Week 04 HW: Protein Design Part I
Part A: Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
1 Dalton = 1.66 x 10-24 grams. 100 Daltons = 1.66 x 10-22 grams. 1 gram = 6.02 x 1023 molecules. 20% of meat is protein, so 100 grams of proteins in 500 grams of meat. Therefore: 100 x 6.02 x 1023 = 6.02 x 1025 molecules of amino acids.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We digest proteins into amino acids rather than absorbing proteins into our genomes.
3. Why are there only 20 natural amino acids?
20 natural amino acids are sufficient to create chemical diversity and efficiency.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes. For example, we can replace the sulfur atom in cysteine with selenium to create selenocysteine.
5. Where did amino acids come from before enzymes that make them, and before life started?
Chemical reactions in the environment.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed.
7. Can you discover additional helices in proteins?
Yes. For example, the pi-helix and the 3-10-helix.
8. Why are most molecular helices right-handed?
Most amino acids in life are L-amino acids => right-handed helices.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Because of the parallel and anti-parallel hydrogen bonding between the strands. The driving force is the formation of hydrogen bonds.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I chose EGFR (Epidermal Growth Factor Receptor), because it is a protein that plays a critical role in cell growth and division, and it is often mutated in various cancers.
2. Identify the amino acid sequence of your protein
BLAST results (BLAST 250 results found in UniProtKB): https://www.uniprot.org/blast/uniprotkb/ncbiblast-R20260301-211408-0195-52595182-p2m/overview
Family: Protein kinase superfamily. Tyr protein kinase family. EGF receptor subfamily.
3. Identify the structure page of your protein in RCSB
There are multiple structures of different EGFR domains in RCSB PDB. The earliest one is 1M14 deposited on 2002-06-17. Resolution is 2.60 Å, so it is a good quality structure.
There are water molecules in the structure, but no ligands or cofactors.
Structure classification family:
- Structural Class: Alpha and beta proteins (a+b)
- Fold: Protein kinase-like (PK-like)
- Superfamily: Protein kinase-like (PK-like)
- Family: Protein kinases catalytic domain-like
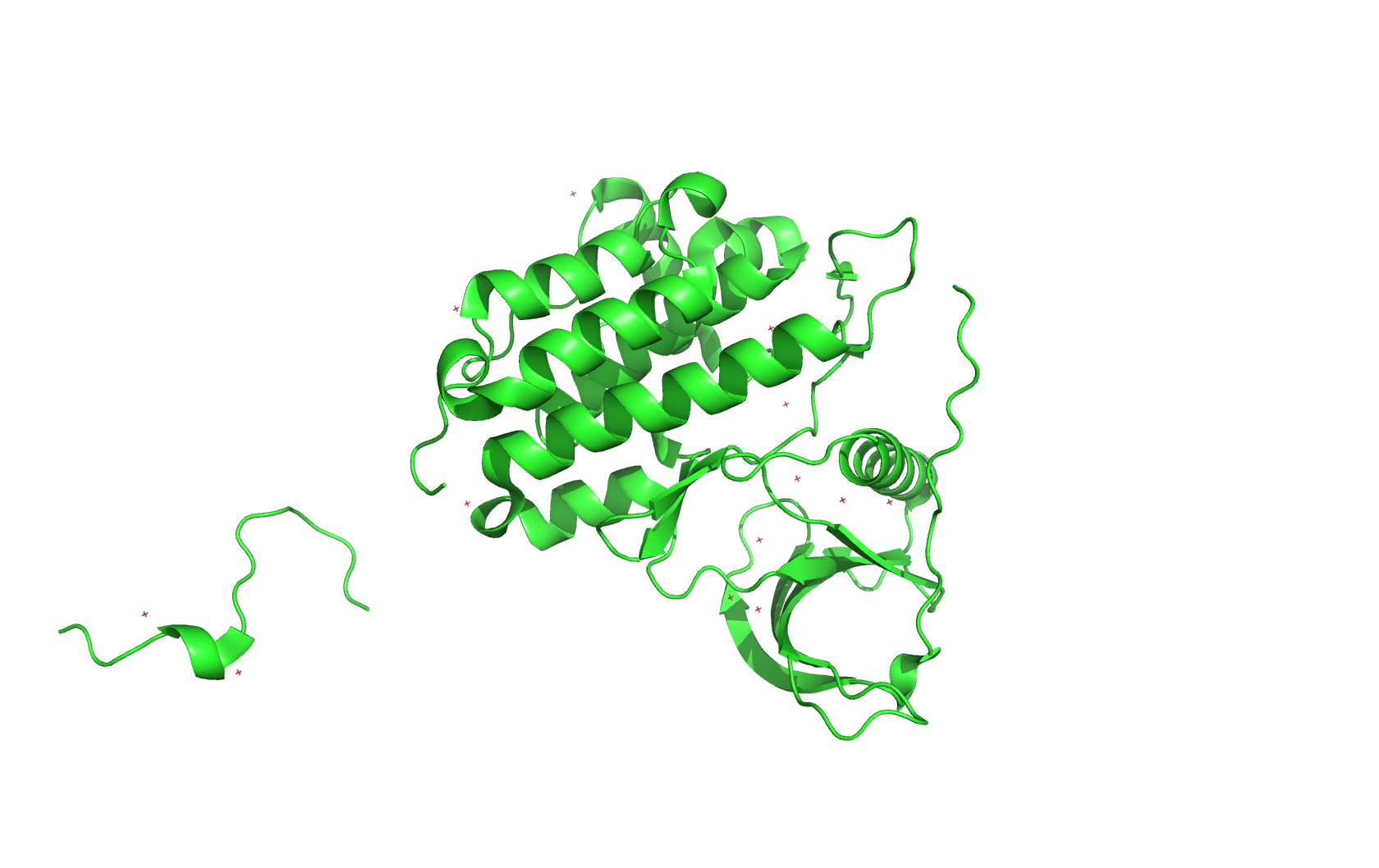
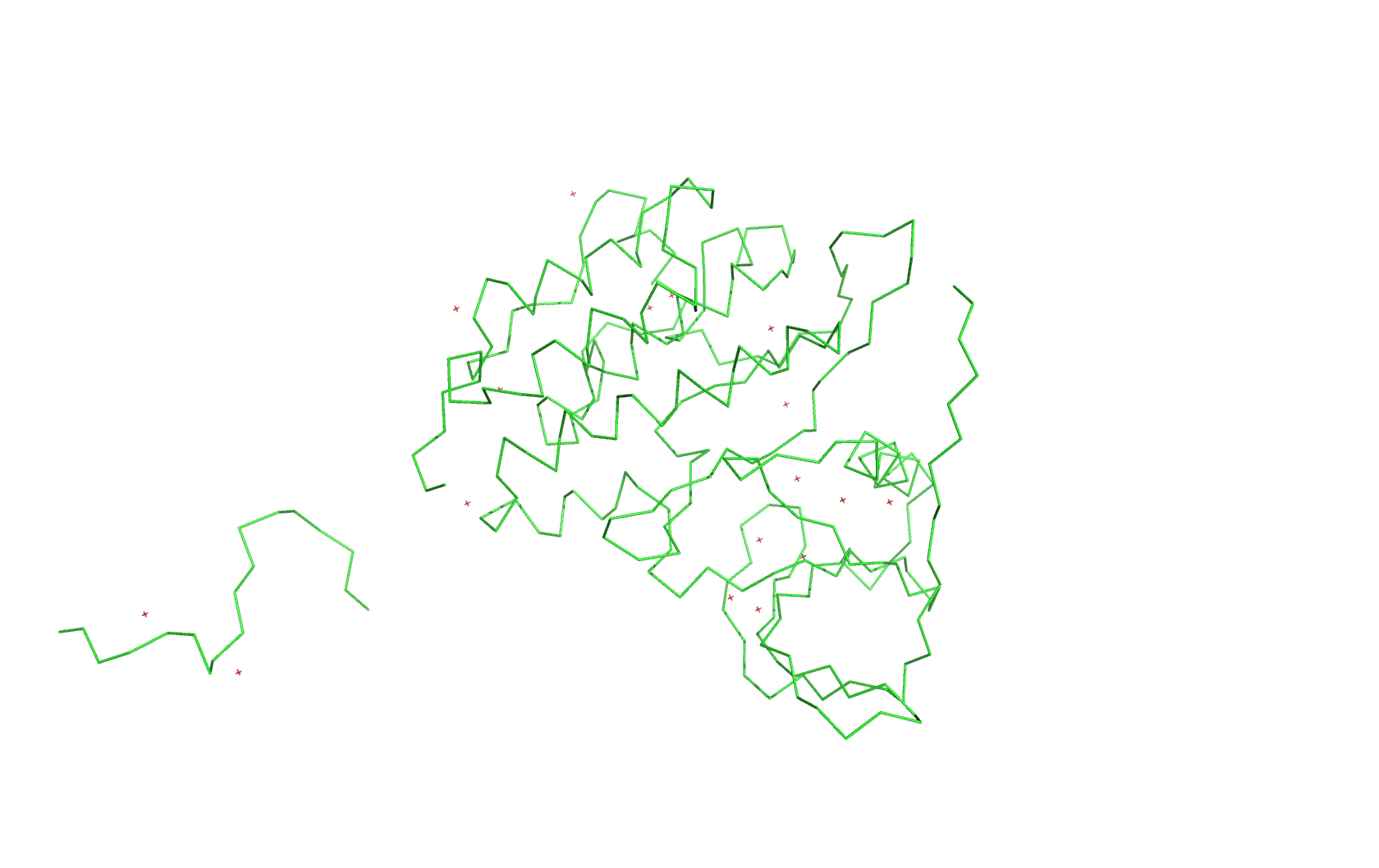
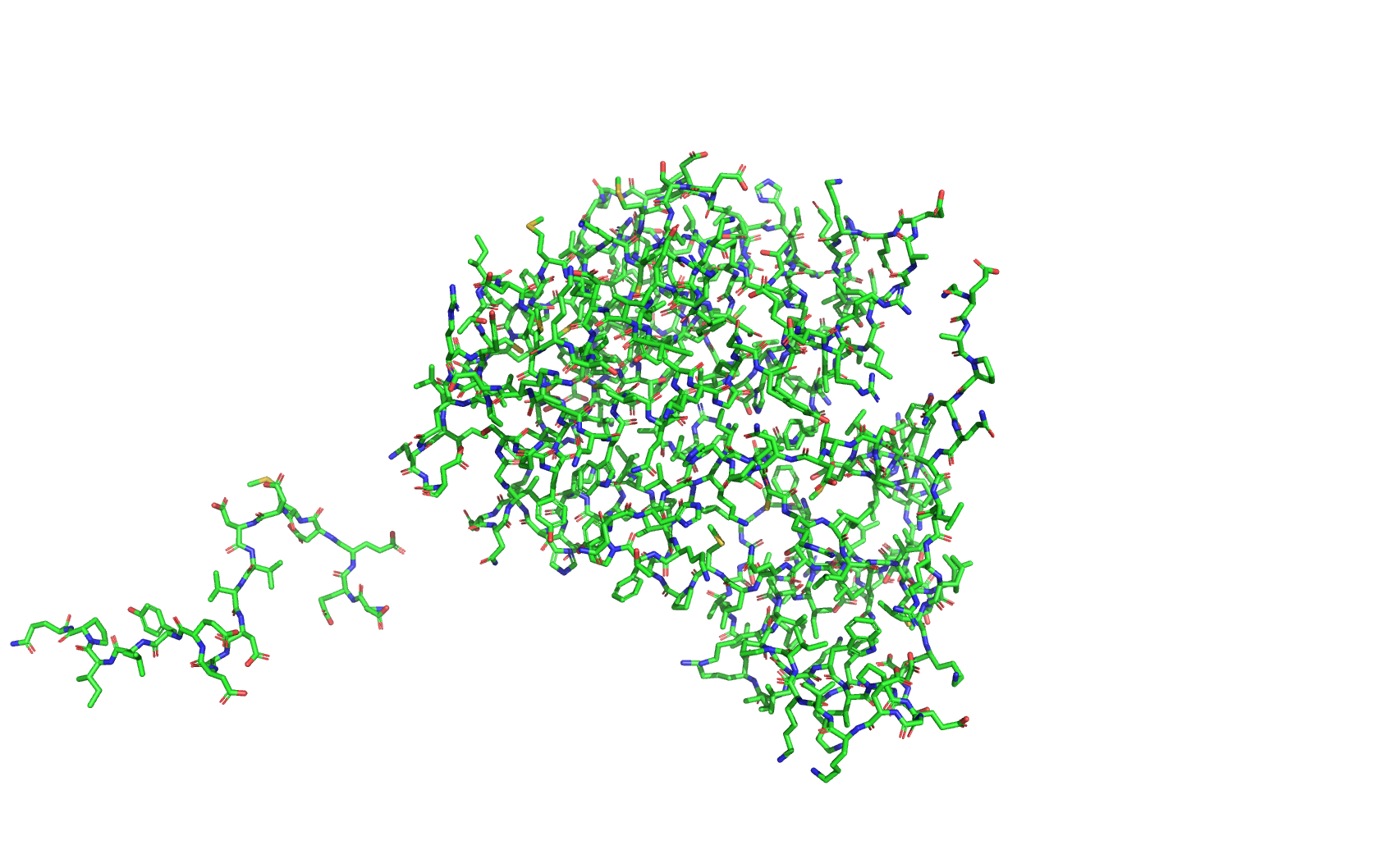
4. Open the structure of your protein in any 3D molecule visualization software:
Cartoon:
Ribbon:
Ball and stick:
Secondary structure (6 alpha helices and 6 beta strands):
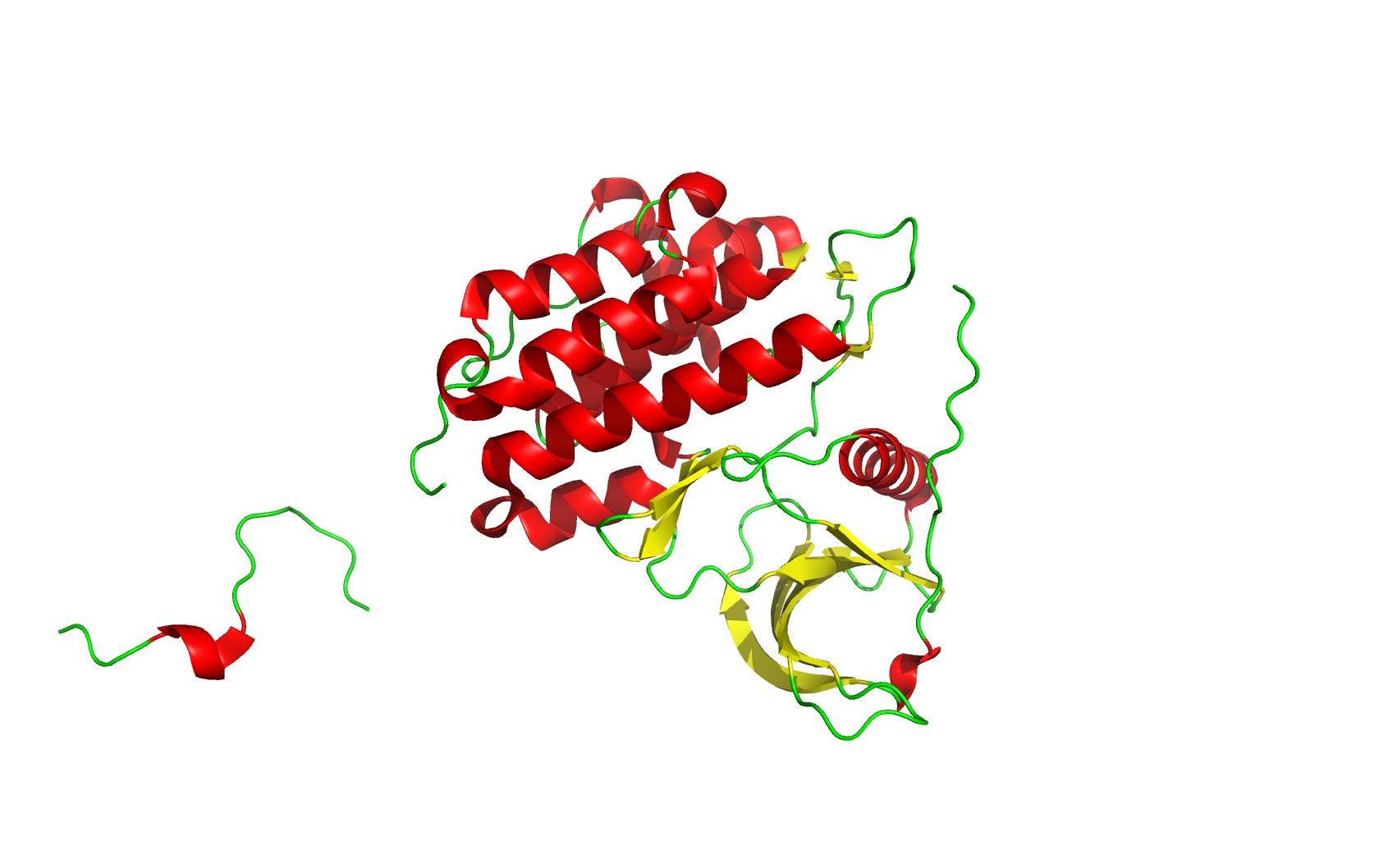
Residue type (green: hydrophilic, gray: hydrophobic. Hydrophilic residues are more on the surface, while hydrophobic residues are more buried inside):
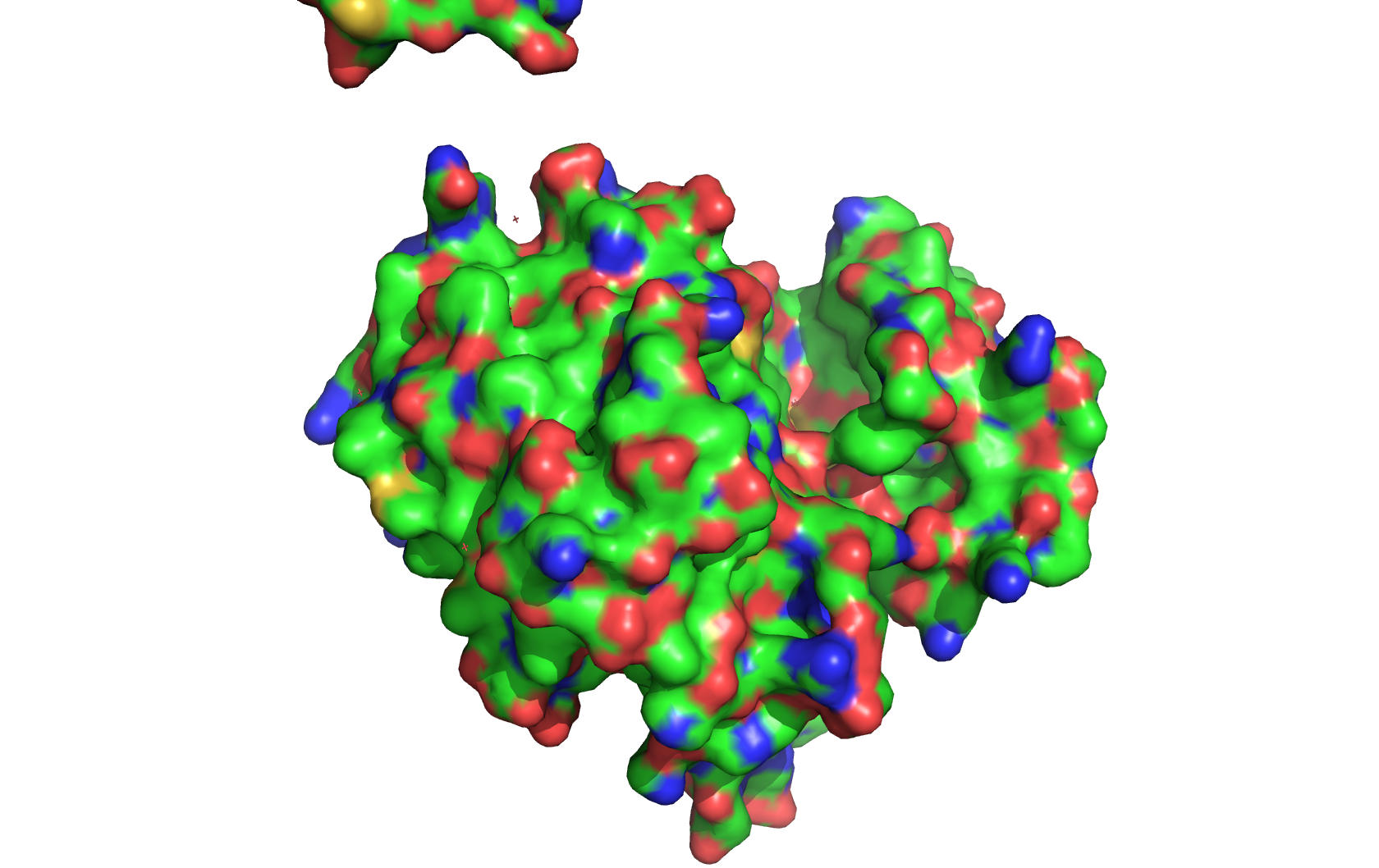
Surface (it has obvious pockets on the surface, as it is a receptor):
Part C. Using ML-Based Protein Design Tools
In this part, I will use a different protein sequence: Poly(ethylene terephthalate) hydrolase (PETase) (https://www.uniprot.org/uniprotkb/A0A0K8P6T7/entry), which is an enzyme that can degrade PET plastics. The PDB access code is 5XFY.
C1. Protein Language Modeling
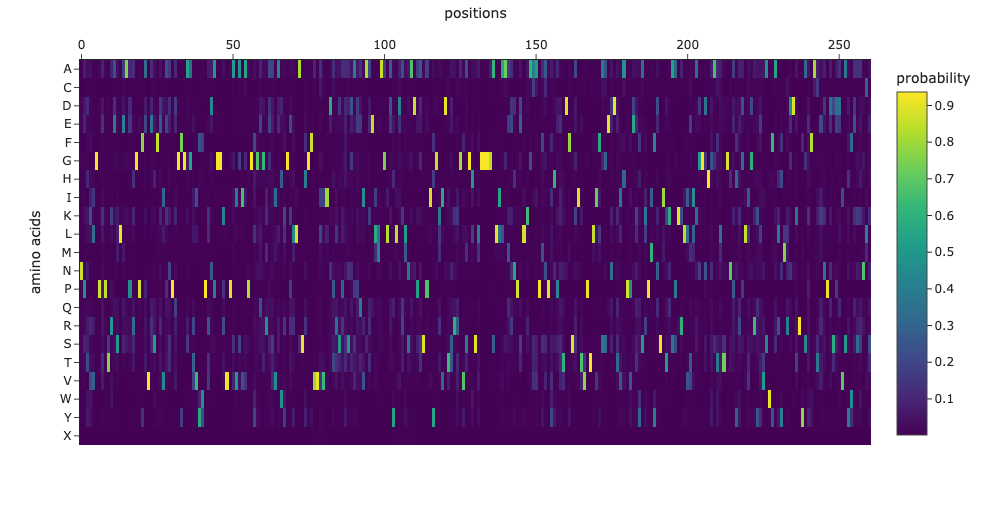
1. Deep Mutational Scans
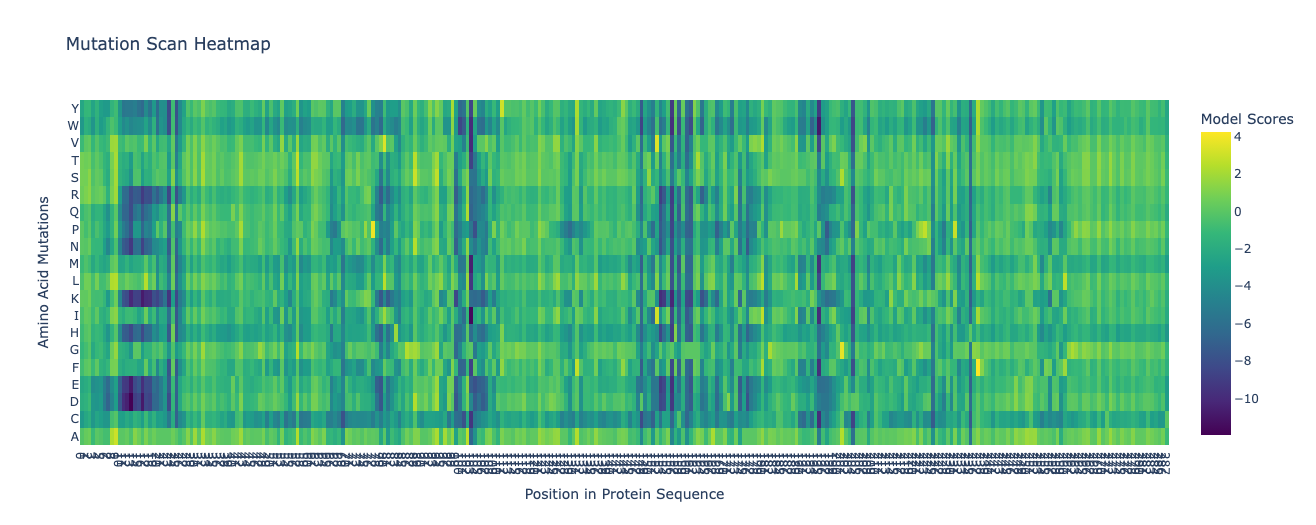
Columns with more dark cells (the wild-type amino acid is strongly preferred) indicate more conserved residues, which are likely important for the structure and function of the protein.
2. Latent Space Analysis
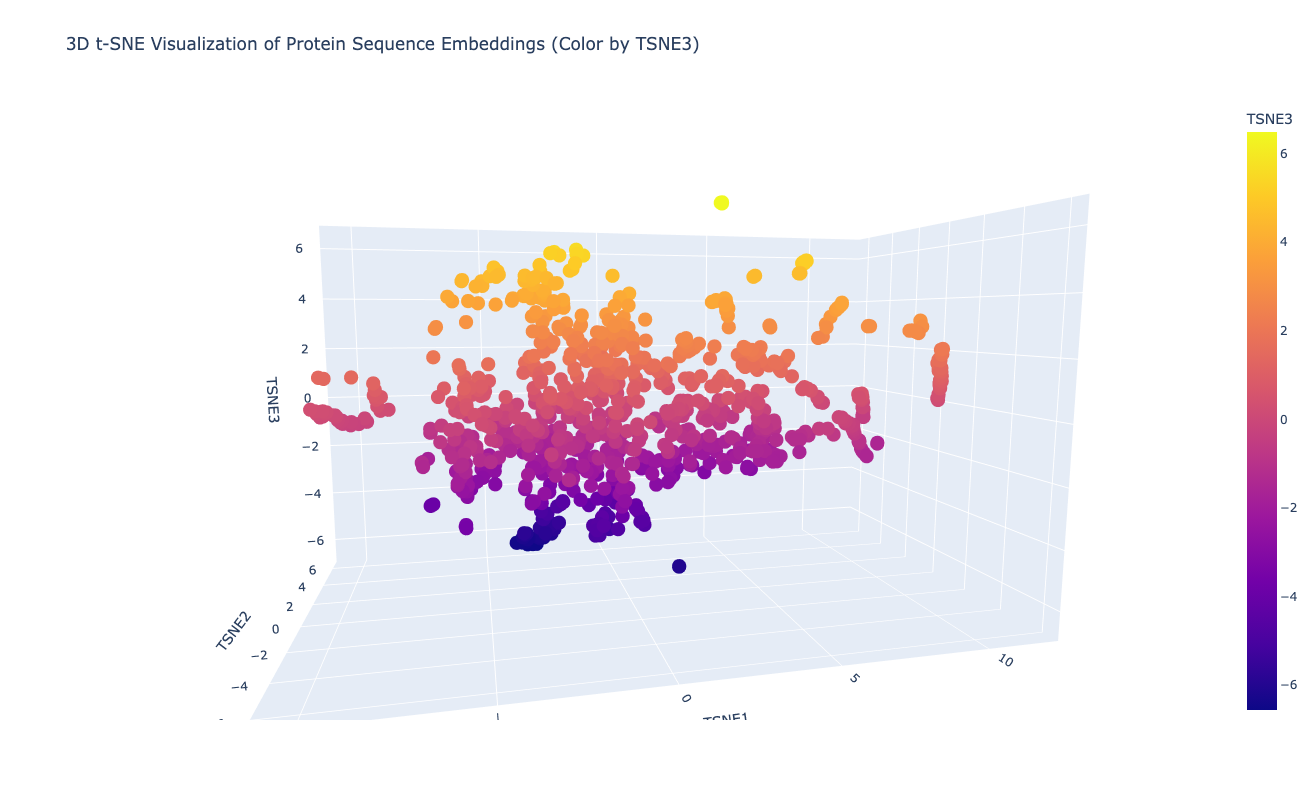
Neighboring proteins usually share the same SCOPe structural class and superfamily number, which indicates that the latent space captures structural and functional similarities between proteins.
When placing my PETase sequence embedding into the dimensionality reduction plot, many of its neighbors belong to c.69: alpha/beta-Hydrolases.
C2. Protein Folding
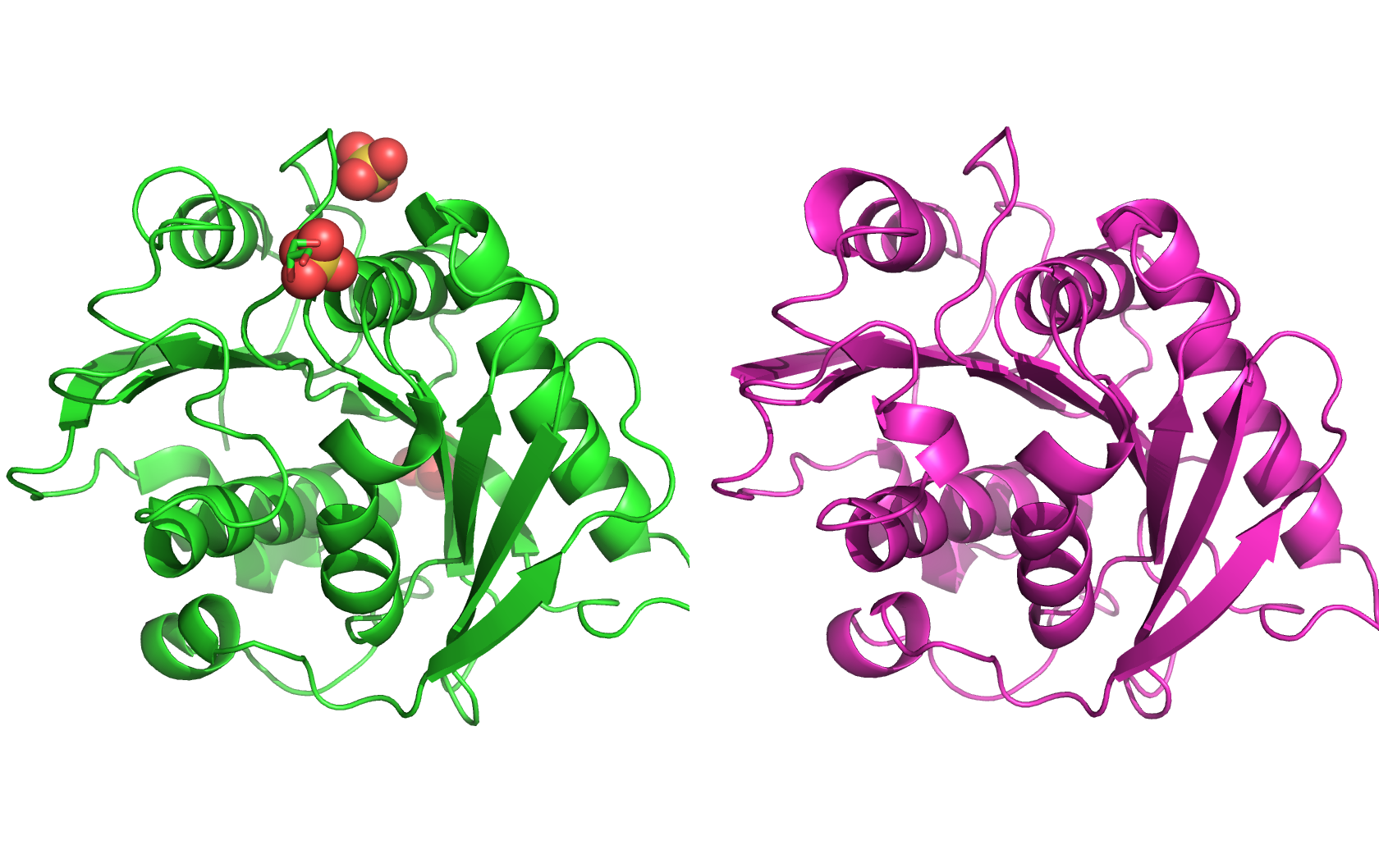
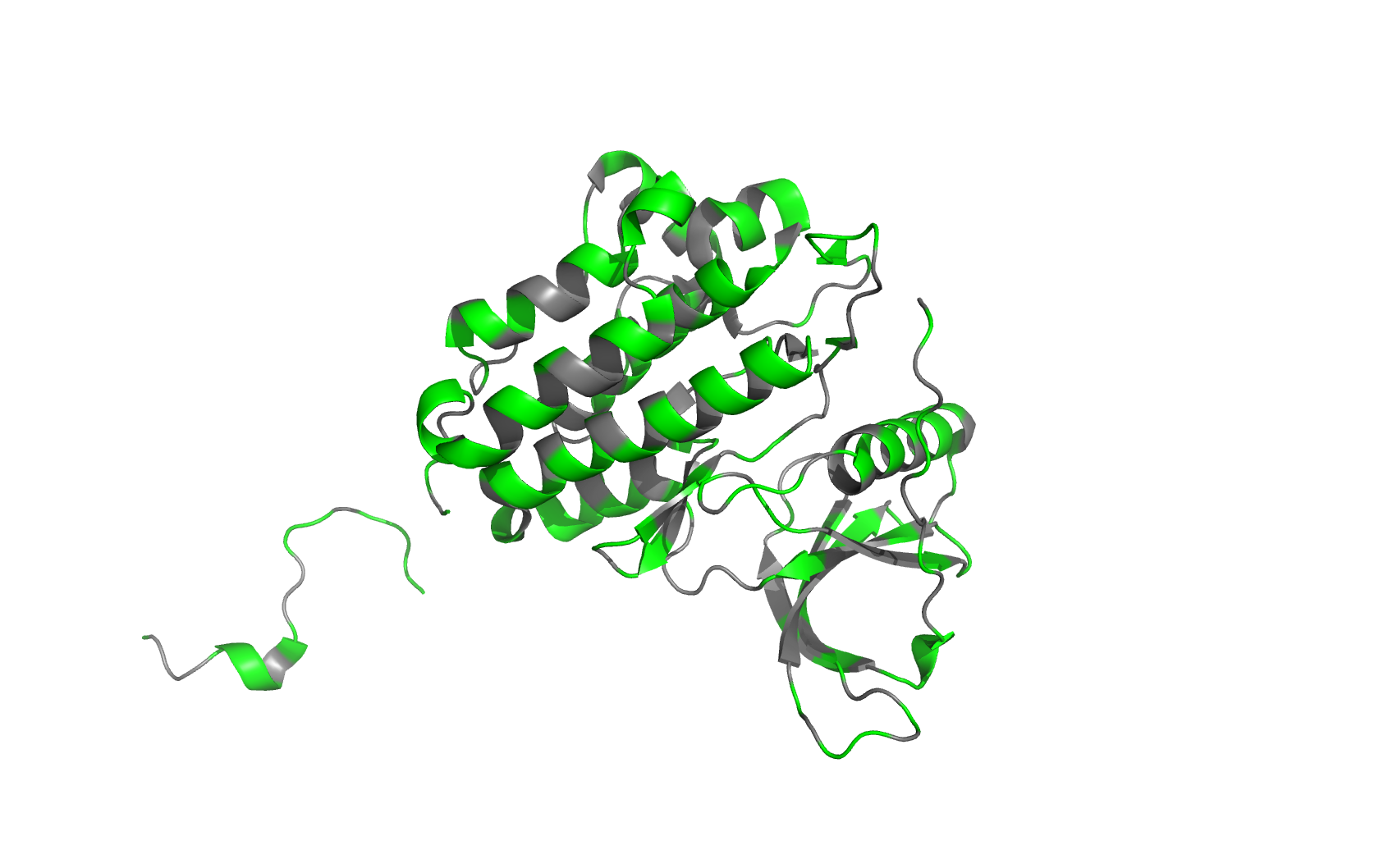
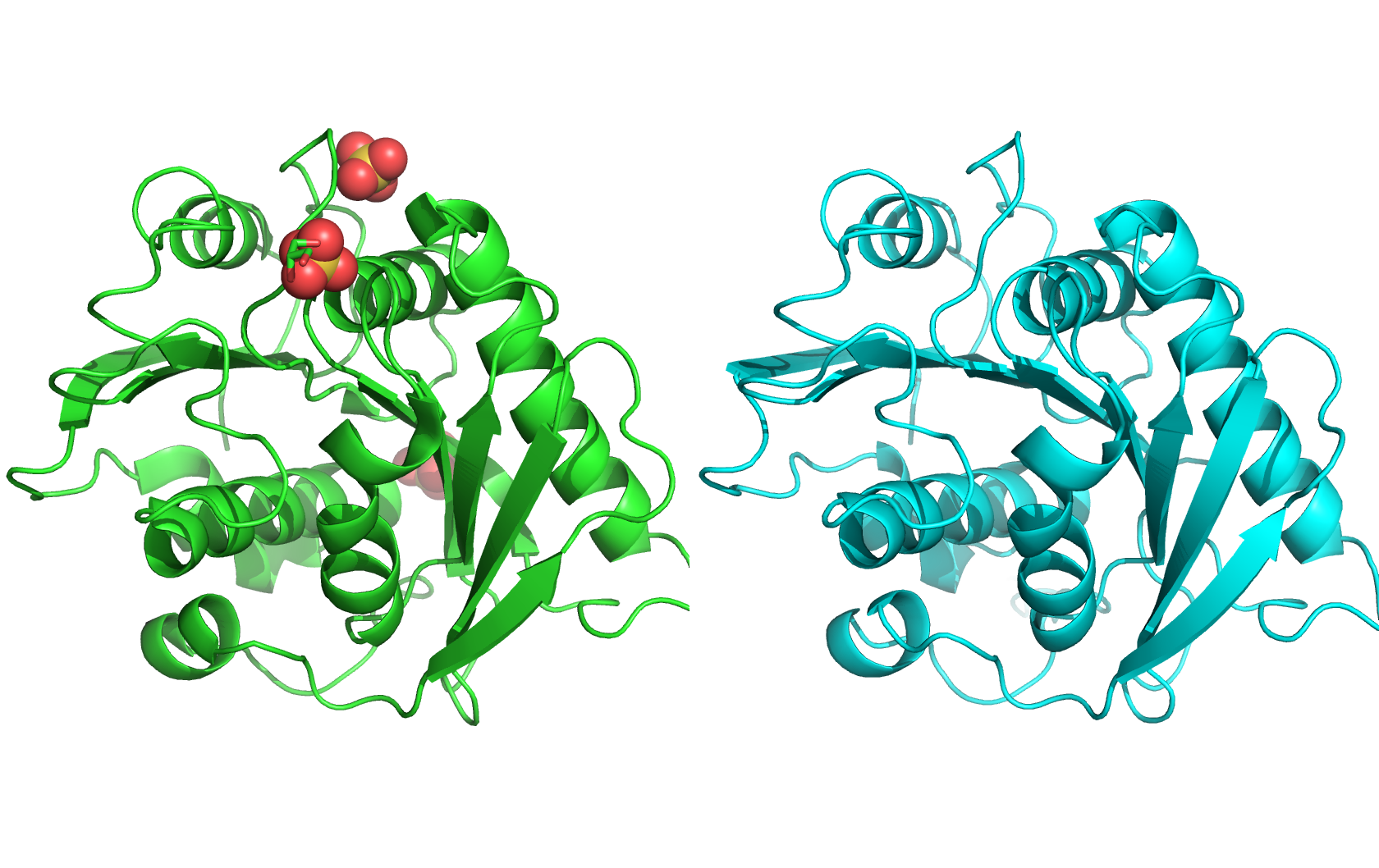
The protein on the left is the experimental structure of PETase (PDB: 5XFY), and the protein on the right is the predicted structure by ESMFold. The two structures are very similar, with a predicted TM score of 0.913. The RMSD reported by PyMOL is 0.540Å.
Changing the sequence with mutations does not have impact on the predicted structure. However, changing large fragments sometimes blows up the predicted structure.
C3. Protein Generation
I used ProteinMPNN to design a sequence based on the structure of the PETase I chose (PDB: 5XFY). The designed sequence is:
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
For about half of the sequence, the highest probability amino acid is the same as the original sequence. For the other half, the predicted amino acid is different from the original one, but they often have similar properties (e.g., both are hydrophobic or both are polar).
2. Input this sequence into ESMFold and compare the predicted structure to your original.
The protein on the left is the experimental structure of PETase (PDB: 5XFY), and the protein on the right is the predicted structure by ESMFold based on the ProteinMPNN-designed sequence. The predicted structure of the designed sequence is very similar to the original structure (0.756Å), and ESMFold is very confident about the prediction (predicted TM score = 0.944).