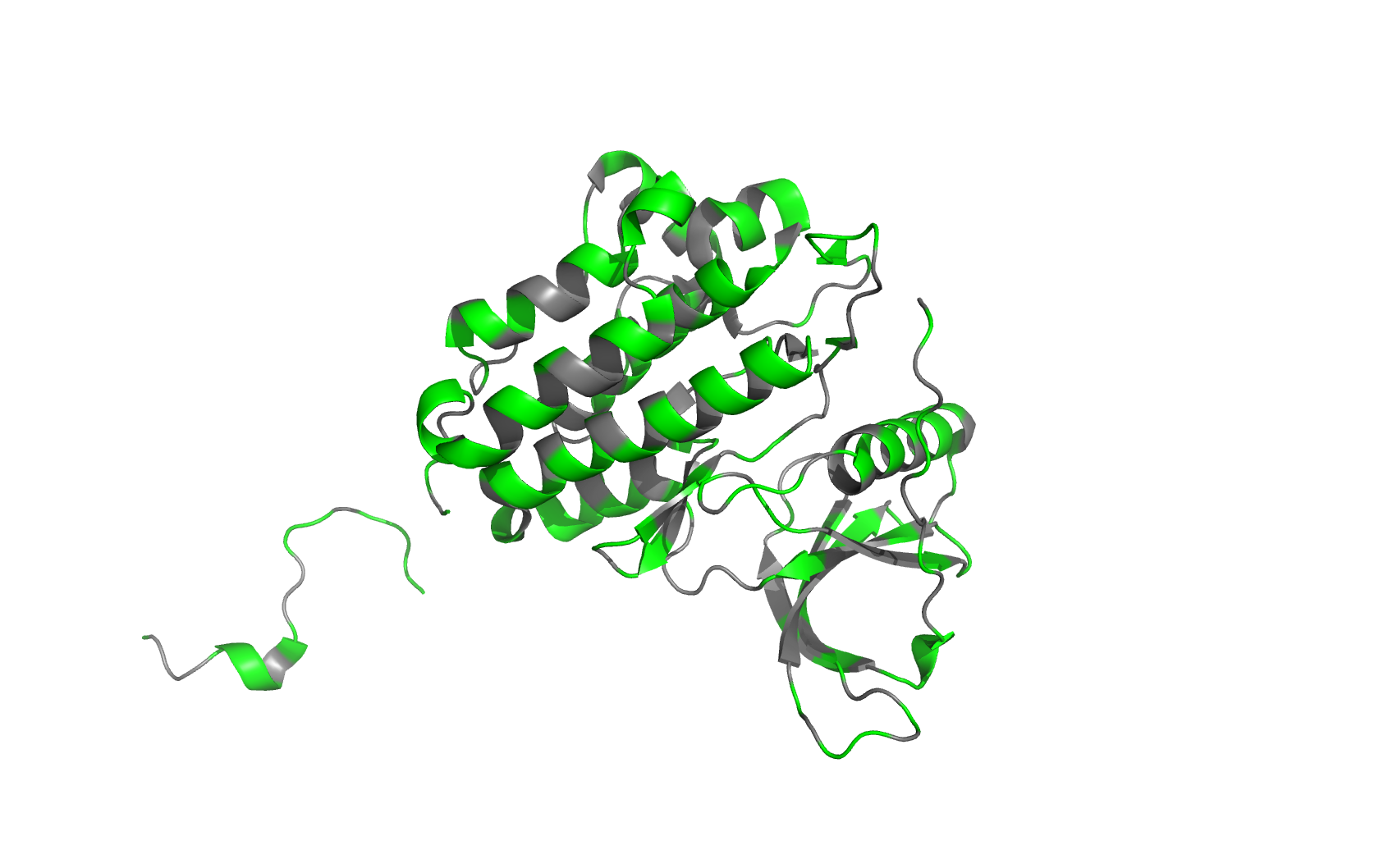
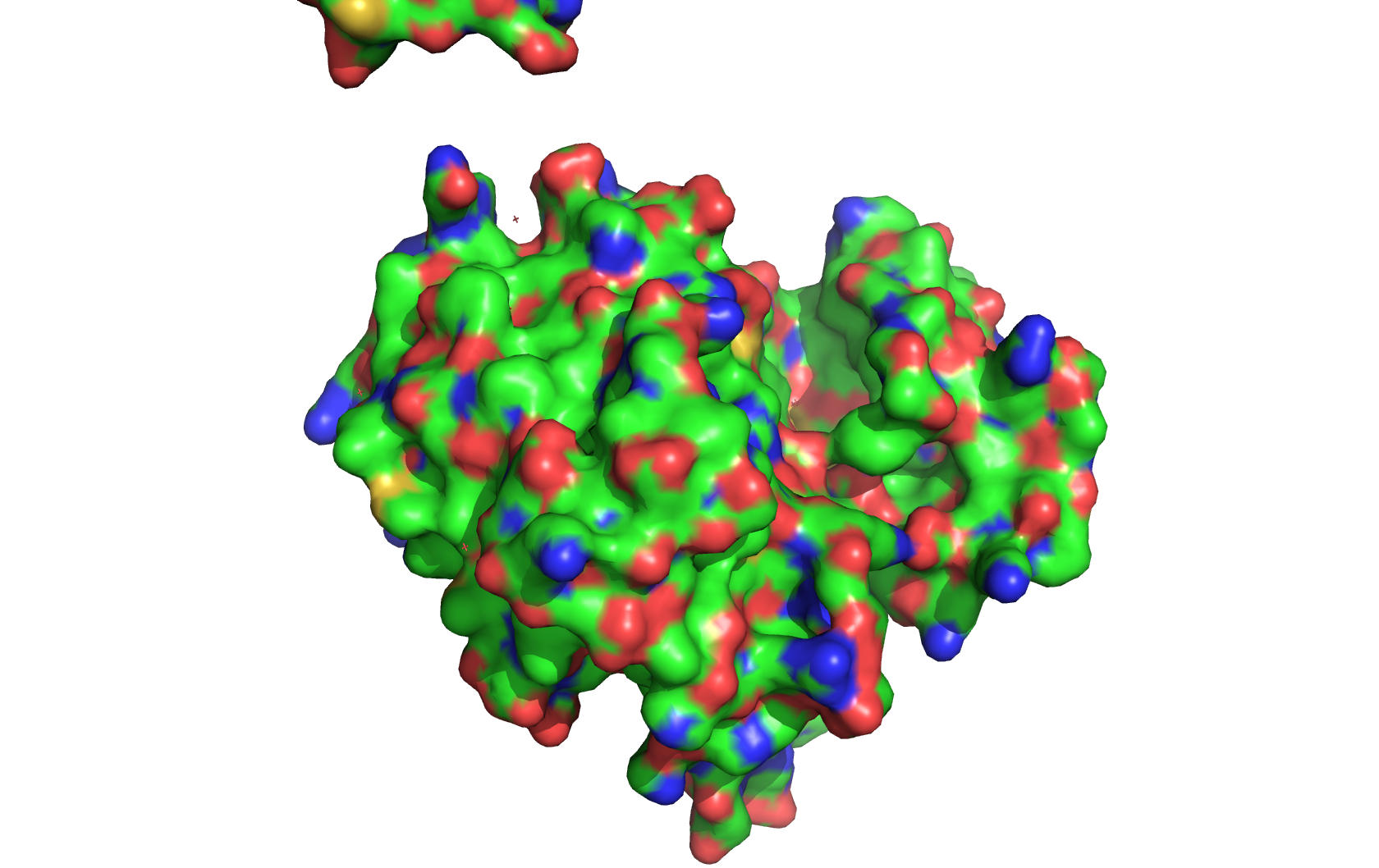
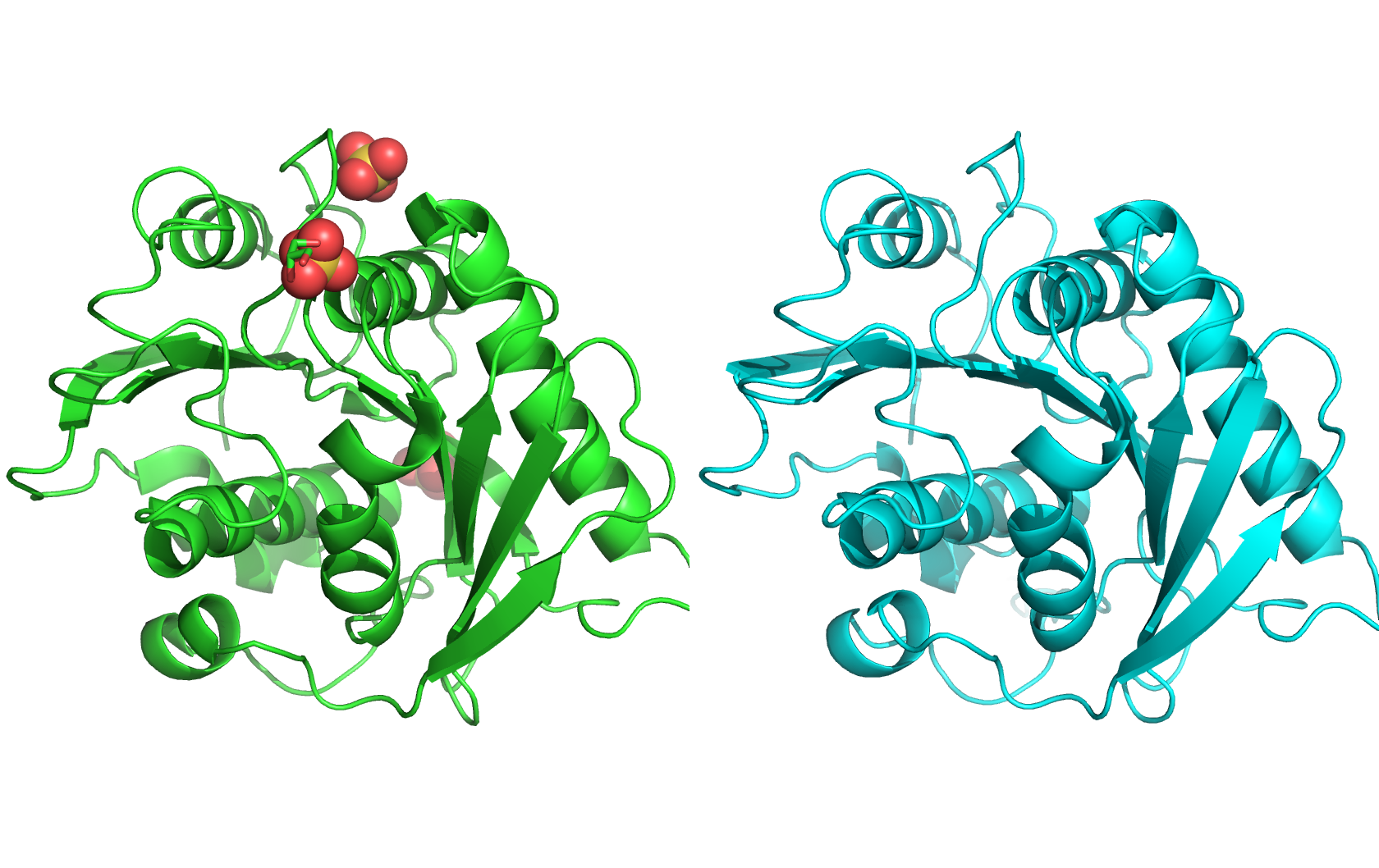
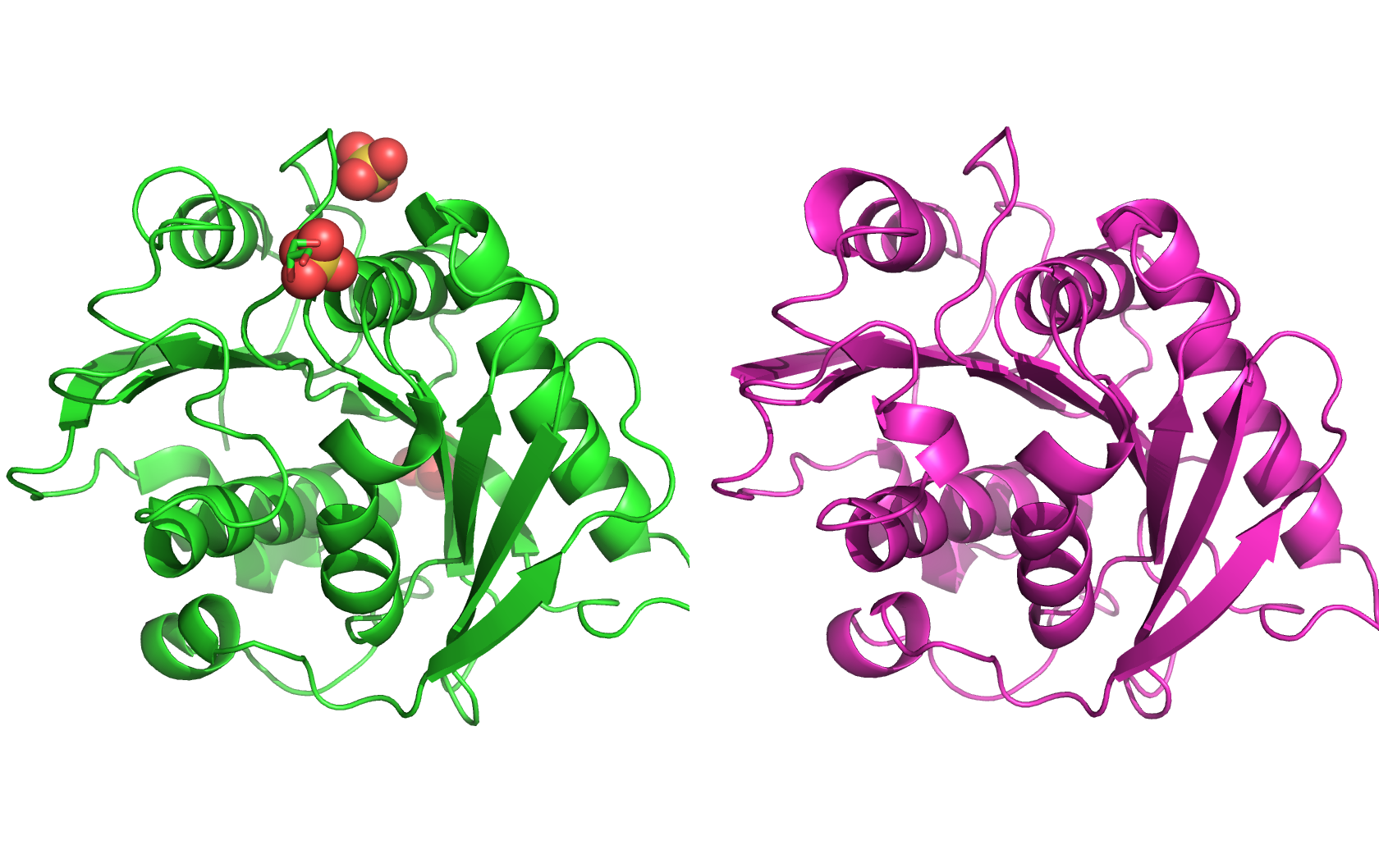
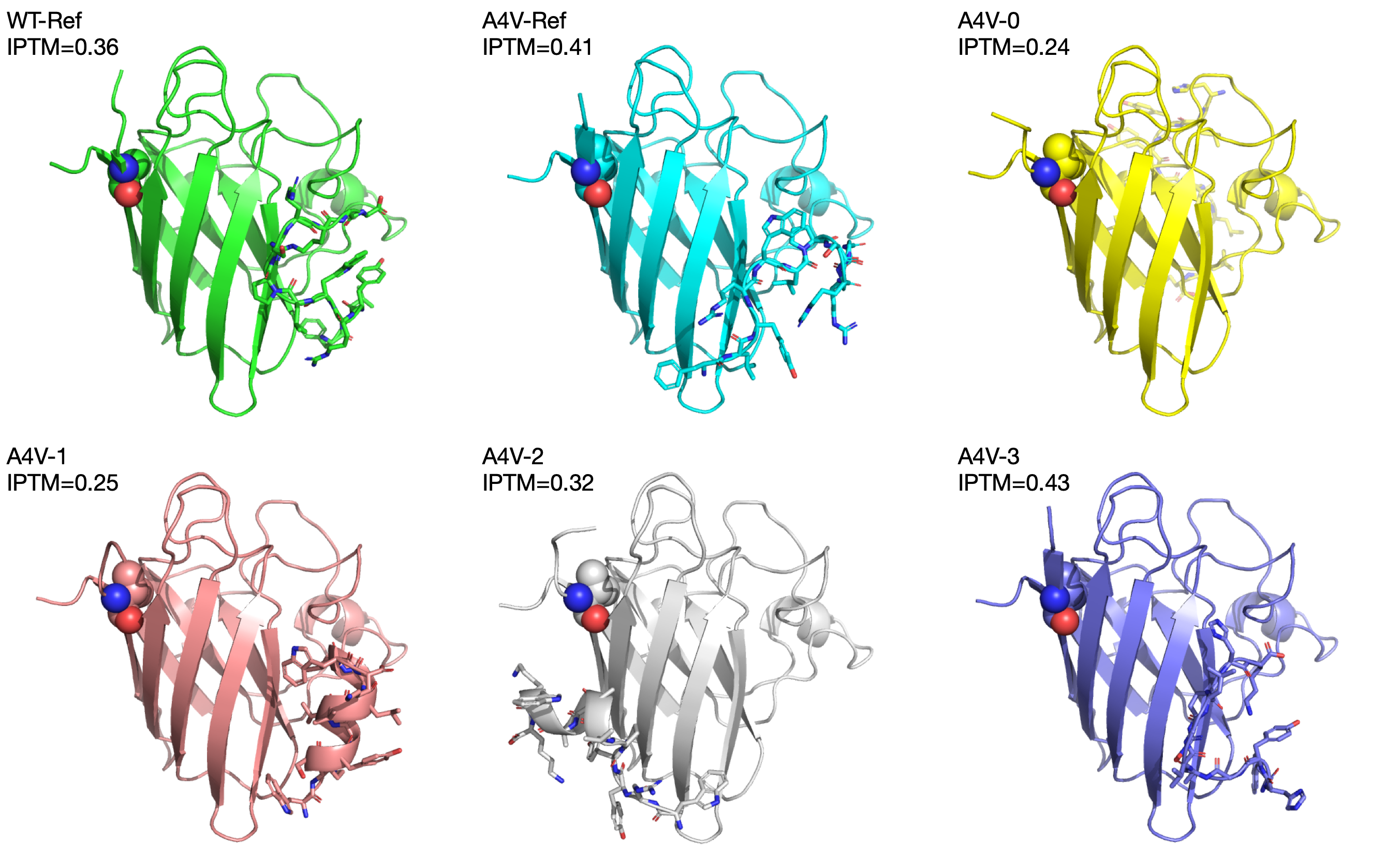
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM The original sequence of SOD1 is: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ Mutate the 4th amino acid A to V (A4V): MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence:
Week 06 HW: Genetic Circuits Part I
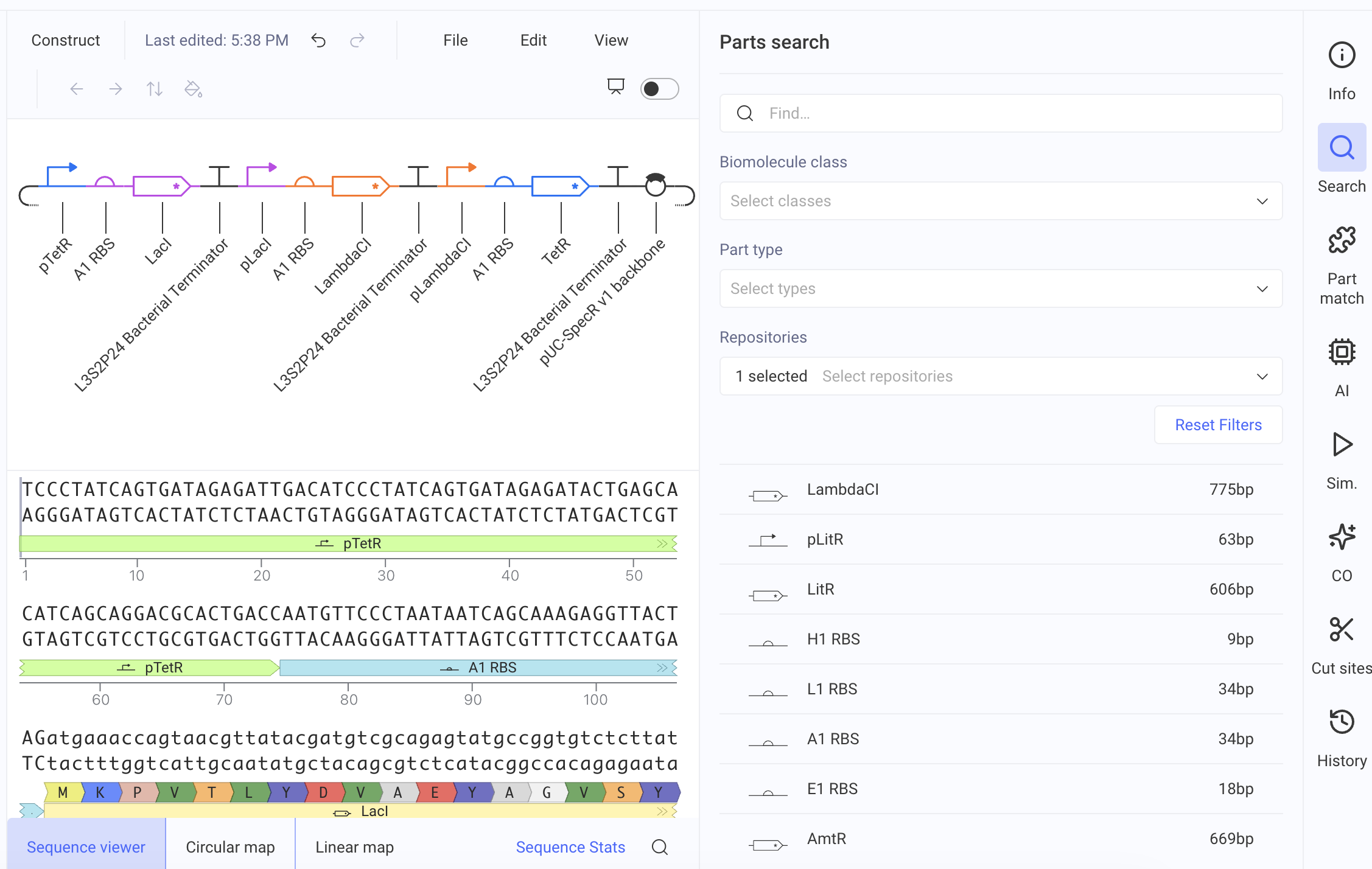
DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion DNA Polymerase: synthesizes new DNA strands by adding nucleotides to the template strand during PCR dNTPs (deoxynucleotides): building blocks of DNA Buffer: provides the optimal conditions 2. What are some factors that determine primer annealing temperature during PCR? Tm (melting temperature) of the primer: the temperature at which half of the DNA duplex dissociates Primer length: longer primers generally require higher annealing temperatures GC content: higher GC content increases the Tm and may require higher annealing temperatures Salt concentration: higher salt concentrations can stabilize the DNA duplex and may require higher annealing temperatures 3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. PCR:
Week 07 HW: Genetic Circuits Part II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs can perform more complex computations than traditional genetic circuits, which are limited to Boolean functions. IANNs can process continuous inputs and produce continuous outputs, allowing for more nuanced control of gene expression. Additionally, IANNs can learn and adapt over time, making them more versatile and capable of handling dynamic environments.
General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Advantages: cell-free systems allow for precise control over the reaction conditions, such as temperature, pH, and the concentration of substrates and cofactors. This can lead to higher yields and faster protein production compared to in vivo methods.
Week 10 HW: Imaging and Measurement
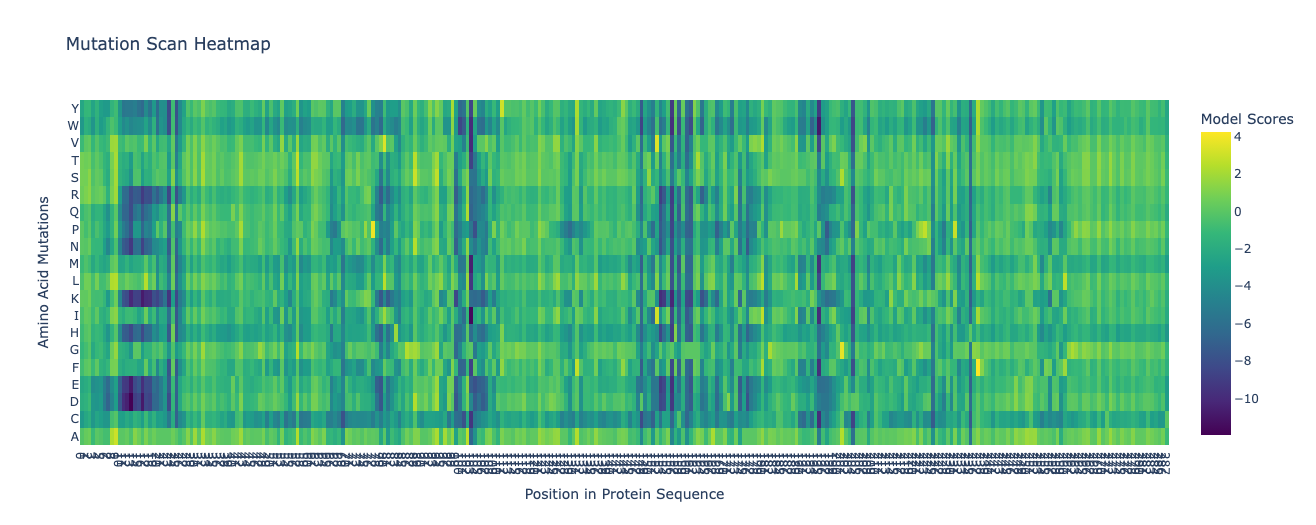
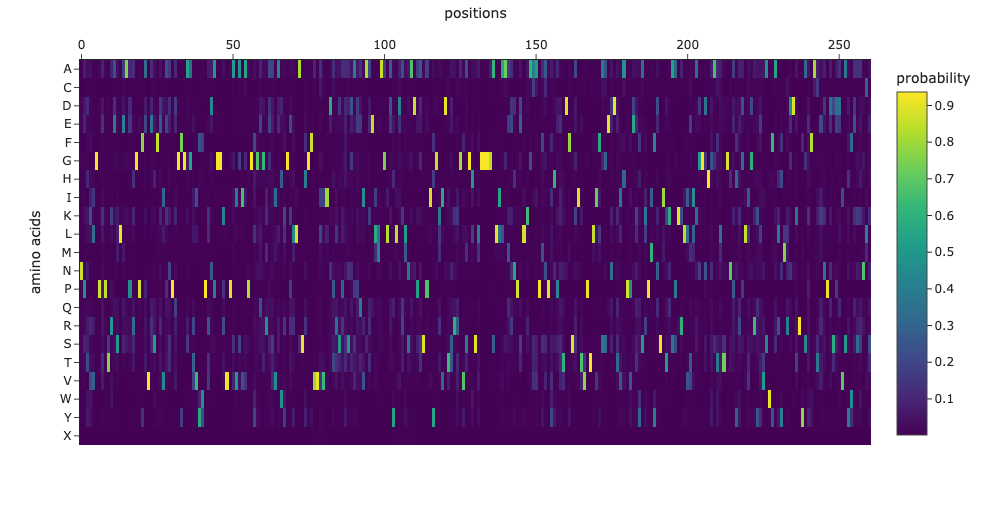
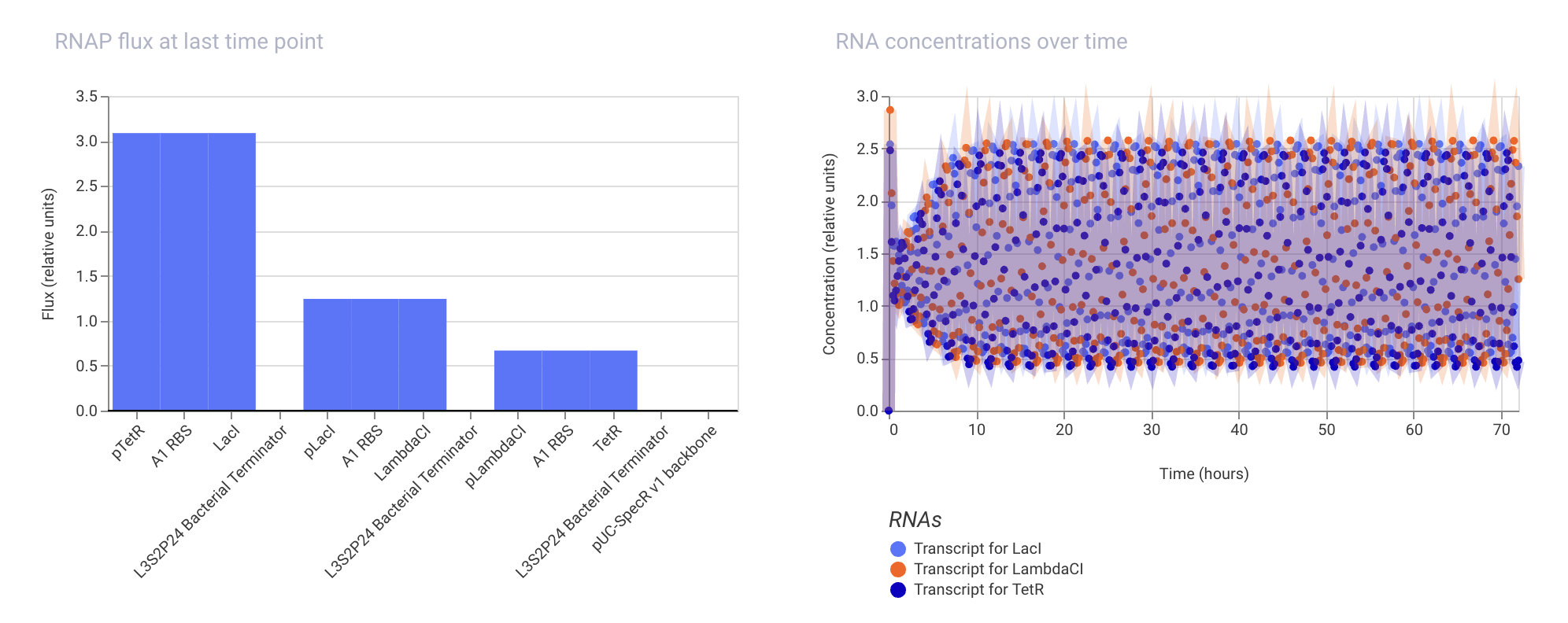
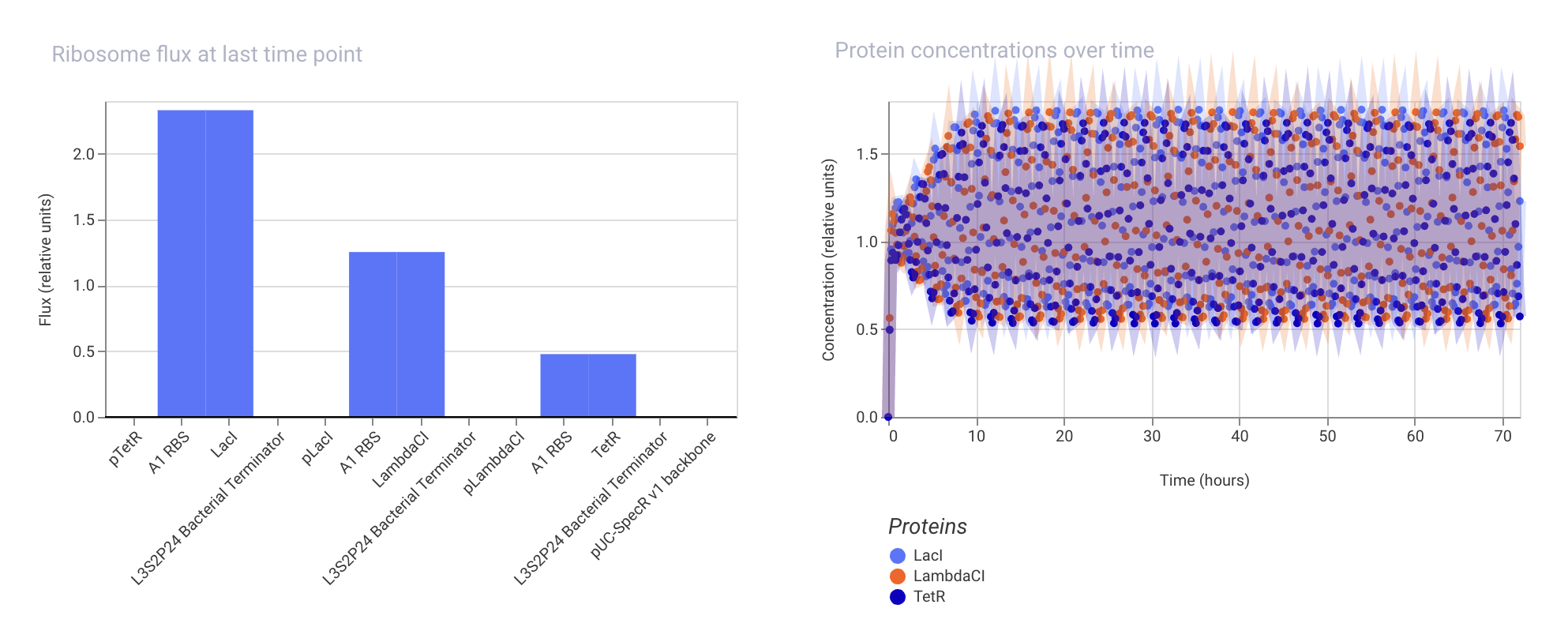
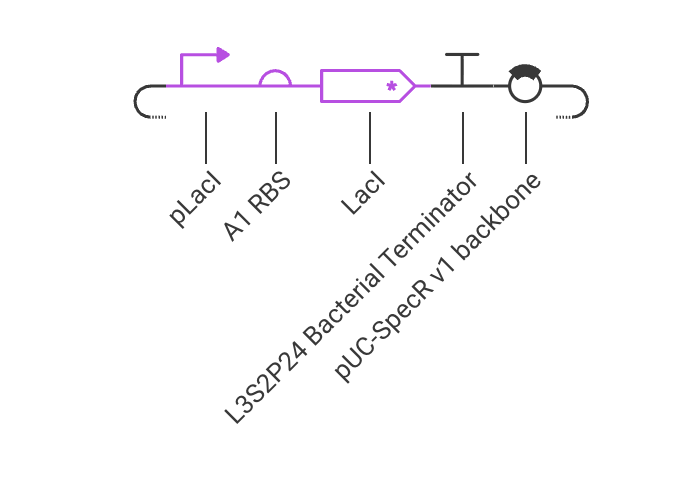
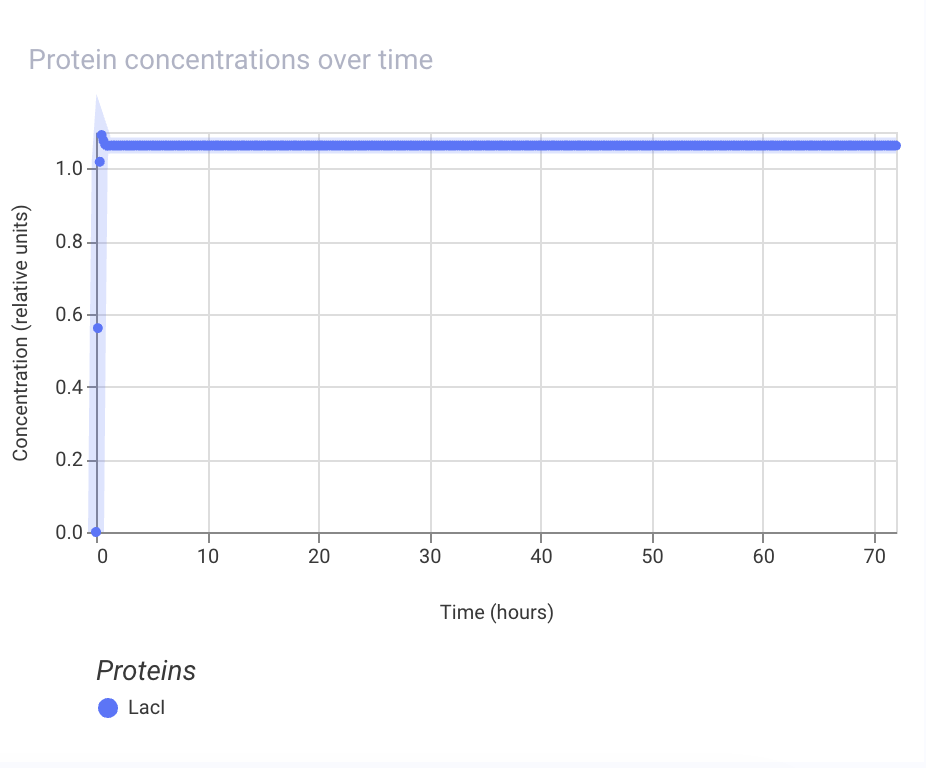
Final Project 1. Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. We will measure PETase thermostability (Tm), residual activity after heat challenge, expression yield, and PET-degradation rate for each variant.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork 1. Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. Done.
- Make a note on your HTGAA webpages including: what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”), what you liked about the project, and what about this collaborative art experiment could be made better for next year. I contributed a little dot on the bottom right plate, but it was overlapped by other contributions later. I liked the interactivity of the project. It’s cool to see how the plates evolve over time as more people contribute.
No homework assignment for this week. Working on the final project.
Week 13 HW: Bio-Design & Living Materials
No homework assignment for this week. Working on the final project.
No homework assignment for this week. Working on the final project.