Week 05 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
The original sequence of SOD1 is:
Mutate the 4th amino acid A to V (A4V):
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence:
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | HLYYAVALELKX | 13.299815648347872 |
| 1 | WRSYAVVLELWK | 17.97100111129112 |
| 2 | WRYYPVAAAWKK | 11.081842724779028 |
| 3 | WHYGAVGLRHKX | 13.983770011694478 |
The perplexity of the reference SOD-1 binding sequence FLYRWLPSRRGG is 20.63523127283615:
Part 2: Evaluate Binders with AlphaFold3
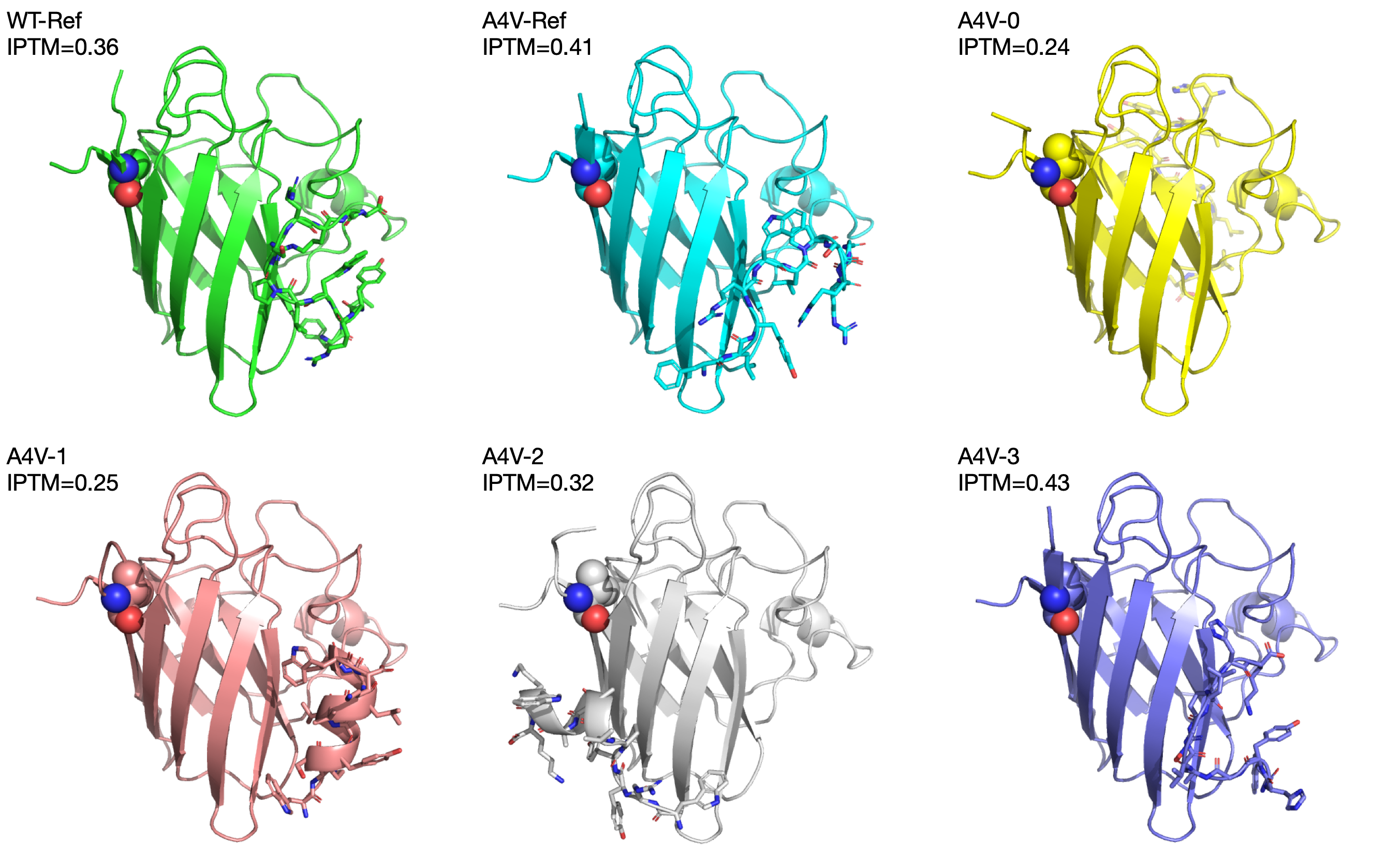
The IPTM scores for the reference peptide against the wild-type and mutant SOD1 are both pretty low (0.36 and 0.41 respectively), indicating that AlphaFold is not very confident about the predicted binding structure. The first three generated peptides have IPTM scores of 0.24, 0.25, and 0.32, which are lower than the reference. The last generated peptide has an IPTM score of 0.43, which is higher than the reference.
Only the third (A4V-2) generated peptide binds to the dimerization interface of SOD1.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Predicted properties of the generated peptides:
| Sequence | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| HLYYAVALELK | 💧 Solubility | Soluble | 1.000 | Probability |
| HLYYAVALELK | 🩸 Hemolysis | Non-hemolytic | 0.081 | Probability |
| HLYYAVALELK | 🔗 Binding Affinity | Weak binding | 6.052 | pKd/pKi |
| HLYYAVALELK | 📏 Length | 11 | aa | |
| HLYYAVALELK | ⚖️ Molecular Weight | 1319.5 | Da | |
| HLYYAVALELK | ⚡ Net Charge (pH 7) | -0.15 | ||
| HLYYAVALELK | 🎯 Isoelectric Point | 6.75 | pH | |
| HLYYAVALELK | 💦 Hydrophobicity (GRAVY) | 0.55 | GRAVY | |
| WRSYAVVLELWK | 💧 Solubility | Soluble | 1.000 | Probability |
| WRSYAVVLELWK | 🩸 Hemolysis | Non-hemolytic | 0.130 | Probability |
| WRSYAVVLELWK | 🔗 Binding Affinity | Weak binding | 6.818 | pKd/pKi |
| WRSYAVVLELWK | 📏 Length | 12 | aa | |
| WRSYAVVLELWK | ⚖️ Molecular Weight | 1549.8 | Da | |
| WRSYAVVLELWK | ⚡ Net Charge (pH 7) | 0.76 | ||
| WRSYAVVLELWK | 🎯 Isoelectric Point | 8.59 | pH | |
| WRSYAVVLELWK | 💦 Hydrophobicity (GRAVY) | 0.17 | GRAVY | |
| WRYYPVAAAWKK | 💧 Solubility | Soluble | 1.000 | Probability |
| WRYYPVAAAWKK | 🩸 Hemolysis | Non-hemolytic | 0.021 | Probability |
| WRYYPVAAAWKK | 🔗 Binding Affinity | Weak binding | 6.124 | pKd/pKi |
| WRYYPVAAAWKK | 📏 Length | 12 | aa | |
| WRYYPVAAAWKK | ⚖️ Molecular Weight | 1538.8 | Da | |
| WRYYPVAAAWKK | ⚡ Net Charge (pH 7) | 2.76 | ||
| WRYYPVAAAWKK | 🎯 Isoelectric Point | 10.00 | pH | |
| WRYYPVAAAWKK | 💦 Hydrophobicity (GRAVY) | -0.72 | GRAVY | |
| WHYGAVGLRHK | 💧 Solubility | Soluble | 1.000 | Probability |
| WHYGAVGLRHK | 🩸 Hemolysis | Non-hemolytic | 0.023 | Probability |
| WHYGAVGLRHK | 🔗 Binding Affinity | Weak binding | 5.442 | pKd/pKi |
| WHYGAVGLRHK | 📏 Length | 11 | aa | |
| WHYGAVGLRHK | ⚖️ Molecular Weight | 1323.5 | Da | |
| WHYGAVGLRHK | ⚡ Net Charge (pH 7) | 1.93 | ||
| WHYGAVGLRHK | 🎯 Isoelectric Point | 9.99 | pH | |
| WHYGAVGLRHK | 💦 Hydrophobicity (GRAVY) | -0.73 | GRAVY |
Predicted properties of the reference peptide:
| Sequence | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| FLYRWLPSRRGG | 💧 Solubility | Soluble | 1.000 | Probability |
| FLYRWLPSRRGG | 🩸 Hemolysis | Non-hemolytic | 0.047 | Probability |
| FLYRWLPSRRGG | 🔗 Binding Affinity | Weak binding | 5.968 | pKd/pKi |
| FLYRWLPSRRGG | 📏 Length | 12 | aa | |
| FLYRWLPSRRGG | ⚖️ Molecular Weight | 1507.7 | Da | |
| FLYRWLPSRRGG | ⚡ Net Charge (pH 7) | 2.76 | ||
| FLYRWLPSRRGG | 🎯 Isoelectric Point | 11.71 | pH | |
| FLYRWLPSRRGG | 💦 Hydrophobicity (GRAVY) | -0.71 | GRAVY |
The peptide WHYGAVGLRHK has the highest ipTM score of 0.43, but it has a relatively low predicted binding affinity (5.442 pKd/pKi). The peptide WRSYAVVLELWK has a lower ipTM score of 0.25 but a higher predicted binding affinity (6.818 pKd/pKi). None of the generated peptides are predicted to be hemolytic or poorly soluble. The peptide WRSYAVVLELWK best balances predicted binding and therapeutic properties, as it has a reasonably high ipTM score and the highest predicted binding affinity among the generated peptides.
Part 4: Generate Optimized Peptides with moPPIt
Generated peptide sequence with predicted solubility score, affinity score, and hemolysis score:
The moPPIt-generated peptide DFRQSTTYQY has a higher predicted binding affinity score and solubility score compared to the PepMLM-generated peptides. Before advancing this peptide to clinical studies, I would evaluate its binding affinity experimentally in vitro, and further assess its stability, toxicity, and pharmacokinetic properties in cell and animal models.
Part C: L-Protein Mutants
I first used Boltz-2 to predict the complex structure of the wild-type L-protein and DnaJ protein:
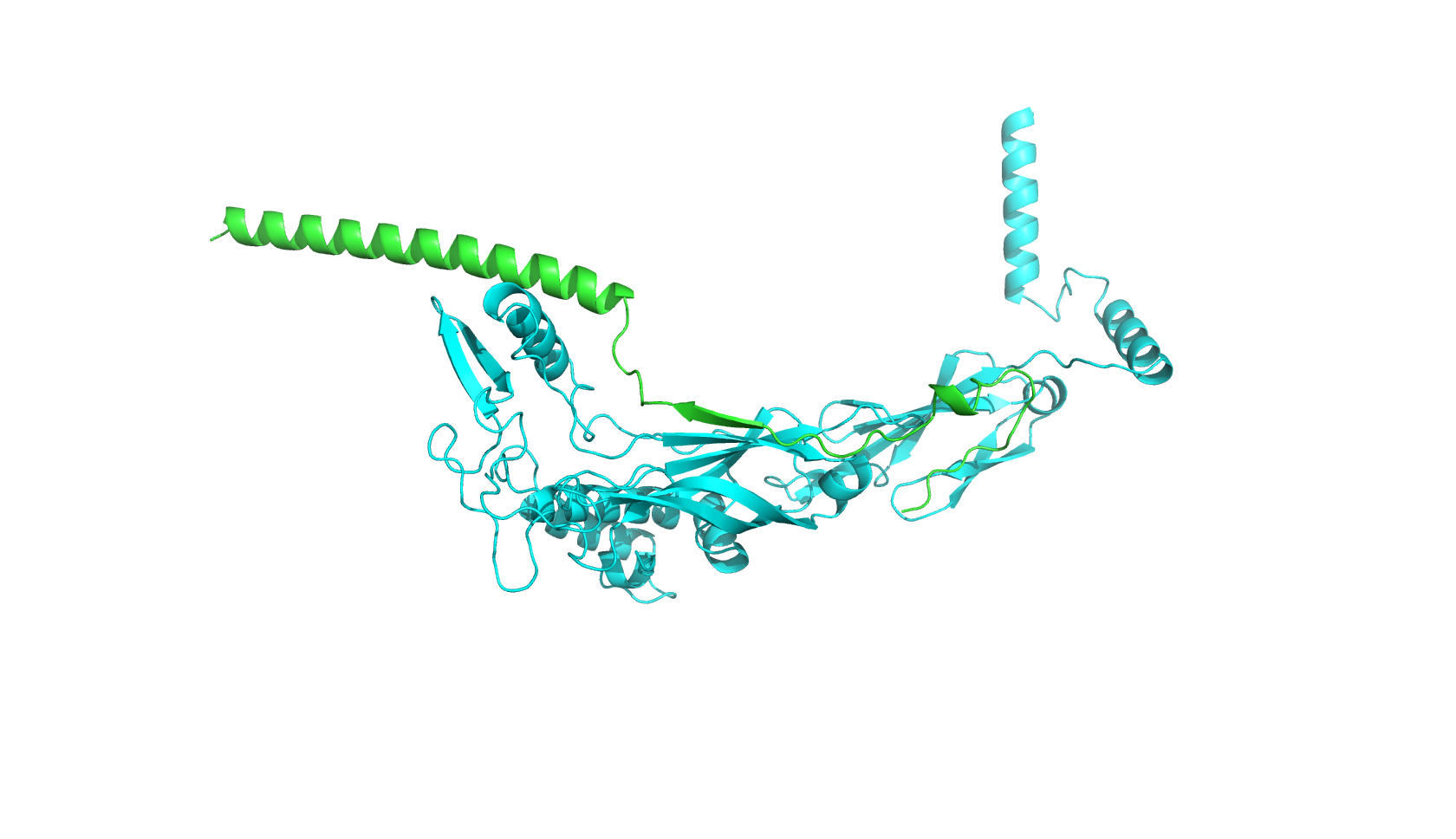
Next, I used FoldX, a force field-based protein design tool that can predict the effects of mutations on protein-protein interfaces. The goal is to identify mutations in the L-protein that are energetically favorable to stabilize the interaction with DnaJ.
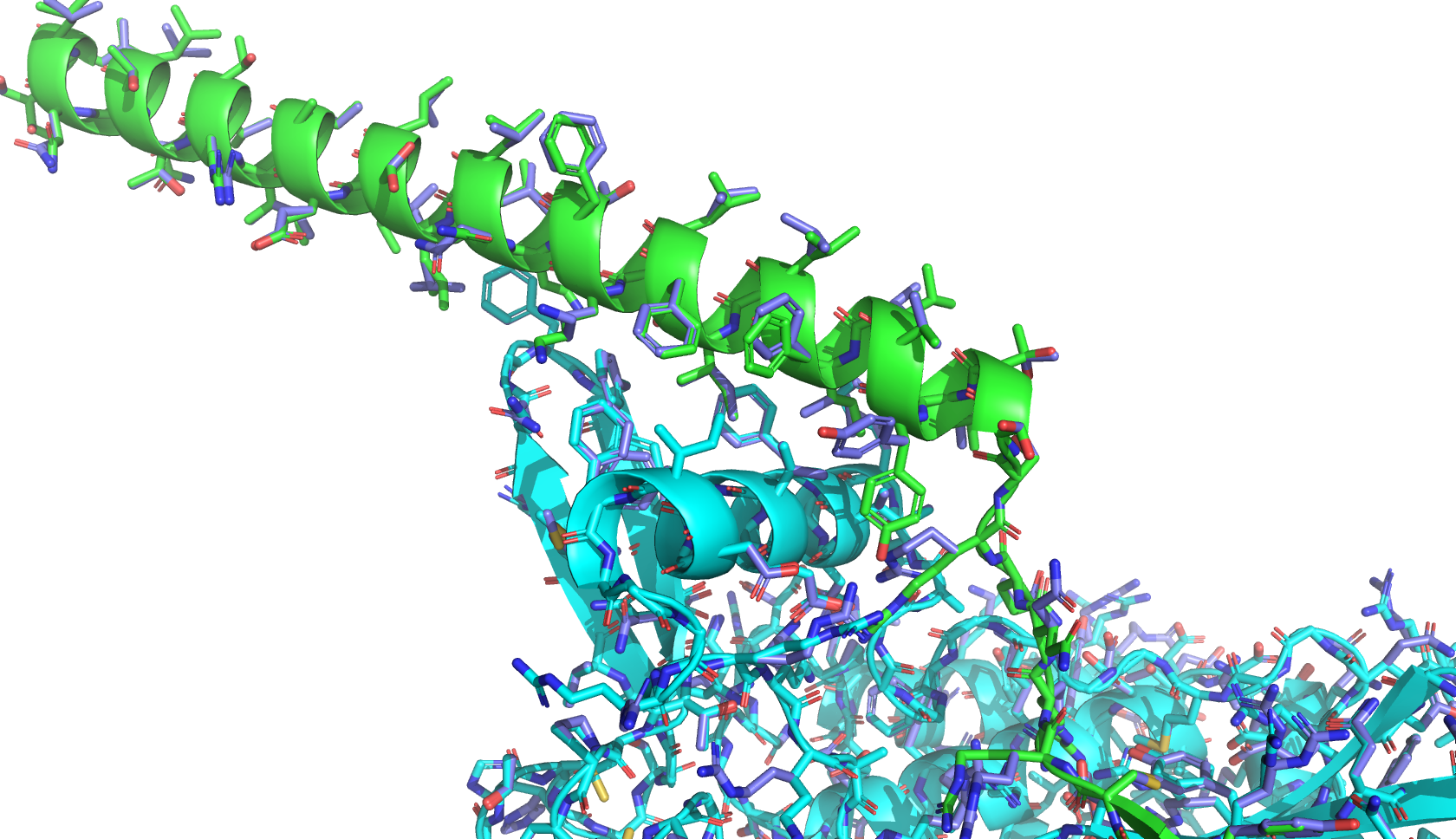
To do this, I first relax the sidechain structure of the L-protein using the following command:
The relaxation process slightly adjusts sidechain conformations to minimize steric clashes and optimize interactions. The resulting relaxed structure is shown below in blue, (green and cyan are the original structure predicted by Boltz-2):
Next, I scan through L-protein and mutate each residue to all 20 amino acids, and compute the change in binding energy (ΔΔG) for each mutation using the following command:
The result of mutations on soluble region is shown below, green indicates mutations that are predicted to stabilize the interaction (negative ddG), while red indicates mutations that are predicted to destabilize the interaction (positive ddG):
The result of mutations on TM region is shown below:
Based on the results above, I would propose the following multi-site mutations in the soluble region:
- DA26L + NA17W + CA29W: sum = −9.23
- DA26L + EA25P + QA8T + FA22H: sum = −9.03
- DA26L + NA17W + RA4E + SA9F: sum = −9.95
- DA26L + EA2D + FA5M + RA20N: sum = −8.69
- DA26L + HA24P + PA28K + RA34M: sum = −7.63
My rationale is that combining single stabilizing mutations will have an additive effect on the overall binding affinity. However, this assumption ignores potential epistatic interactions between mutations (non-additive effects).