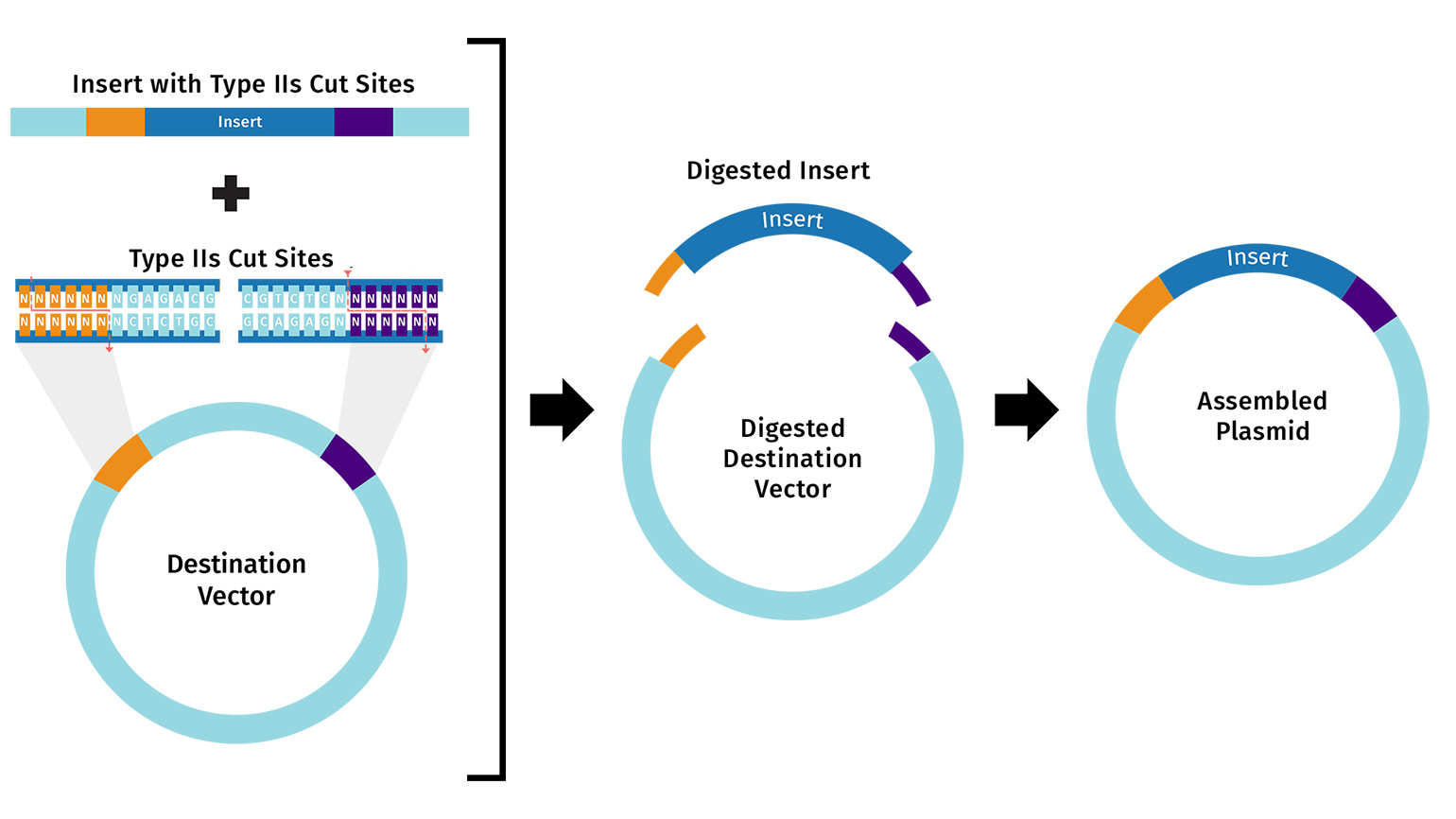###
### YOUR CODE HERE to create your design
###
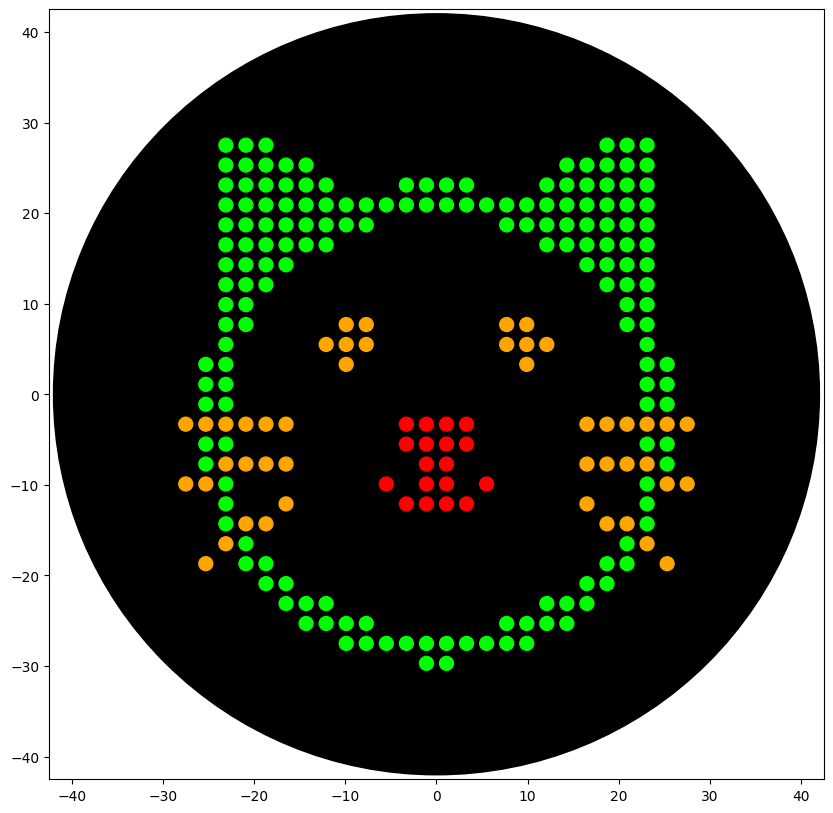
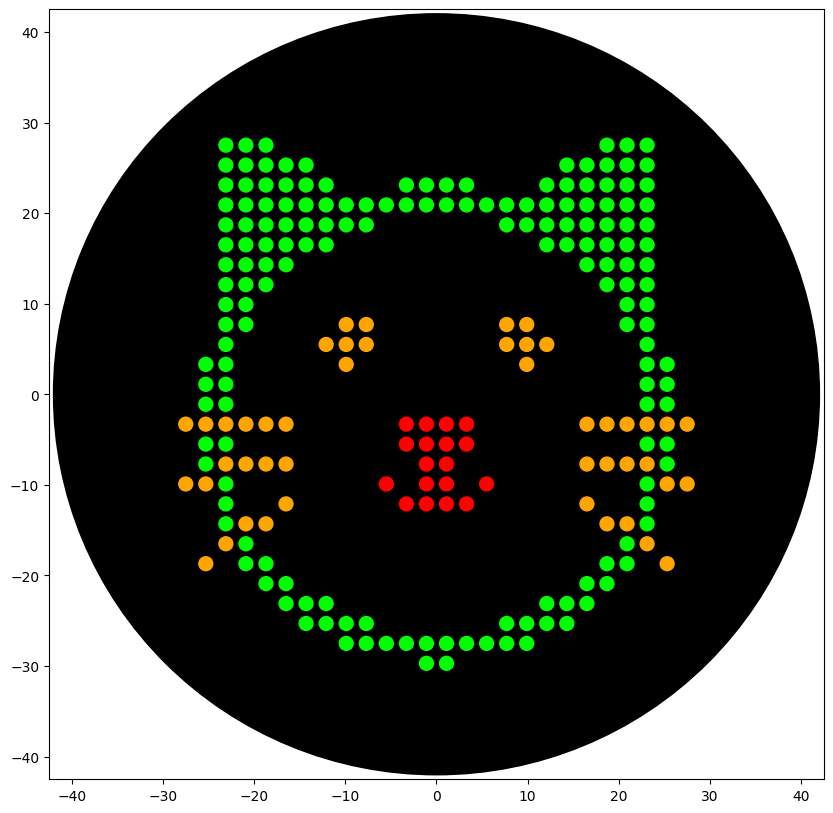
sfgfp_points = [(-3.3, -3.3),(-1.1, -3.3),(1.1, -3.3),(3.3, -3.3),(-3.3, -5.5),(-1.1, -5.5),(1.1, -5.5),(3.3, -5.5),(-1.1, -7.7),(1.1, -7.7),(-5.5, -9.9),(-1.1, -9.9),(1.1, -9.9),(5.5, -9.9),(-3.3, -12.1),(-1.1, -12.1),(1.1, -12.1),(3.3, -12.1)]
mrfp1_points = [(-23.1, 27.5),(-20.9, 27.5),(-18.7, 27.5),(18.7, 27.5),(20.9, 27.5),(23.1, 27.5),(-23.1, 25.3),(-20.9, 25.3),(-18.7, 25.3),(-16.5, 25.3),(-14.3, 25.3),(14.3, 25.3),(16.5, 25.3),(18.7, 25.3),(20.9, 25.3),(23.1, 25.3),(-23.1, 23.1),(-20.9, 23.1),(-18.7, 23.1),(-16.5, 23.1),(-14.3, 23.1),(-12.1, 23.1),(-3.3, 23.1),(-1.1, 23.1),(1.1, 23.1),(3.3, 23.1),(12.1, 23.1),(14.3, 23.1),(16.5, 23.1),(18.7, 23.1),(20.9, 23.1),(23.1, 23.1),(-23.1, 20.9),(-20.9, 20.9),(-18.7, 20.9),(-16.5, 20.9),(-14.3, 20.9),(-12.1, 20.9),(-9.9, 20.9),(-7.7, 20.9),(-5.5, 20.9),(-3.3, 20.9),(-1.1, 20.9),(1.1, 20.9),(3.3, 20.9),(5.5, 20.9),(7.7, 20.9),(9.9, 20.9),(12.1, 20.9),(14.3, 20.9),(16.5, 20.9),(18.7, 20.9),(20.9, 20.9),(23.1, 20.9),(-23.1, 18.7),(-20.9, 18.7),(-18.7, 18.7),(-16.5, 18.7),(-14.3, 18.7),(-12.1, 18.7),(-9.9, 18.7),(-7.7, 18.7),(7.7, 18.7),(9.9, 18.7),(12.1, 18.7),(14.3, 18.7),(16.5, 18.7),(18.7, 18.7),(20.9, 18.7),(23.1, 18.7),(-23.1, 16.5),(-20.9, 16.5),(-18.7, 16.5),(-16.5, 16.5),(-14.3, 16.5),(-12.1, 16.5),(12.1, 16.5),(14.3, 16.5),(16.5, 16.5),(18.7, 16.5),(20.9, 16.5),(23.1, 16.5),(-23.1, 14.3),(-20.9, 14.3),(-18.7, 14.3),(-16.5, 14.3),(16.5, 14.3),(18.7, 14.3),(20.9, 14.3),(23.1, 14.3),(-23.1, 12.1),(-20.9, 12.1),(-18.7, 12.1),(18.7, 12.1),(20.9, 12.1),(23.1, 12.1),(-23.1, 9.9),(-20.9, 9.9),(20.9, 9.9),(23.1, 9.9),(-23.1, 7.7),(-20.9, 7.7),(20.9, 7.7),(23.1, 7.7),(-23.1, 5.5),(23.1, 5.5),(-25.3, 3.3),(-23.1, 3.3),(23.1, 3.3),(25.3, 3.3),(-25.3, 1.1),(-23.1, 1.1),(23.1, 1.1),(25.3, 1.1),(-25.3, -1.1),(-23.1, -1.1),(23.1, -1.1),(25.3, -1.1),(-25.3, -5.5),(-23.1, -5.5),(23.1, -5.5),(25.3, -5.5),(-25.3, -7.7),(25.3, -7.7),(-23.1, -9.9),(23.1, -9.9),(-23.1, -12.1),(23.1, -12.1),(-23.1, -14.3),(23.1, -14.3),(-20.9, -16.5),(20.9, -16.5),(-20.9, -18.7),(-18.7, -18.7),(18.7, -18.7),(20.9, -18.7),(-18.7, -20.9),(-16.5, -20.9),(16.5, -20.9),(18.7, -20.9),(-16.5, -23.1),(-14.3, -23.1),(-12.1, -23.1),(12.1, -23.1),(14.3, -23.1),(16.5, -23.1),(-14.3, -25.3),(-12.1, -25.3),(-9.9, -25.3),(-7.7, -25.3),(7.7, -25.3),(9.9, -25.3),(12.1, -25.3),(14.3, -25.3),(-9.9, -27.5),(-7.7, -27.5),(-5.5, -27.5),(-3.3, -27.5),(-1.1, -27.5),(1.1, -27.5),(3.3, -27.5),(5.5, -27.5),(7.7, -27.5),(9.9, -27.5),(-1.1, -29.7),(1.1, -29.7)]
azurite_points = [(-9.9, 7.7),(-7.7, 7.7),(7.7, 7.7),(9.9, 7.7),(-12.1, 5.5),(-9.9, 5.5),(-7.7, 5.5),(7.7, 5.5),(9.9, 5.5),(12.1, 5.5),(-9.9, 3.3),(9.9, 3.3)]
mwasabi_points = [(-27.5, -3.3),(-25.3, -3.3),(-23.1, -3.3),(-20.9, -3.3),(-18.7, -3.3),(-16.5, -3.3),(16.5, -3.3),(18.7, -3.3),(20.9, -3.3),(23.1, -3.3),(25.3, -3.3),(27.5, -3.3),(-23.1, -7.7),(-20.9, -7.7),(-18.7, -7.7),(-16.5, -7.7),(16.5, -7.7),(18.7, -7.7),(20.9, -7.7),(23.1, -7.7),(-27.5, -9.9),(-25.3, -9.9),(25.3, -9.9),(27.5, -9.9),(-16.5, -12.1),(16.5, -12.1),(-20.9, -14.3),(-18.7, -14.3),(18.7, -14.3),(20.9, -14.3),(-23.1, -16.5),(23.1, -16.5),(-25.3, -18.7),(25.3, -18.7)]
scale = 1
def draw_points(points, color="Red"):
segments = []
for i in range(0, len(points), 20):
segments.append(points[i : i+20])
for seg in segments:
pipette_20ul.pick_up_tip()
pipette_20ul.aspirate(len(seg), location_of_color(color))
for x, y in seg:
adjusted_location = center_location.move(types.Point(x=x*scale, y=y*scale))
dispense_and_detach(pipette_20ul, 1, adjusted_location)
pipette_20ul.drop_tip()
draw_points(sfgfp_points, "Red")
draw_points(mrfp1_points, "Green")
draw_points(azurite_points, "Orange")
draw_points(mwasabi_points, "Orange")
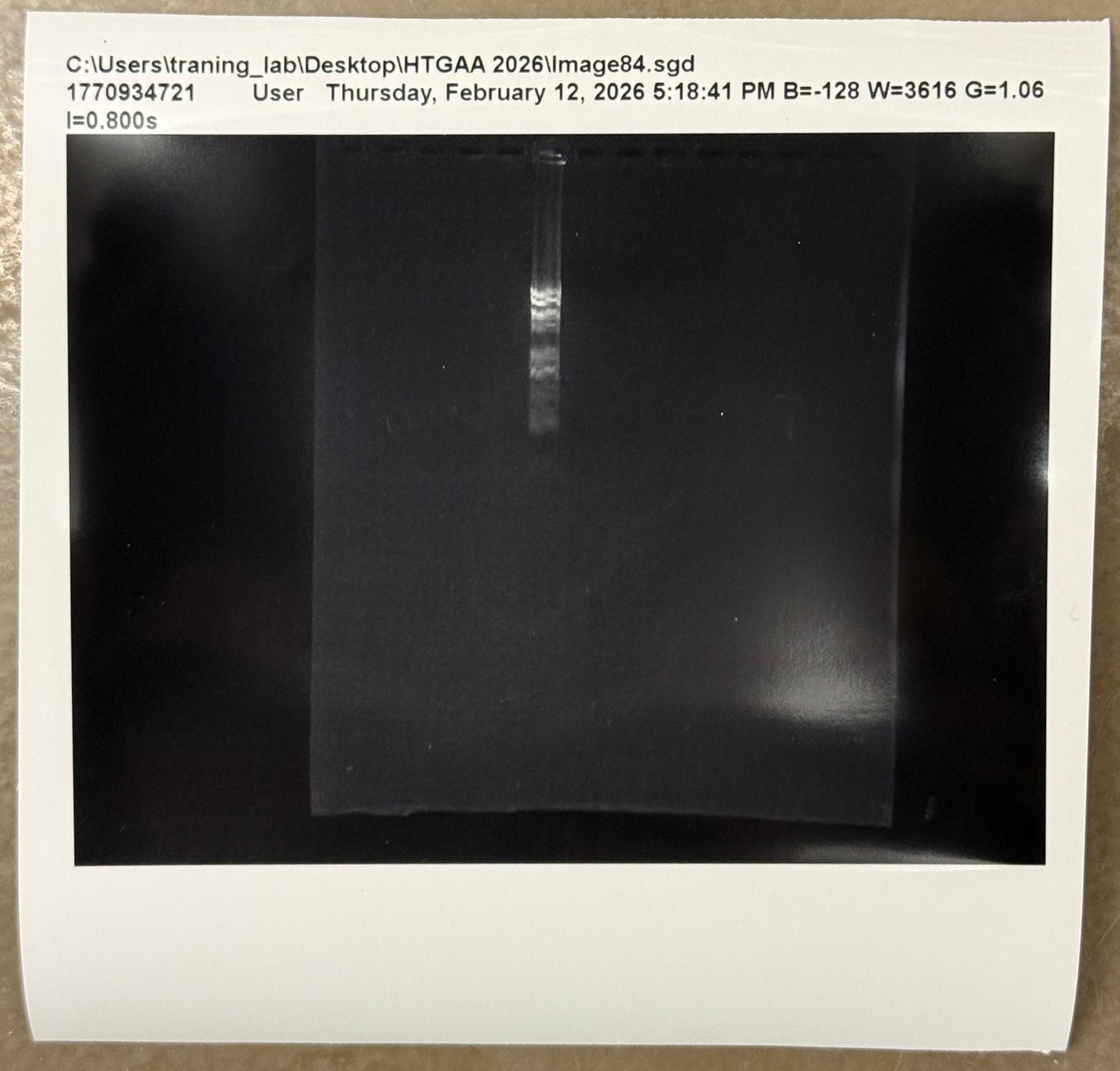
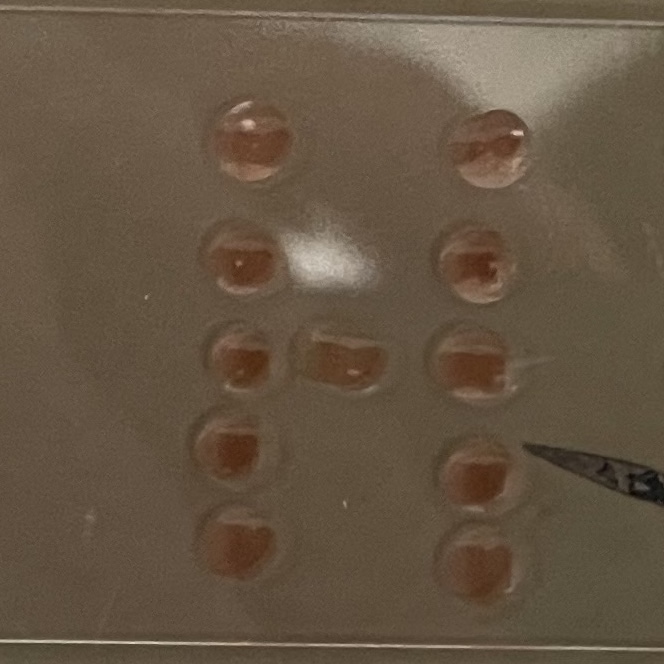
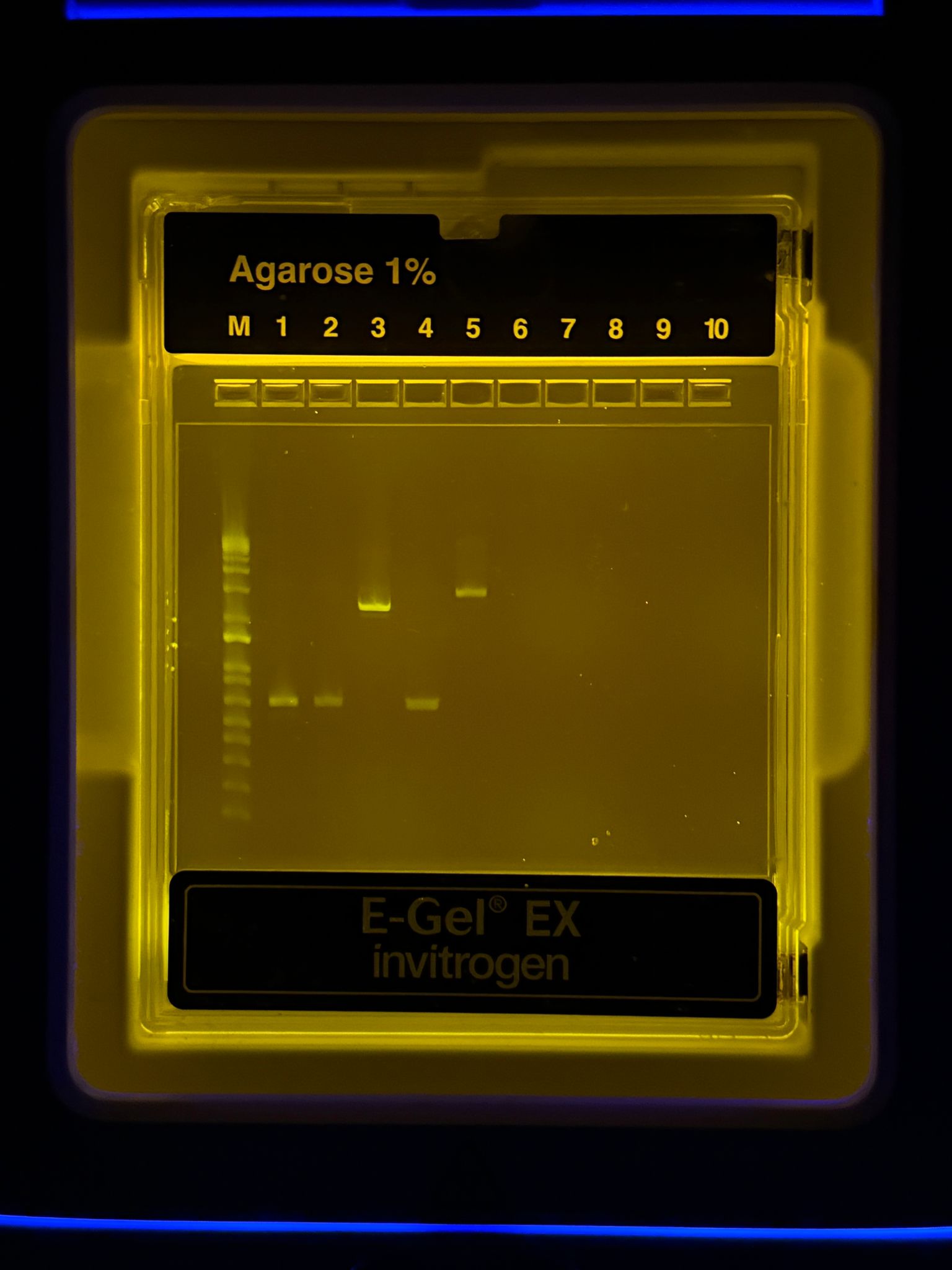
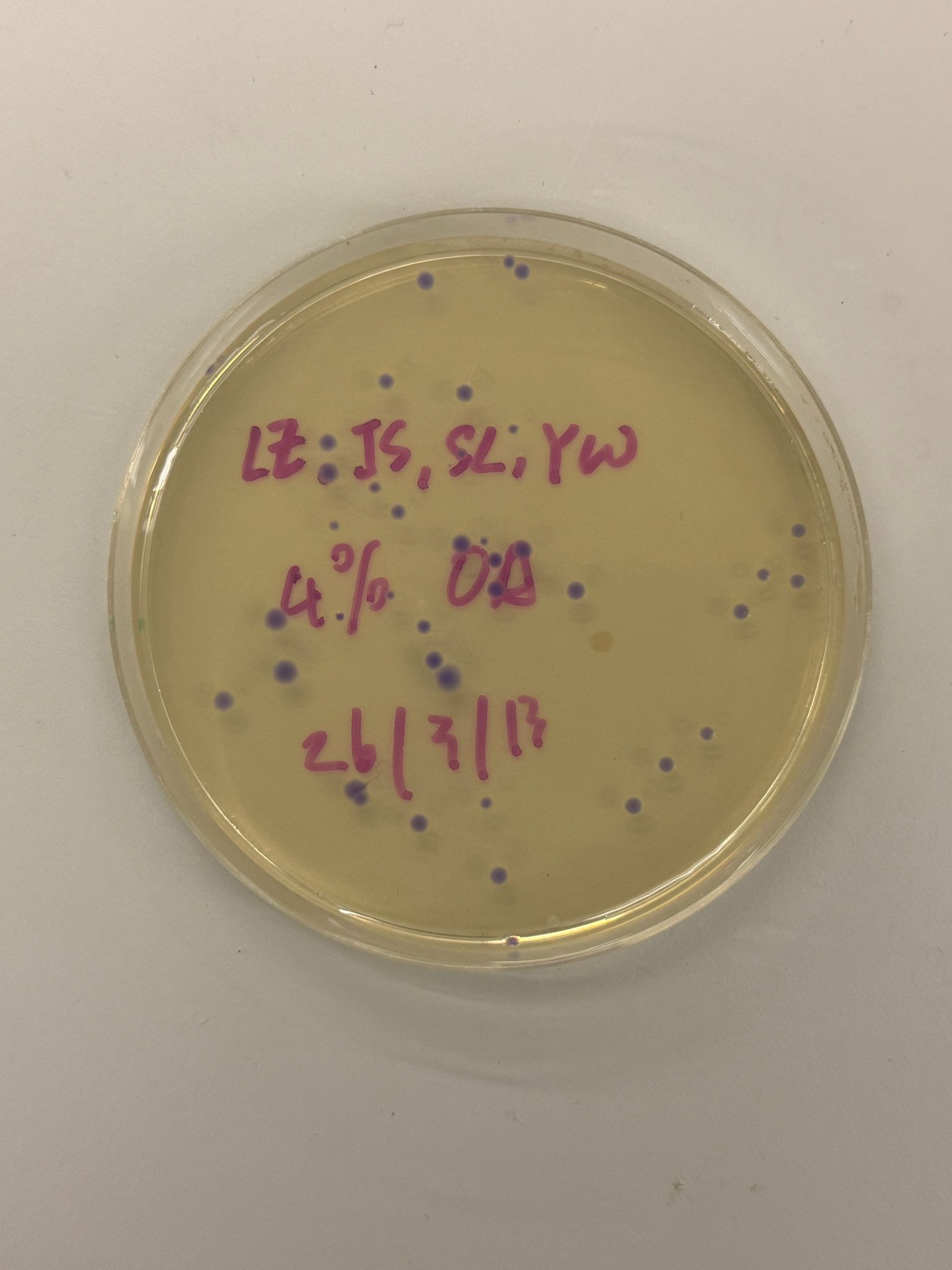
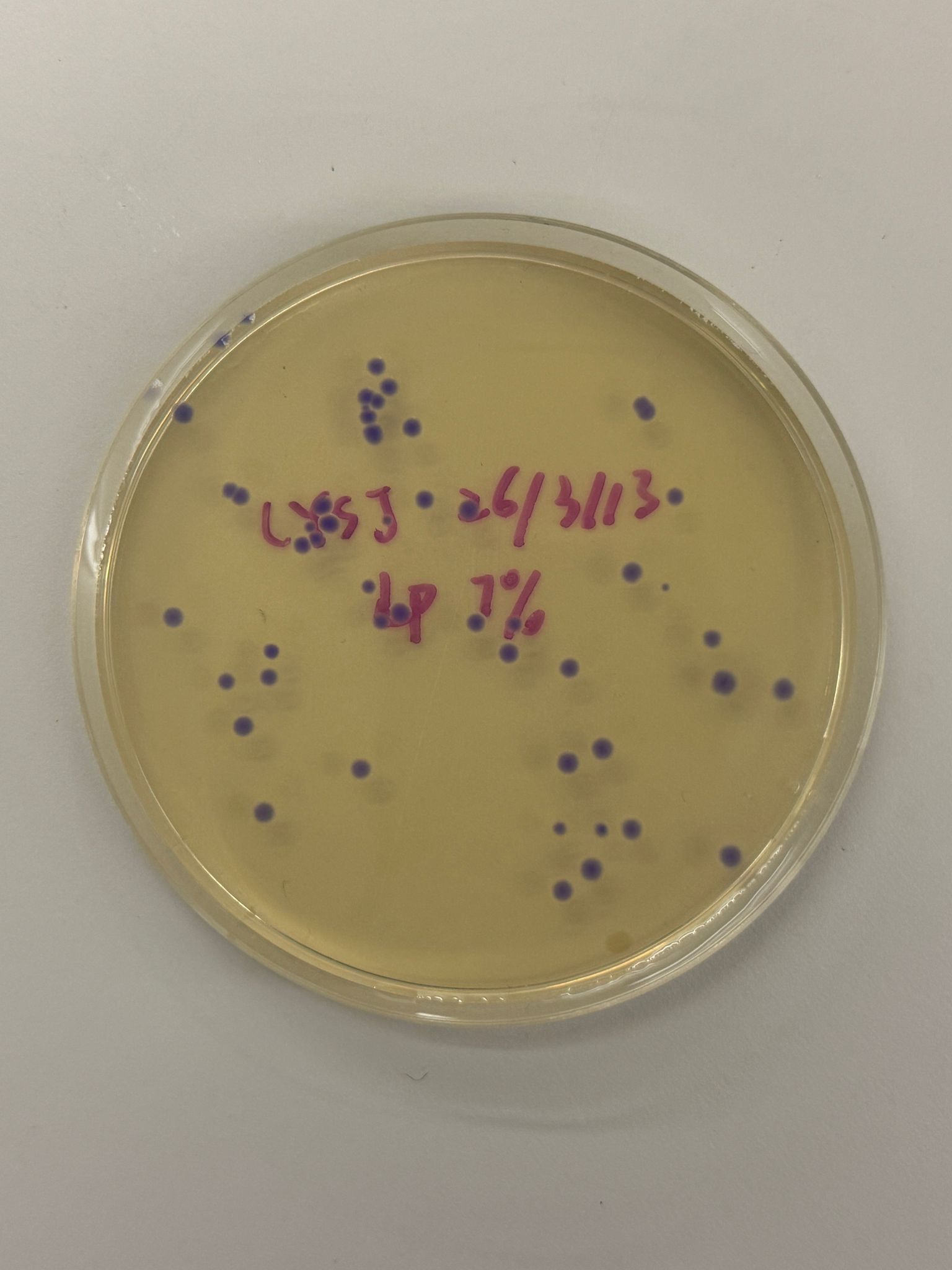
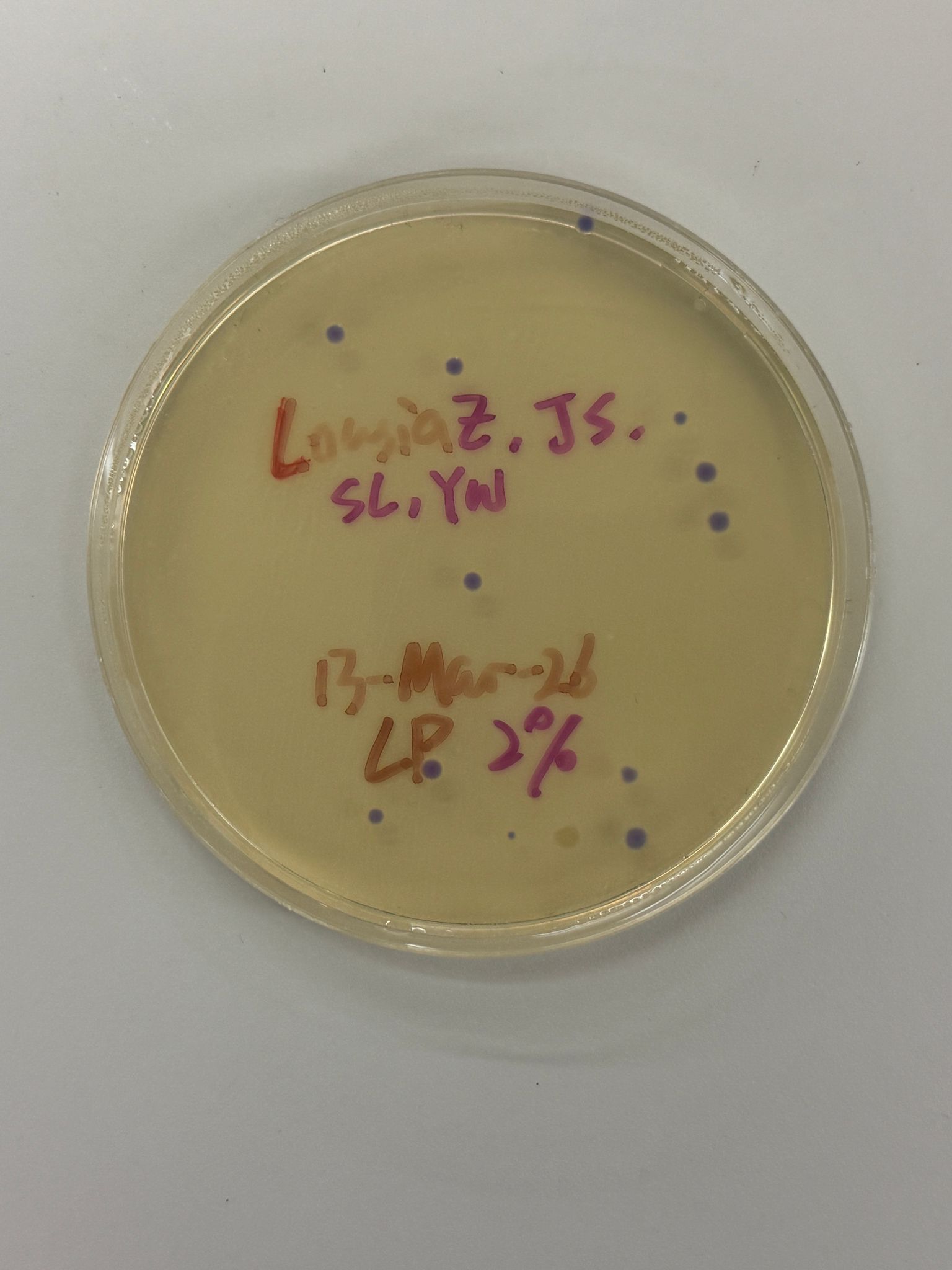
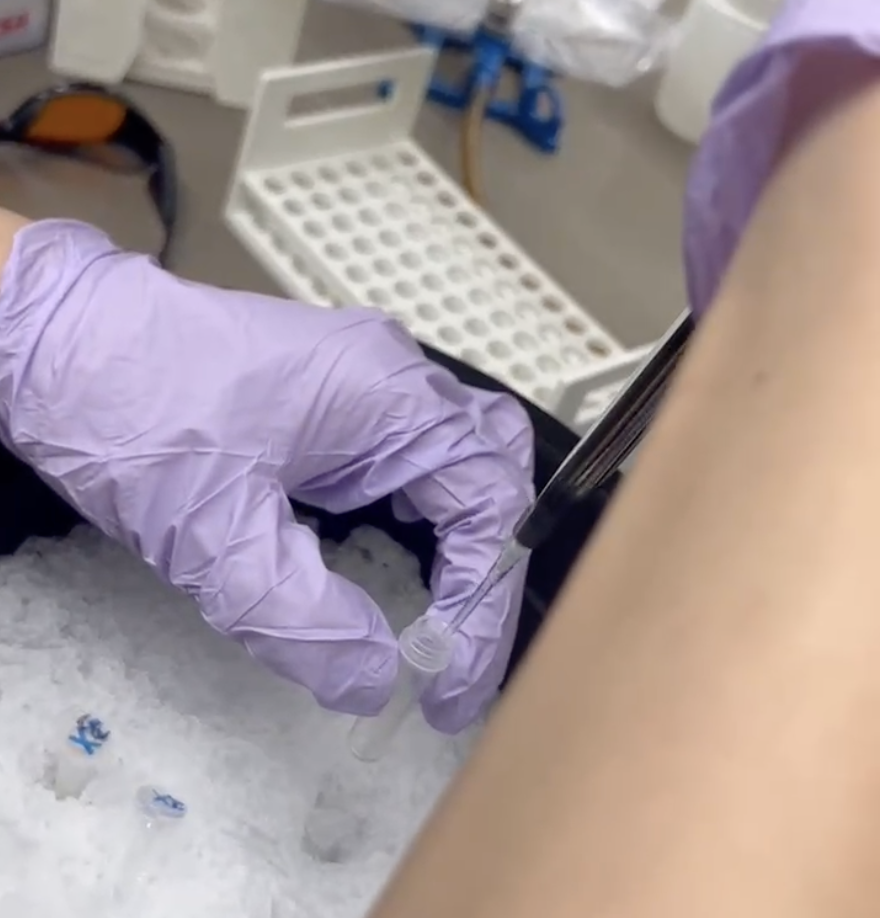
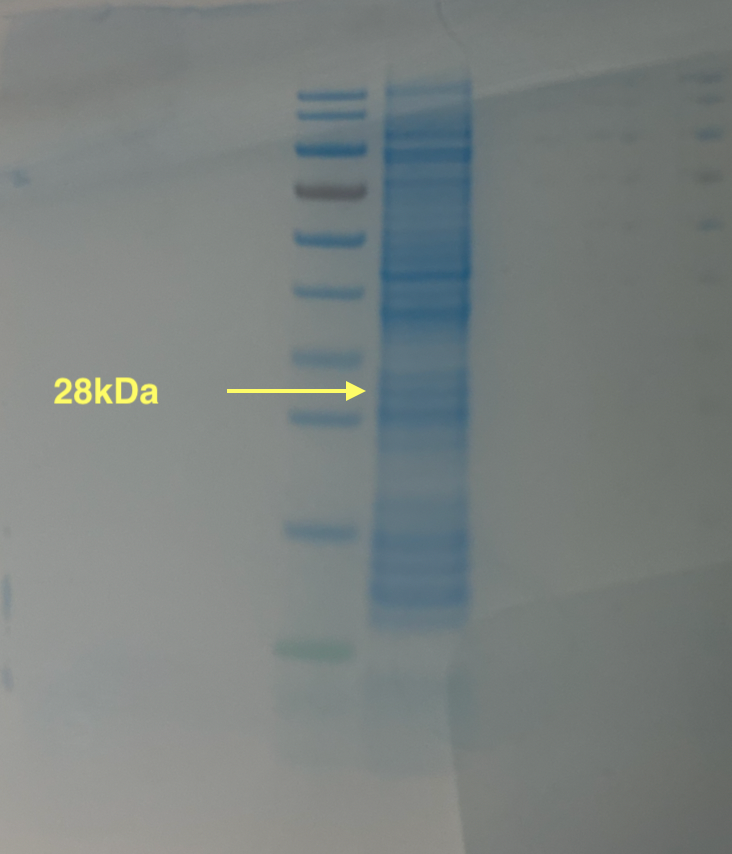
With the help of our TA Ronan, the art was printed with an Opentrons robot. The result is shown below:
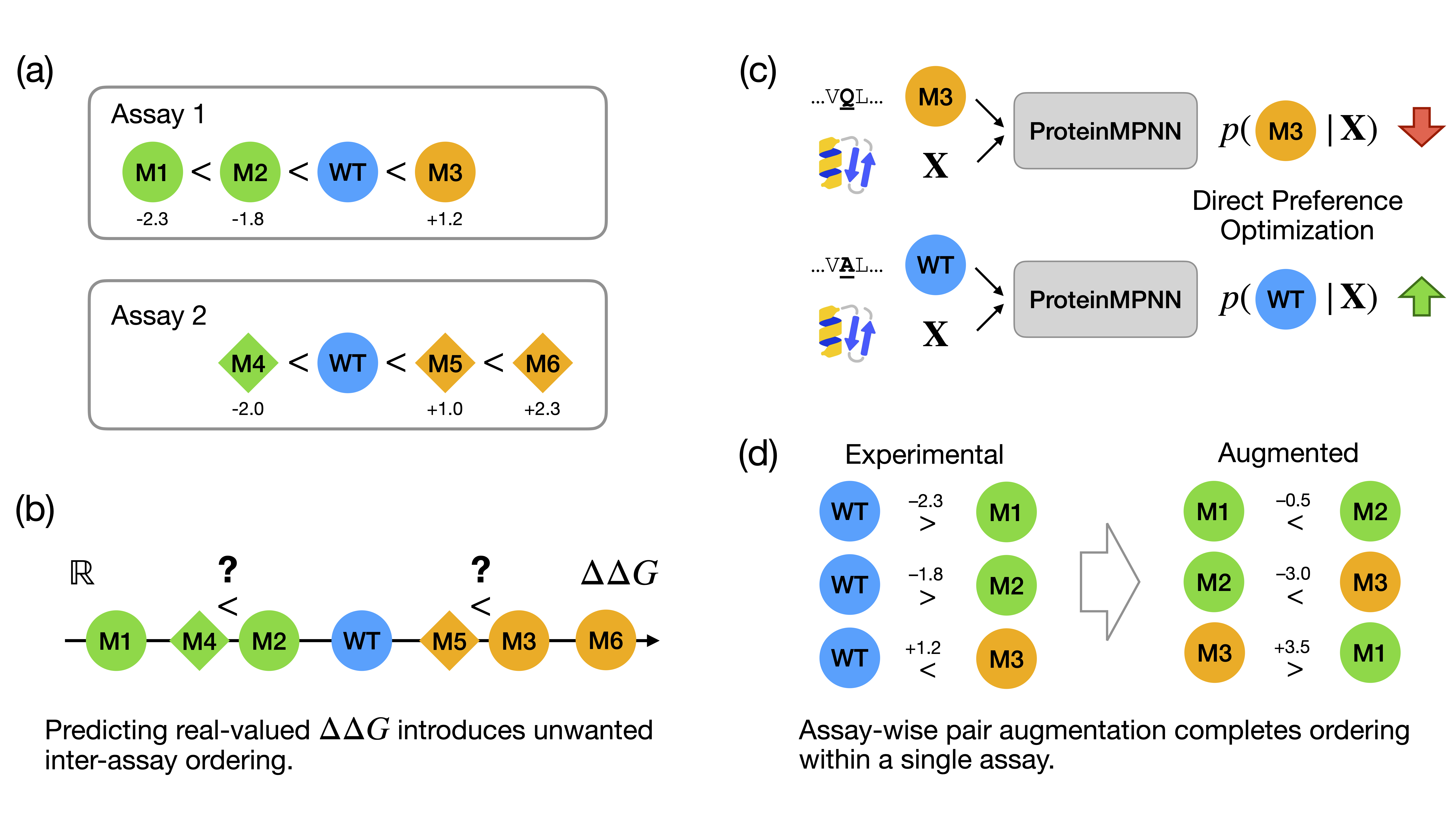
This paper introduces PyLabRobot, an open-source Python library that provides a unified interface for controlling various liquid-handling robots and accessories, including Opentrons.
PyLabRobot also includes a simulator (like the OpentronMock provided in this homework’s Google Colab notebook), which allows users to test and debug their protocols without needing access to the physical robot.
Further, this paper also demonstrates the integration with LLMs, allowing users who are not familiar with programming to create protocols using natural language instructions, which are then translated into executable code for the robot.
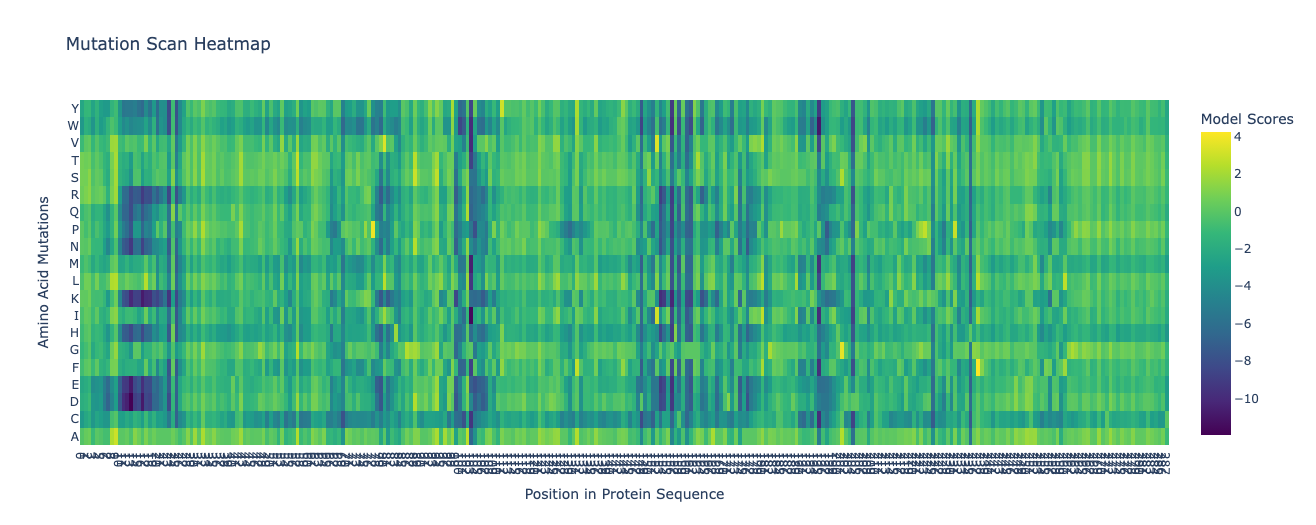
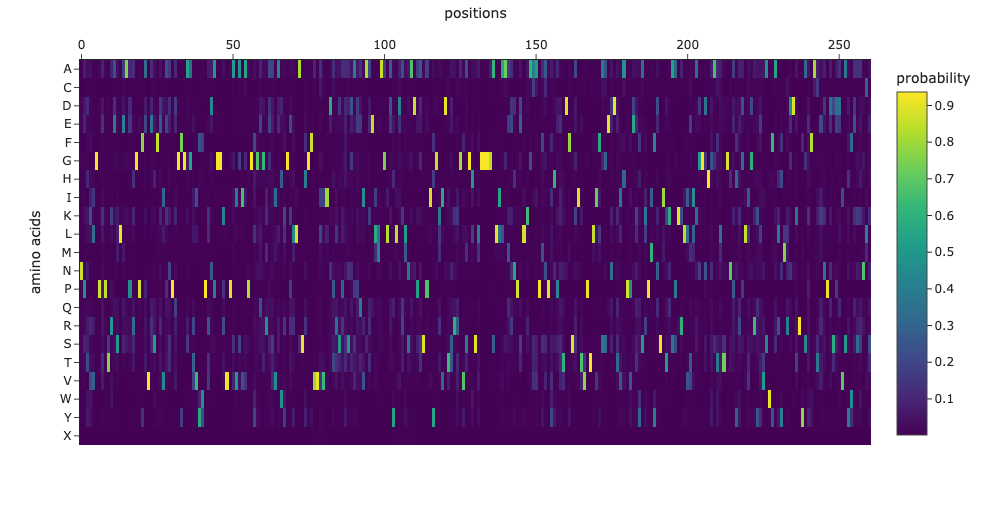
I am interested in using lab automation to do machine-learning guided directed evolution of PET-ase (PET plastic degradation enzyme).
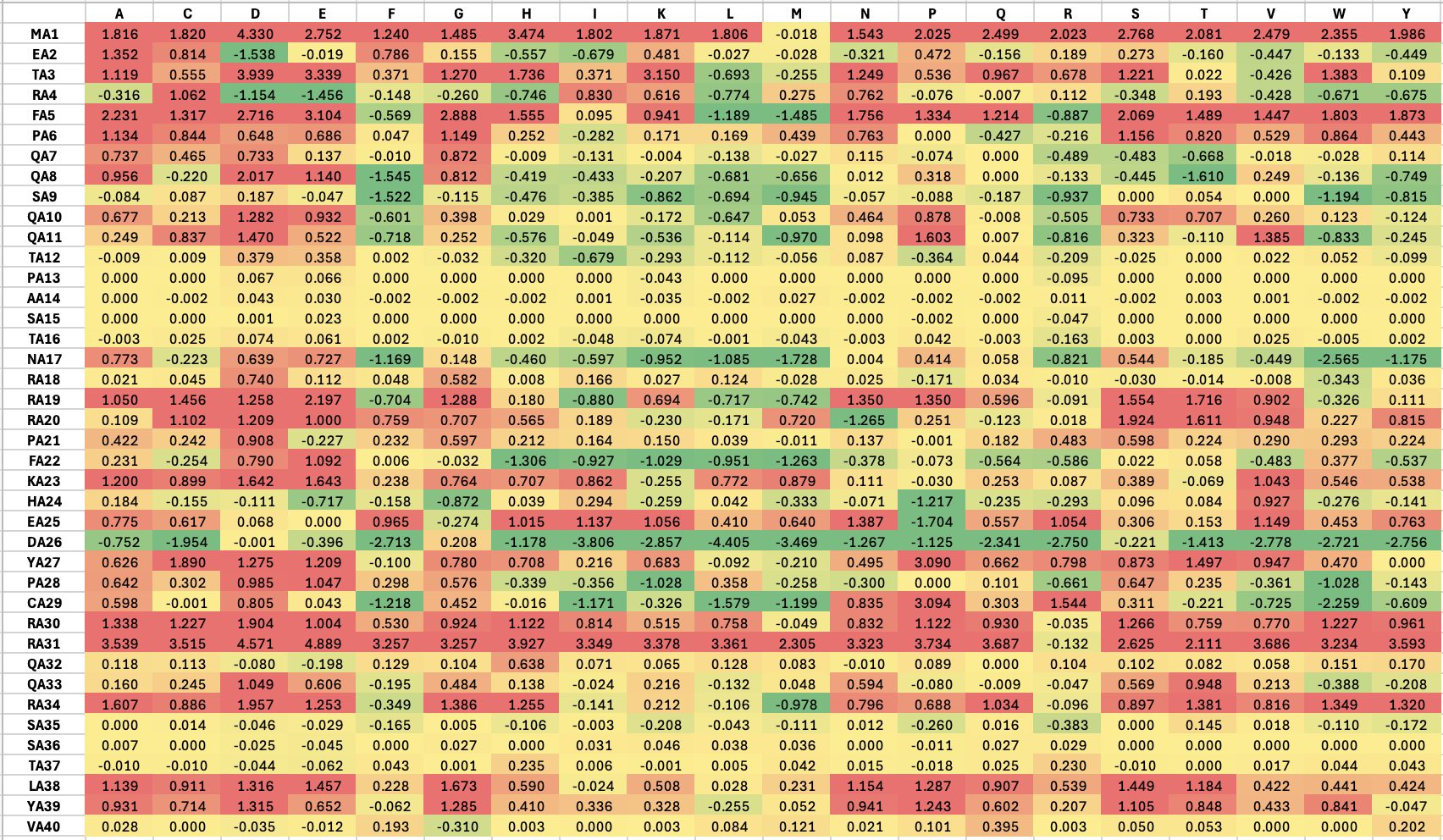
First, I will need to use machine learning models such as ProteinMPNN to design an initial library of PET-ase variants. I will place orders for the DNA fragments of these variants from Twist Bioscience.
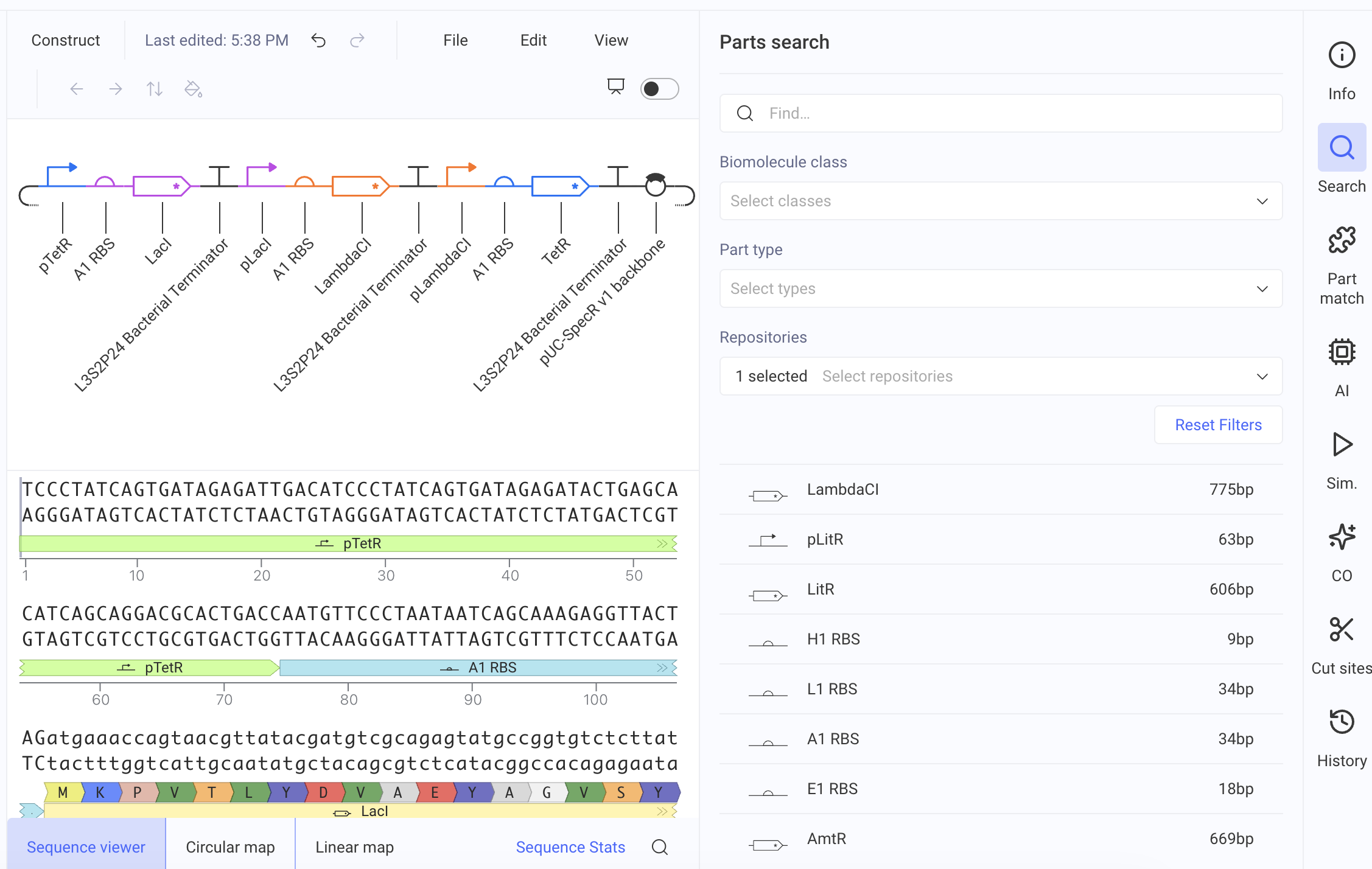
Second, I will use liquid handler to assemble the DNA fragments into plasmids, and then transform the plasmids into E. coli cells.
Then, I will use a plate reader to measure the activity of the PET-ase variants in degrading PET plastic. This can also be done in a high-throughput manner using 96-well or 384-well plates with an automation robot.
Finally, I will use the activity data to train a machine learning model to predict the activity of new PET-ase variants, and then use the model to design the next round of variants for testing. This iterative process can be repeated until we find highly active PET-ase variants for degrading PET plastic.