Class Assignment Application: SoilBuddy With climate change set to disrupt global agriculture and the unsustainability of commercial monocultures threatening to render the arable land left unusable, humanity urgently needs solutions facilitating soil stewardship to fortify our food supply. Existing solutions are either too expensive for small-to-mid farms, worsening inequality in the developing world, impractical for massive commercial farms, necessitating guesswork set to be upended by the sea change wrought by climate change, or provide, at best, noisy, time-lagged feedback through remote sensing.
Part 0 Done.
Part 1 Since it’s passe to create “MIT” with the electrophoresis gel, I decided to reverse the order of the letters to spell “TIM” instead.
Part 2 I don’t have in-person access to a node, so can’t perform the wet lab component.
Part 1 Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
The paper I’ll be analyzing is titled “Development of a high-throughput minimum inhibitory concentration (HT-MIC) testing workflow” published in Frontiers in Microbiology.
In summary, the researchers used an Opentrons OT-2 robot to automate an assay used to test the safety-efficacy profile of an antibiotic. They accomplished this by programmatically preparing a serial dilution of the antibiotic candidate across multiple 96-well plates with replicates, innoculating the wells with the test bacteria, then measuring the optical density of each well with a plate reader to determine bacterial viability after the antibiotic challenge.
Part A Number of amino acids = 500g/(100 Da) = 5 mol = 3.0 x 10^24 molecules of amino acids (approx). Over the course of digestion, the polynucleotides are broken down into constituent nucleobases prior to absorption. Considering the degeneracy of codons, these 20 amino acids were sufficient to produce a diversity of proteins, and were evolutionarily conserved due to a lack of strong selection pressure against them (ie. they were “good enough”). . They could’ve formed abiotically given the presence of organic precursors such as carboxylic acids, amines and small molecule side groups, catalyzed by zeolites, and triggered by lightning and heat on the primordial earth or extraterrestrial asteroids respectively. Left-handed spirals. Yes, some examples are $\pi$ helices and $3_{10}$ helices. It’s more energetically stable given L-amino acids and D-saccharides predominate natural protein helices and the sugar-phosphate backbone of polynucleotides respectively. In aqueous environments, the hydrophobic portions of beta sheets are driven together by steric interactions while the hydrophilic groups facilitate relatively strong intermolecular hydrogen bonding. Beta sheets allow for strong intermolecular bonding, in hydrophobic zippers, that renders misfolded proteins more stable than their functional conformations and facilitates the addition of more proteins to the aggregate fibril - the biochemical basis of amyloid pathogenesis. Yes, you could make materials out of amyloid beta sheets but they’d only be stable under aqueous conditions and would be potential biotoxins. . Part B I chose to investigate an antifreeze protein from an arctic bacterium as a potential solution for winter snow-clearing.
Part A Part 1: Generate Binders with PepMLM Binder Sequence Perplexity Score WRYYATVARHKE 15.38014901 WLYYVVVLRHGE 32.45105037 WRYYAAGARLKE 11.75101427 WRYYATAVELKG 10.44080960 Part 2: Evaluate Binders with AlphaFold3 Peptide ID Binder Sequence ipTM Score Location Peptide 0 WRYYATVARHKE 0.35 Approaches the $\beta$ barrel; surface-bound Peptide 1 WLYYVVVLRHGE 0.33 Approaches the $\beta$ barrel; surface-bound Peptide 2 WRYYAAGARLKE 0.35 Approaches the $\beta$ barrel; surface-bound Peptide 3 WRYYATAVELKG 0.22 Conforms to the dimer interface; binds to a pocket Control FLYRWLPSRRGG 0.31 Surrounding the $\beta$ barrel; surface-bound While peptides 0-2 exceed the control (known binder)’s ipTM scores, indicating stronger protein-protein interaction, they still represent low confidence values. I would not conclude that they produce stronger protein-protein interactions for use as potential molecular glues.
Assignment: DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The NEB 2X master mix includes engineered thermostable DNApol to synthesize complimentary strands during the extension phase of PCR, with low error rate and high expressivity to enable faster, more accurate DNA replication. It includes a buffer to maintain optimum pH for enzymatic catalysis and prevent the denaturation of enzyme or nucleic acids, as well as ${Mg}{Cl}_{2}$ to provide $Mg^{2+}$ as a cofactor for DNApol.
Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs more easily provides for graded responses and can scale intelligent responses; they can approximate every mathematical function and not just step functions.
Homework Part A Assignment 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production. It lacks the stress response, proteostasis and detoxification of an in vivo environment, allowing for much higher expression of protein including toxic varieties or those incorporating xenobiotic compounds, as in bio-orthogonal experiments. Reaction conditions are standard and can be externally controlled/varied (eg temperature) more precisely than in in vivo systems. The progress of reaction is more easily followed using imaging techniques, allowing for rapid optimization and sequential syntheses. Two cases where cell-free expression is better than cell production are the expression of protein toxic to bacterial/fungal chassis (eg antibacterial/antifungal compounds) as well as proteins intended for use in human medicine (free of endotoxins).
Waters Lab Part I 28006.60Da
z=32 based on consecutive m/z’s (875.4421, 848.9162); MW = 27982Da; Accuracy = 99.9%
No, its amplitude above background could mean it’s not reliably distinguished from background noise.
Waters Lab Part II When a protein unfolds, several more ionizable sites on the polypeptide are exposed. As such, the denatured protein will exhibit several more m/z peaKs, and typically at higher m/z values than the native protein conformation.
Part A I enjoyed the sense of oneness and how it spoke to the power of science to transcend divisions of identity and geography
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents E. coli Lysate: Provides biological medium and machinery (eg ribosomes) Salts/Buffer: Optimal pH, ionic atmosphere and osmolarity Energy / Nucleotide System: Power transcription/translation Translation Mix (Amino Acids): Allow for translation to produce mRNA from DNA Additives: Nicotinamide in particular allows for the regeneration of NAD Backfill: Maintain appropriate volume for manipulation of the biological systems
With climate change set to disrupt global agriculture and the unsustainability of commercial monocultures threatening to render the arable land left unusable, humanity urgently needs solutions facilitating soil stewardship to fortify our food supply. Existing solutions are either too expensive for small-to-mid farms, worsening inequality in the developing world, impractical for massive commercial farms, necessitating guesswork set to be upended by the sea change wrought by climate change, or provide, at best, noisy, time-lagged feedback through remote sensing.
I propose SoilBuddy as a low-cost, real-time, large-area biomachine platform to supercharge the soil microbiome using nature’s own solutions. The crux of SoilBuddy is intelligent biosensing that incorporates human-readable signals, provides an attack surface for genetic circuit-based intervention, and can integrate with existing, non-biological soil monitoring/fertilization workflows. At the same time, SoilBuddy features means to intervene in the soil microbiome by orchestrating microorganisms, and modulating nitrogenation, oxygenation and ion levels in the soil.
Getting to brass tacks, I outline its sensor/effector features below, with core functions necessary for a MVP in bold:
While I was inspired by the four tenets of biomedical ethics, I reckoned the most important goals relevant to SoilBuddy are Non-Maleficence and Justice. Given the echoes of the GMO debate from earlier in the century, and the proliferative capacity of bacterial systems, prioritizing safeguards against ecological contamination are crucial for governing our bacteria-based platform. At the same time, given the long history of agricultural intervention being wielded as a political tool to harm access to food and development, it’s crucial that we prioritize justice in the administration of SoilBuddy to ensure its applications prove to be a tool for good.
Goal 1: Non-Maleficence
In the context of SoilBuddy, a commitment to non-maleficence - articulated in the credos “first, do no harm” - involves a rejection of utilitarian logic to instead foreground safeguards against proliferation and mitigation of unintended consequences before the launch of SoilBuddy.
1.1: Counter-Proliferation
SoilBuddy must be governed to prevent runaway proliferation in wild ecosystems and spillover events should be minimized as far as possible.
1.2: Mitigation
Before applying SoilBuddy, practitioners in the SoilBuddy ecosystem must enact measures that mitigate the harm caused by both foreseen unintended consequences (ie. known risks of biotech in agriculture per se, such as gene contamination) as well as unforeseen unintended consequences (ie. put in place inherent safeguards or plans of action to manage unknown unknowns).
Goal 2: Justice
In the context of SoilBuddy, our construction of justice pertains to its distribution and development into perpetuity – I mean to open-source soil health.
2.1: Material Equity
Governance should focus on ensuring geographic and socioeconomic equity in access to the materiel of SoilBuddy, administering its distribution on the basis that food ought to be a human right, in line with the UN’s SDGs.
2.2: IP Equity
Governance should guard against the privatization of developments on SoilBuddy as a platform, recognizing that, again, access to food as a human right contributes to the common good of the species. Insomuch as SoilBuddy will start off open-source, its derivative art should also be accessible to all.
Governance Actions
Option One: Building-in Self-Limitation as a Technical Strategy (“Self-Limitation”)
Similar to how nuclear power plants exported under technical assistance programs are designed to make weaponization impossible, one governance action for industrial, academic and governmental practitioners in the SoilBuddy ecosystem to undertake would be stressing self-limitation when designing SoilBuddy: incorporating failsafes (eg robust, immutable genetic kill-switches) within SoilBuddy to mitigate the risks of unknown unknowns when it comes to mitigating accidental proliferation; and only including biosensor and effector modalities amenable which negate or greatly minimize the risk of deliberate proliferation.
Academic practitioners will need to self-police their research into SoilBuddy to avoid the development of high-risk features, as will industrial stakeholders, despite the potential upside of risky features in contravention of safety-first design principles. National governments must agree to avoid research into safeguard-free SoilBuddy platforms despite the agricultural productivity they may enjoy, not to speak of avoidig the weaponization of SoilBuddy to wage agricultural war. International governments will have to dedicate scarce resources, and scarcer funding to coercive as well as persuasive measures to promote this technical strategy, promoting global buy-in despite political differences as well as counterproliferative interdiction.
A great uncertainty remains as to whether such kill-switches remain biologically feasible, and robust to accidental/natural degradation (eg mutations, horizontal gene transfer). Pessimistically speaking, this strategy also assumes an alignment of academic-industrial complexes away from private interests, not to speak of strategic apparatuses of power. Given how anything short of absolute, collective alignment will rapidly corrode the incentive of collective security that motivates cooperation on this technical strategy, it is also unclear exactly how stable such an arrangement will be without active enforcement against rogue states and non-state actors.
Beyond the failure modes analyzed while elucidating the policy’s assumptions, it is also possible this technical strategy of self-limitation could dampen the research momentum required to make headway on a key technology of humanity’s food future; that, shirking risk altogether might prevent a substantially greater benefit that outweighs even the existential risk of SoilBuddy’s proliferation and inequitable distribution (ie. concentration within centers of comparative advantage) which could actualy be a more efficacious state of affairs.
Option Two: Mandating Equitable Access as an Explicit Policy Measure (“Legislating Equity”)
Switching gears from food, let’s consider the state of California’s Assembly Bill 685 as a piece of legislation enshrining access to water as a human right; in a similar fashion, option two would involve (inter)national legislation (and requisite enforcement) guaranteeing equitable access to the material and IP of SoilBuddy as the key to a more secure food supply across geopolitical borders and strata of development. This stands in stark contrast to the oligopolistic pricing of economic factor inputs and complementary agritech/biotech goods used in the production of food crop in certain major agricultural markets, as well as the fervent privatization of IP governing crop cultivation methods – much like the denial by John Deere of farmers’ right to repair their equipment, but in contraposition to Norman Borlaug’s methods of knowledge-sharing that underpinned the green revolution.
Given the bottlenecks manufacturing inequity – such as mass production, marketing and distribution – lie in industry, academia does not figure in Option Two as much insofar as we discount the role of university corporations that might own key patents and licenses withholding access to SoilBuddy platforms and developments thereof. Taking them to constitute industrial actors for the sake of argument, we thus find that the key stakeholders in Option Two are the industry players charged with balancing the profit motive and corporate responsibility, national regulators that must maintain the free market incentive to innovate with an interest in delivering on their duty to citizens, and international government faced with material limitations in enforcing violations of legislated equity despite national interests in shaping the competitiveness of their own agricultural productivity, or those of others.
Specifically, taking this legislation to be an international treaty guaranteeing mechanisms of cross-subsidization pegged to national income levels viz debt and requiring the registration of SoilBuddy IP with a INGO, we find that Option Two also requires the assumption of total cooperation to negate private incentives to cheat (in an economic sense). Beyond overlapping similarities with Option One’s modes of failure at the industrial and national levels, Option Two relies even more on state and international capacity for the enforcement of this law since there isn’t a structural incentive to ensure equity, as much as there is one to maintain technical non-proliferation due to salutary effects on SoilBuddy metabolic budgets, the avoidance of legal liability or sidestepping openings that might facilitate corporate espionage.
Apart from the failure of legislated equity to take off given the precedent of global pharmaceutical distribution, there is a distinct risk that this legislation may be weaponized for hybrid contestation on the international level. On the other hand, should there be comprehensive good-faith buy-in, the runaway success of mandated equity might paradoxically entrench corollary inequities in the distribution of R&D capacity across the Global South by eliminating the incentive to develop indigenous agritech capabilities, or entrench inter-nation trade dependencies, that if not adequately de-risked, could be disrupted by trade disruptions - innocuous as they might be, such as the maritime trade disruption caused by the Ever Given’s freak beaching in the Suez Canal.
Option Three: Engineering Markets to Incentivize Equity (“Incentivized Equity”)
Apart from altruism and compulsion, a third drive worth exploring is that of economic incentives that might appeal to naturally self-interested, rational economic agents. Similar to the ethos underpinning the FDA orphan drugs scheme, I propose the disbursement of strings-attached market incentives such as tax holidays, targeted research funding, export preferences, indirect subsidies and fast-track approval to govern the nonproliferative development and equitable distribution of SoilBuddy.
Here, a finer dissection of industry is necessary. We lump industry’s R&D side of house with academia in the form of the academic-industrial complex, which will naturally be incentivized by the targeted fiscal outlays promoting responsible SoilBuddy platform development to incorporate biosafety features in the IP that will head into commercialization; this can be verified by funding bodies during progress reviews and the tiered disbursement of funding tranches subject to the satisfactory fulfilment of performance metrics germane to non-maleficent and just SoilBuddy platforms. Next, industrial players overseeing commercialization, production and marketing will be incentivized by corporate incentives such as tax holidays and indirect subsidies to stay faithful to safety-first IP, as well as allow regulatory oversight of their products to ensure they meet the counterproliferative and equitable design features articulated above. On the international level, member fee waivers at INGOs as well as the enactment of diplomatic commons would promote good faith adherance to biosafety in the use and distribution of SoilBuddy, just as the UN has slowly but steadily driven development across the board, and the WTO common prosperity through responsible trade.
Option Three rests heavily on governmental capacity for the microeconomic to international administration of the scheme, from the efficient disbursement of funds, to the capacity for responsible statesmanship promoting the common good. Aside from the obvious failure mode should government failure (ie. rent-seeking) distort incentives for the responsible development and distribution of SoilBuddy, there is the crucial issue of national strategic interest in the performance of the biotech firms developing SoilBuddy in the high-income biotech exporters likely to muster the industrial and governmental heft to implement Option Three in the first place; importing nations might indeed suffer dumping should the subsidies provided for the responsible development and disbursement of SoilBuddy be anti-competitive. Thus, strong international governance of SoilBuddy’s trade is pivotal in maintaining the sustainability of SoilBuddy’s application worldwide.
Evaluation of Governance Actions
We evaluate the governance actions on their efficacy in meeting the governance outcomes articulated above, as well as policy desirability, feasibility and sustainability.
Governance Outcome / Policy Feature
Option One “Self-Limitation”
Option Two “Legislated Equity”
Option Three “Incentivized Equity”
Non-Maleficence (Counterproliferation)
1
2
2
Non-Maleficence (Mitigation)
1
2
3
Justice (Material Equity)
3
2
3
Justice (IP)
3
2
3
Desirability (Scale of drawbacks)
1
1
2
Feasibility (Policy implementation)
3
2
2
Sustainability (Policy sustainment)
2
1
3
Selection of Governance Action(s)
I would select Option One (“Self-Limitation”) and Option Two (“Legislated Equity”) as complementary policies that offer both a high-uncertainty, high-reward as well as moderately-achievable option comprehensively guaranteeing biosecurity on both counts, of non-maleficence and justice.
Option One (“Self-Limitation”) alone would seem the most promising governance action barring its inability to directly achieve justice. It is also highly contingent on the biological underpinnings of SoilBuddy, which is an unfortunately uncertain, if rewarding bet. Given the shortfall of a single policy, we pursue either Options Two (“Legislated Equity”) or THree (“Incentivized Equity”) as a synergistic policy that, in a swiss cheese fashion, provides better coverage of biosecurity objectives.
The key differentiator between Option Two (“Legislated Equity”) and Option Three (“Incentivized Equity”) lay in the unsustainability of fiscal outlays at both the national and international levels as required for the latter, as well as the greater alignment of coercive incentive in the former to adhere to non-maleficence. As a whole, Option Two is attainable, though contingent on the tedious, if achievable negotiation of international interests to settle on a suitable piece of biosecurity legislation.
Crucially, both policies hinge on the fair assumption that there is sufficient technical as well as state capacity to see SoilBuddy through in the first place in a responsible manner. At the same time, we qualify that, pending further research, the technical research underpinning first-of-its-kind killswitches and safety measures required for Option One is more uncertain than international cooperation on the governance of a risky technology, which has precedent in the historical governance of nuclear energy, as well as more contemporary discussions of human germline editing and mirror life.
Ethical Concerns and Appropriate Governance Actions
This week’s class and this exercise as a whole have brought to mind two worries, one deontological and the other of a more utilitarian turn. Firstly, I questioned if the governance of biosecurity might err on the side of caution and unduly impinge on the freedom of sovereign individuals to pursue their SoilBuddy-related curiosities, or nations to self-actualize their agricultural potetial. Applying a Lockean construction of the tradeoff between governance and freedom, however, one realizes that the necessary surrender of a few freedoms pertaining to the riskiest of technologies, especially ones that might have existential implications for the species and planet as a whole, really is a freedom from, to channel Isaiah Berlin, a greater evil; the freedom to feed our individual or collective curiosities is contingent on the responsible stewardship of the commons which make it possible.
Secondly, from the standpoint of effective altruism, I wondered if governance might be enacting longtermist harm by impeding the pace of technological research necessary to mitigate the existential threat the species faces to its food supply. Again, on the balance of probabilities this more consequentialist reasoning enables, we conclude that the risk of massive ecological contamination in the image of an Atwood dystopia requires that we factor in guardrails against irreversible harm borne of an irresponsibly Pandoran scientific accelerationism.
On a metacognitive level, inasmuch as one might worry about the balance of caution and speed required to safely realize the promise of SoilBuddy for food security across the species, it is this very debate that constantly refines humanity’s construction of this negotiation between risk and reward.
Week 2 Lecture Prep
Professor Jacobson
Polymerase has an error rate of 1:10^6. Given the length of the human genome, ~3.2 Gbp, the number of expected errors (3200) is substantial; the error rate of polymerase is significant for the length of the human genome. The error rate of polymerase is mitigated by proofreading during DNA replication, as well as MutS-mediated mismatch repair after replication.
Taking the average human protein to contain ~400 amino acids in its primary structure, this means ~400 mRNA codons would be necessary to code for it. Given the degeneracy of the genetic code, working backwards and assuming a roughly equal proportion of different amino acids for the sake of argument, the codon table implies 4^(820) * 3^(120) * 2^(1220) = 1.3210^178 different sets of DNA triplet codes to encode the typical human protein. In practice, certain DNA triplet code sequences encode mRNAs that are more structurally stable, degradation-resistent, and amenable to translational regulatory elements necessary for mRNA translation and protein folding.
Dr. LeProust
Solid-phase phosphoramidite synthesis.
Due to accumulated errors, the yield for fully-synthesized sequences is impractically low, especially since they have to be purified and isolated. For long sequences, steric hindrance near the substrate as well as chemical degradation further reduce yield.
For similar reasons as above, direct synthesis’ stepwise inefficiency would cause the yield to be impractically low.
George Church
They are: Phenylalanine, Threonine, Tryptophan, Leucine, Lysine, Methionine, Arginine, Histidine, Isoleucine and Valine [1]. To use the Jurassic Park example at surface value, I think picking lysine was a poor choice for omnivorous/carnivorous dinosaurs since they were able to obtain the essential amino acid from their diet, or the environment. At the same time, it offers a broader lesson on building safeguards into our biotechnology; even if we replaced lysine with a xenobiotic amino acid, for example, simply relying on one failsafe doesn’t provide a reasonable amount of biosecurity.
Since it’s passe to create “MIT” with the electrophoresis gel, I decided to reverse the order of the letters to spell “TIM” instead.
Part 2
I don’t have in-person access to a node, so can’t perform the wet lab component.
Part 3
Part 3.1
I chose to investigate an inorganic pyrophosphatase-driven proton pump because it’s the most relevant to the SoilBuddy MVP. Here are other candidates I considered and why I rejected them:
Bacteriorhodopsin: +Prokaryote-native -Light-driven rxn is impractical for a soil-dwelling bacterium
Membrane H+-ATPase: +Native cellular localization at membrane is favorable -High risk of cross-talk with native membrane proton pumps and may preclude bacterial survival
Methanogenic proton pump: - Presupposing an electron acceptor for the correct functioning of the proton pump limits our choices of target organism and may hinder the systems integration of other effector means for SoilBuddy
We need to optimize codons because different model organisms have aminoacyl-tRNAs in different abundances. Certain organisms may not have enough, or any, tRNAs with the anticodons corresponding to our nucleotide sequence, while for others, certain tRNAs may be more abundant, and thus facilitate more efficient translation of mature mRNA.
I’ll be optimizing my sequence for E. coli since it’s both a well-characterized transformation and plasmid amplification platform, and amenable to SoilBuddy’s target application. Given the length of the target sequence, I’ll hedge my bets on Golden Gate Assembly, and optimize my codons without the restriction sites for common type IIS enzymes such as BsaI, BbsI, BsmBI, and FokI. I used Twist Bioscience’s codon optimization tool:
Once the target sequence is spliced into a cloning vector via golden gate assembly and a competent strain of E. coli is obtained, the bacteria could be transformed using heat shock treatment. After selection for expression of the cloning vector, transformed bacteria would be incubated in broth.
Cells would be extracted via centrifugation and lysis with detergent to release the membrane protein. The membrane protein would be purified using chromatography and verified using Western blot.
A cell-free protocol would use E. coli lysate to conduct the in vitro transcription then translation of the plasmid vector containing the target sequence. Purification and verification of the protein would similarly involve chromatography and Western blot.
Part 3.5
Upon transcription of a DNA sequence to pre-mRNA, post-transcriptional modification of the pre-mRNA involves alternative splicing, which allows for different combinations of exons to be present in the mature mRNA, and subsequently translated into protein isoforms.
Alternatively, transcription initiation complexes may form on different promoters within the gene, causing different transcripts to begin with.
Part 4
Part 4.1
Done
Part 4.2
Part 5
Part 5.1: DNA Read
(i) I’d like to sequence the eDNA of the soil microbiome to gain an understanding of the organisms SoilBuddy would have to co-exist with.
(ii)
I’d use Illumina NGS sequencing because it balances read-length - more than sufficient for analyzing eDNA fragments - with cost and thoroughput. It is classified as a 2nd-generation sequencing technology since it utilizes sequencing-by-synthesis, albeit in a massively parallel architecture.
My input is purified DNA fragments obtained from soil solution. Depending on the distribution of fragment sizes, a coarse reading of which may be obtained with a pilot gel electrophoresis using a standard ladder, I will first use enzymatic fragmentation to produce suitable DNA fragments, then ligate Illumina NGS-specific adapters containing barcode sequences, and finally run a low number of PCR cycles to amplify my DNA fragments.
After PCR amplification, I’ll pool my samples to ensure equal representation in my library and verify their purity before loading them into the sequencer.
Under Illumina NGS, the fragments of DNA in my library hybridize sparsely over the surface of the sequencer’s flow cell, thanks to the adapters ligated to the eDNA samples. The fragments first undergo bridge amplification PCR wherein multiple copies of the fragments (both forward and reverse strands corresponding to the fragment sequence) are generated in small clusters within the flow cell’s nanowells. Reverse strands are enzymatically cleaved and washed off the flow cell.
Once clusters are produced, the cell is flooded with a primer, DNA polymerase and modified fluorescently-tagged nucleotides which can only be incorporated by the DNA polymerase one base at a time to the oligomers surrounding the clusters. Unbound nucleotides are washed away, then a picture of the flow cell is taken, which reveals the specific base incorporated in each cluster by virtue of the unique wavelength of fluorescent light given off by each modified nucleotide (ie. A, T, C or G). The fluorescent tag blocking DNA synthesis is chemically inactivated, then the chip is again flooded with fluorescently-tagged nucleotides. This process repeats till the full length of DNA fragments has been replicated.
Specifically, Illumina NGS identifies the specific bases using the unique fluorescent signature emitted by the fluorescent tag attached to each of the four possible nucleotides.
After demultiplexing, the output is short read data. I’d subsequently process the raw read data using a metagenomic analysis pipeline to identify specific taxa and species present in the original soil solution sample.
Part 5.2: DNA Write
(i) I’d like to synthesize DNA for a genetic circuit that regulates bacterial membrane protein pumps depending on external pH as part of my SoilBuddy MVP. I’d choose a set point optimized for food crops such as corn.
(ii)
I’d use Gibson Assembly to put my genetic circuit together given the simplicity and reliability of the protocol. First, I’d identify appropriate restriction enzyme combinations for the BioBricks parts I’ll require. Next, I’ll pick two BioBricks parts at a time for insertion into the target plasmid.
Each BioBricks part and the vector will be digested in respective reaction chambers with the appropriate restriction enzymes, followed by thermal denaturation (and thus inactivation) of the restriction enzymes. Thereafter, the digested BioBricks parts and vector will be incubated together in molar amounts designed to optimize formation of the target construct along with a DNA ligase. After transformation into a bacterial platform, selection, amplification and extraction, repeat the process till all the BioBricks parts are incorporated within the recombinant plasmid vector.
Needless to say, the process is extremely time-intensive due to the multiple, repetitive transformations and amplifications involved. While laboratory automation could allow the process to scale, it would be very resource-intensive. Lastly, while each particular step might be lossy, regular purification, selection and verification steps would ensure the desired vector is formed at the end of the day.
Part 5.3: DNA Edit
(i) One possible development of SoilBuddy would be a process rather than an organism that transforms native soil microbiota to enhance their salutary functions (eg nitrogen-fixing, pH buffering). To that end, I would like to edit prokaryotic bacteria and eukaryotic fungal DNA considering the particular niche of commensal microbiota near plant roots. This would involve, mainly, gene insertion to introduce novel gene products or base edits to enhance existing regulatory mechanisms in the microorganisms.
(ii)
Given the range of target systems (both prokaryotic and eukaryotic) as well as payloads desired (ie. novel genes and edits), I would leverage the versatility of CRISPR. Taking a CRISPR-based gene knock-in of yeast as an example, we first design a DNA sequence encoding a gRNA that recognizes our target insertion site and has minimal off-target effects. Then, we select a yeast plasmid with an antibiotic selection marker and which contains a sequence encoding Cas9 endonuclease. Using Gibson assembly, we first digest the vector and gRNA-encoding sequence with appropriate restriction enzymes, then ligate the sequence into the plasmid using DNA ligase, transform the recombinant vector into competent yeast cells, select and amplify the plasmid through incubation.
We prepare a donor template containing the gene to be inserted and transform the exogenous donor template into yeast. Now, the yeast contains Cas9 endonuclease from expression of the plasmid, which hybridizes with gRNA (also expressed from transcrption of the plasmid DNA) to form ribonucleoprotein complex. The crRNA in the gRNA binds to the recognition site before Cas9 domain activity causes a double-stranded break in the yeast DNA. Thereafter, the donor template provides for Homology-Directed Repair that knocks in the intended gene.
The preperatory work involves designing the gRNA, selecting yeast cells that are competent and do not suffer from damage to their Homology-Directed Repair mechanism, and selecting a donor template incorporating a reporter (eg GFP) such that unintended edit products are minimized. At the same time, edited yeast cells have to be selected due to the low efficiency of gene knock-in through fluorescence-based flow-cytometry for example.
While a well-designed gRNA and donor template will minimize indels, off-target edits and maximize the chances of successful gene knock-in, statistically speaking, the odds of a cell being successfully edited are low. Hence, care needs to be taken in selecting for edited cells, which can then be expanded to counteract the low efficiency of knock-in
Week 3 HW: Lab Automation
Part 1
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
In summary, the researchers used an Opentrons OT-2 robot to automate an assay used to test the safety-efficacy profile of an antibiotic. They accomplished this by programmatically preparing a serial dilution of the antibiotic candidate across multiple 96-well plates with replicates, innoculating the wells with the test bacteria, then measuring the optical density of each well with a plate reader to determine bacterial viability after the antibiotic challenge.
The method outlined in the paper minimizes human error, standardizes the innoculation pattern used for each plate well and allows for a massive range of antibiotic concentrations, as well as antibiotics to be tested in parallel. This allows for massive improvements in thoroughput, accuracy and replicability of results.
Part 2
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
Considering my main plan, SoilBuddy, lab automation serves primarily to augment the speed at which my experiments may be run, and allow for overlapping scheduling to optimize lab time.
There are a few key steps where lab automation will come in handy:
Building: plasmid digestion, transformation, and selection of bacteria
Testing: Testing the performance of the reporter system against multiplexed stimulus conditions
Building
Roughly, here are the steps I’ll be following along with the corresponding pieces of lab equipment required for each:
(if the plasmid isn’t ordered with gene construct and genetic switch incorporated) ATC Thermal cycler for PCR amplification of casette, Plate incubator for plasmid digestion
Plate incubator for heat shock transformation
Plate incubator for bacterial recovery, expansion and selection
Testing
Roughly, here are the steps I’ll be following along with the corresponding pieces of lab equipment required for each:
Opentrons OT-2 for preparing serial dilutions of N2 agent and pH
Opentrons OT-2 for preparing different innoculations of culture and replicate plates
Spark plate reader for reading out reporter results
Week 4 HW: Protein Design Part I
Part A
Number of amino acids = 500g/(100 Da) = 5 mol = 3.0 x 10^24 molecules of amino acids (approx).
Over the course of digestion, the polynucleotides are broken down into constituent nucleobases prior to absorption.
Considering the degeneracy of codons, these 20 amino acids were sufficient to produce a diversity of proteins, and were evolutionarily conserved due to a lack of strong selection pressure against them (ie. they were “good enough”).
.
They could’ve formed abiotically given the presence of organic precursors such as carboxylic acids, amines and small molecule side groups, catalyzed by zeolites, and triggered by lightning and heat on the primordial earth or extraterrestrial asteroids respectively.
Left-handed spirals.
Yes, some examples are $\pi$ helices and $3_{10}$ helices.
It’s more energetically stable given L-amino acids and D-saccharides predominate natural protein helices and the sugar-phosphate backbone of polynucleotides respectively.
In aqueous environments, the hydrophobic portions of beta sheets are driven together by steric interactions while the hydrophilic groups facilitate relatively strong intermolecular hydrogen bonding.
Beta sheets allow for strong intermolecular bonding, in hydrophobic zippers, that renders misfolded proteins more stable than their functional conformations and facilitates the addition of more proteins to the aggregate fibril - the biochemical basis of amyloid pathogenesis. Yes, you could make materials out of amyloid beta sheets but they’d only be stable under aqueous conditions and would be potential biotoxins.
.
Part B
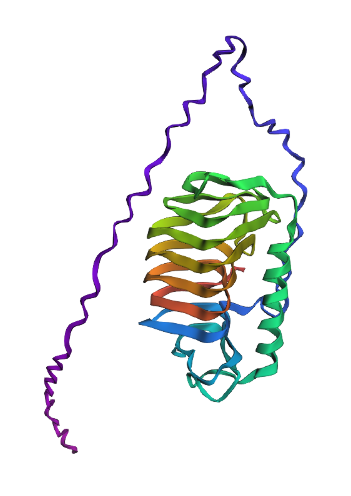
I chose to investigate an antifreeze protein from an arctic bacterium as a potential solution for winter snow-clearing.
The sequence, as obtained from UniProt is as follows:
It is 280 residues long. Threonine is the most common AA residue, appearing a cumulative 33 times.
There are 250 homologs from across the tree of life.
It belongs to the ice-binding protein superfamily.
The structure was modelled in Apr 2014. The model is high-resolution, providing details down to 2.10Å. There are no non-protein components in the final protein structure. It belongs to the ice-binding protein superfamily
Cartoon View
Ribbon View
Ball-and-Stick View
Labelling by Secondary structure
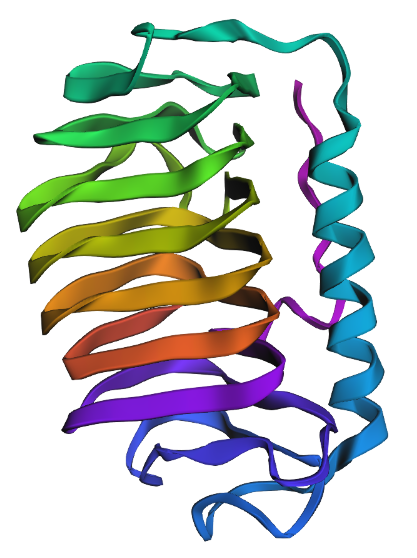
The protein has more sheets than helices, though the helices are substantially larger.
Labelling by residue hydropathy.
Blue represents hydrophilicity while red represents hydrophobicity
Hydrophobic residues are encountered sparingly, on outward-oriented branches, while hydrophilic residues are found on the main beta sheets accessible to the protein’s surroundings.
Surface View
There are a few pockets for water to bind to.
Part C
C1
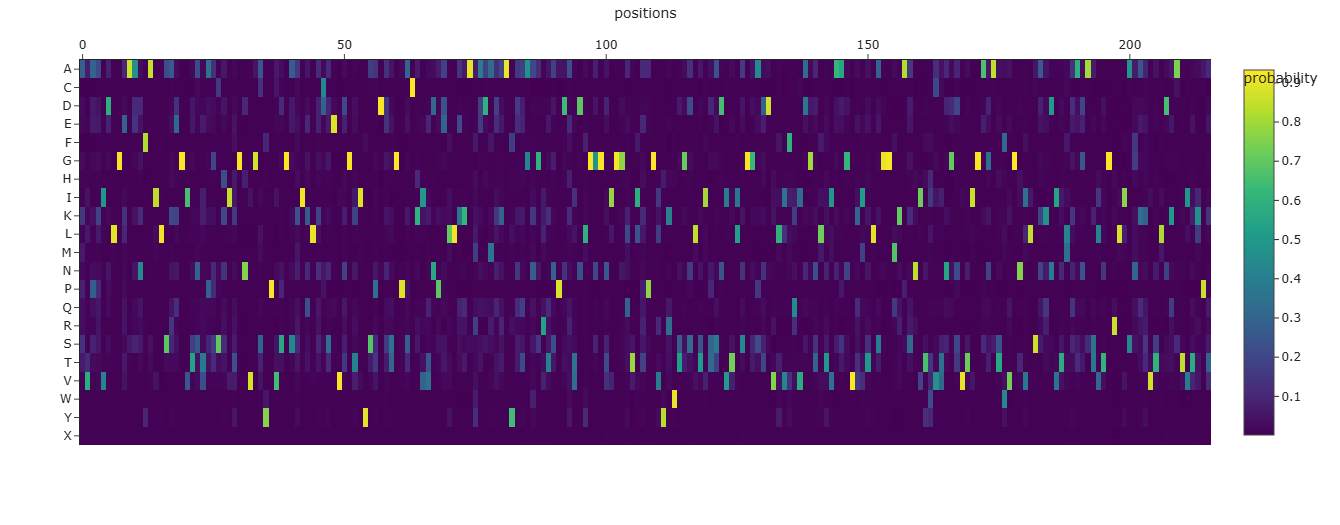
Deep Mutational Scans
Mutational Heat Map
I think the bright bands which occur at regular intervals signify residues involved in linking helices together since they offer some flexibility in binding nucleation sites.
Latent Space Analysis
Yes, they tend to be homologs
C2
Folding
It largely matches the original structure including the spiral linking several beta sheets and a large alpha helix. However, it fails to model the hydrophobic side branches of the protein separately and instead connects them using a polypeptide chain.
No, it is quite sensitive to missense mutations. This comports with the earlier heatmap of mutational scans.
C3
Inverse Folding
Comparison of Structures
Original:
Modified Sequence:
In comparison to the original, the predicted structure of the modified protein retains the broad helix and beta sheet spiral structure but its beta sheet spiral is misshapen and the hydrophobic branches are overlarge. Given the difference between experimentally-derived structure and computational folding prediction for the original protein, I would believe that this new sequence would fail to demonstrate ice-binding behavior in a biological environment.
D
Protein Engineering Goals
Enhance the thermal stability of MS2-L peptide derived from the ssRNA Leviviridae phage
Protein Engineering Workflow
Use Protein Language Models to conduct high-thoroughput in silico mutation of the protein
Grade mutants’ thermal stability using Rosetta
Carrying forward the top quintile of mutants as seeds for the next iteration, repeat the process till increments in stability are minimal
Week 5: HW Protein Design Part II
Part A
Part 1: Generate Binders with PepMLM
Binder Sequence
Perplexity Score
WRYYATVARHKE
15.38014901
WLYYVVVLRHGE
32.45105037
WRYYAAGARLKE
11.75101427
WRYYATAVELKG
10.44080960
Part 2: Evaluate Binders with AlphaFold3
Peptide ID
Binder Sequence
ipTM Score
Location
Peptide 0
WRYYATVARHKE
0.35
Approaches the $\beta$ barrel; surface-bound
Peptide 1
WLYYVVVLRHGE
0.33
Approaches the $\beta$ barrel; surface-bound
Peptide 2
WRYYAAGARLKE
0.35
Approaches the $\beta$ barrel; surface-bound
Peptide 3
WRYYATAVELKG
0.22
Conforms to the dimer interface; binds to a pocket
Control
FLYRWLPSRRGG
0.31
Surrounding the $\beta$ barrel; surface-bound
While peptides 0-2 exceed the control (known binder)’s ipTM scores, indicating stronger protein-protein interaction, they still represent low confidence values. I would not conclude that they produce stronger protein-protein interactions for use as potential molecular glues.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide ID
Binder Sequence
Predicted Binding Affinity
Solubility
Hemolysis Probability
Net Charge (pH 7)
Molecular Weight
Peptide 0
WRYYATVARHKE
5.659 pKd/pKi
1.000
0.034
1.85
1579.8 Da
Peptide 1
WLYYVVVLRHGE
6.562 pKd/pKi
1.000
0.158
-0.15
1533.8 Da
Peptide 2
WRYYAAGARLKE
5.878 pKd/pKi
1.000
0.042
1.77
1483.7 Da
Peptide 3
WRYYATAVELKG
5.785 pKd/pKi
1.000
0.060
8.50
1456.6 Da
While there isn’t enough of an ipTM range to draw meaningful conclusions regarding binding affinity, I would think that structural conformation lends itself, by necessity, to the strength of protein-protein interactions. Peptide 1 actually had the highest hemolysis probability and the lower pH; on that note, peptide 3 makes for the best therapeutic candidate considering its net charge at pH 7 and solubility assists in drug delivery while its binding affinity renders it efficacious as a therapeutic target.
Part 4
KKCGKQFKQKQE
This is vastly different in terms of conformation and osmolarity as compared to the other peptides. I would request representative lab samples of the protein and conduct initially binding efficiency assays using MS before advancing either as a candidate.
Part B
Part C
Protein Design Process Overview
We’ll be utilizing random mutagenesis to generate variants of L-protein optimized for both (1) Lysis Activity and (2) Expression Levels. We begin with the ground truth of the experimental data provided, generate combinations of mutations and test them usign Af2-multimer.
Mutation Set
We filter mutations by excluding nonsense mutations and prioritizing missense mutations which enhance both lytic activity and expression levels. Frankly, the synergistic effects of different mutations is not a straightforward problem to solve, so we resort to a brute force attack on different possible combinations of mutations in L protein that affect its binding to DNAJ. In an ideal world, we would create a performance score that assessed its lytic activity and expression as well, but existing models wouldn’t encode these properties especially for a small transmembrane protein.
Mut #
Amino Acid Position
Amino Acid Change
0
13
P->L
1
15
S->A
2
18
R->G
3
18
R->I
4
30
R->Q
5
30
R->L
6
31
R->I
7
44
L->P
8
45
A->P
9
46
I->F
Mathematically, there are 26*32=576 possible sets of mutations that can be made.
Evaluation
In the absence of reliable lysis and expression level metrics, we resort to DNAJ:L Protein binding affinity as predicted by Af2-Multimer instead.
Results
Mutations
iptm value
0_1
0.152 $\pm$ 0.02
0_2
0.16 $\pm$ 0.02
0_3
0.148 $\pm$ 0.02
0_4
0.152 $\pm$ 0.02
0_5
0.144 $\pm$ 0.02
0_6
0.168 $\pm$ 0.02
0_7
0.17 $\pm$ 0.02
0_8
0.178 $\pm$ 0.02
0_9
0.208 $\pm$ 0.04
1_2
0.15 $\pm$ 0.02
1_3
0.152 $\pm$ 0.02
1_4
0.154 $\pm$ 0.03
1_5
0.146 $\pm$ 0.01
1_6
0.168 $\pm$ 0.03
1_7
0.178 $\pm$ 0.03
1_8
0.172 $\pm$ 0.02
1_9
0.178 $\pm$ 0.02
2_4
0.146 $\pm$ 0.02
2_5
0.146 $\pm$ 0.02
2_6
0.156 $\pm$ 0.02
2_7
0.164 $\pm$ 0.03
2_8
0.174 $\pm$ 0.03
2_9
0.196 $\pm$ 0.04
3_4
0.136 $\pm$ 0.02
3_5
0.144 $\pm$ 0.02
3_6
0.156 $\pm$ 0.03
3_7
0.136 $\pm$ 0.02
3_8
0.142 $\pm$ 0.02
3_9
0.144 $\pm$ 0.02
4_6
0.146 $\pm$ 0.03
4_7
0.14 $\pm$ 0.02
4_8
0.144 $\pm$ 0.02
4_9
0.14 $\pm$ 0.02
5_6
0.146 $\pm$ 0.03
5_7
0.14 $\pm$ 0.02
5_8
0.144 $\pm$ 0.02
5_9
0.14 $\pm$ 0.02
6_7
0.142 $\pm$ 0.01
6_8
0.142 $\pm$ 0.02
6_9
0.14 $\pm$ 0.02
7_8
0.14 $\pm$ 0.02
7_9
0.14 $\pm$ 0.02
8_9
0.152 $\pm$ 0.02
Thus, METRFPQQSQQTLASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT (mut #0 and #9) is identified to be the “best” L-protein
Week 6 HW: Genetic Circuits Part I
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The NEB 2X master mix includes engineered thermostable DNApol to synthesize complimentary strands during the extension phase of PCR, with low error rate and high expressivity to enable faster, more accurate DNA replication.
It includes a buffer to maintain optimum pH for enzymatic catalysis and prevent the denaturation of enzyme or nucleic acids, as well as ${Mg}{Cl}_{2}$ to provide $Mg^{2+}$ as a cofactor for DNApol.
What are some factors that determine primer annealing temperature during PCR?
Primer sequence (specifically, the number of complimentary base pairs and how many are C-G as opposed to A-T): If a primer sequence contains more complimentary base pairs (eg because the forward primer is longer, say, or has higher sequence homology with the sense strand) the annealing temperature will be higher. If the primer contains more CG as opposed to AT content, the annealing temperature will be higher.
Buffer: A higher concentration of salt can lead to elevated ion levels, favoring the salting out of proteins and DNA which might, to some extent, facilitate annealing at a lower temperature.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both PCR and Restriction Enzyme Digests produce linear dsDNA and can be highly specific for the target region if designed properly.
However, while PCR aims to amplify a target sequence, restriction enzyme digests aim to isolate a target sequence from a given amount of DNA sample. PCR involves thermocycling while restriction enzyme digests are conducted at a constant optimum temperature. PCR requires DNA primers while restriction enzyme digests do not require additional DNA material on top of the sample itself.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Firstly, we can check their purity using a 260/280 absorbimetry assay, and verify the predominant fragment length using gel electrophoresis.
Secondly, we can use Sanger sequencing the verify the sequence of linear DNA produced.
How does the plasmid DNA enter the E. coli cells during transformation?
Either through chemical means in heat shock treatment, or electrical means in electroporation, the bacterial cell membrane and cell wall are modified to grant plasmid DNA greater permeability - whether through the alteration of membrane thickness, structure and charge in chemical competence, or the electrical stress-induced poration of the bacterial cell membrane in electroporation.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly relies on Type IIS as opposed to Type IIP restriction enzymes to enable one-step syntheses of multipart constructs. In Golden Gate Assembly, the specific type of restriction enzymes used doesn’t depend on palindromic recognition sequences, allowing greater versatility in engineering restriction sites, and doesn’t leave parts of the restriction site in the final construct.
In Golden Gate Assembly, the respective genetic components are first checked to ensure they don’t contain any internal Type IIS recognition sites, and a unique 4-base overhang is engineered to dictate the correct order of assembly. Afterwards, the genetic components are added to a one pot reaction mixture containing the specific Type IIS restriction enzyme used, DNA ligase and buffer solution.
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
^ See above
Model this assembly method with Benchling or Asimov Kernel!
Sure thing!
Assignment: Asimov Kernel
Refer to this notebook for the constructs and repressilator
Lab Homework
I’m a remote committed listener so don’t have access to a physical lab
Week 7 HW: Genetic Circuits Part II
Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs more easily provides for graded responses and can scale intelligent responses; they can approximate every mathematical function and not just step functions.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
IANNs could classify the cell surface of tumor cells and initiate the secretion of the best paracrine messengers to recruit NK and Tc cells. The input would be the MHC of the target cell and the IANN would be a classifier whose output would be one of various preprogrammed cellular responses. The IANN would need to select a particular cellular response robustly without being modified.
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium-based styrofoam, myco-bricks and myco-leather: that’s better than existing styrofoam, bricks and leather because it’s biodegradable, carbon-lighter and cruelty-free.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I’d like to engineer them as an alternative protein source and biofoundry. Fungi are eukaryotic, enabling native eukaryotic gene regulation and post-translational modification, can be grown off of waste material and fungi have much higher translational efficiency.
First DNA Twist Order
lux operon engineered to function as an AND genetic circuit
Week 9 HW: Cell-Free Systems
Homework Part A
Assignment
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production.
It lacks the stress response, proteostasis and detoxification of an in vivo environment, allowing for much higher expression of protein including toxic varieties or those incorporating xenobiotic compounds, as in bio-orthogonal experiments. Reaction conditions are standard and can be externally controlled/varied (eg temperature) more precisely than in in vivo systems. The progress of reaction is more easily followed using imaging techniques, allowing for rapid optimization and sequential syntheses. Two cases where cell-free expression is better than cell production are the expression of protein toxic to bacterial/fungal chassis (eg antibacterial/antifungal compounds) as well as proteins intended for use in human medicine (free of endotoxins).
2. Describe the main components of a cell-free expression system and explain the role of each component.
Cell Extract (specifically, transcription and translation machinery): Allow for the transcription and translation of DNA input
ATP/GTP as well as phosphorylative regeneration system: Energy carrier drives the expression of protein
Nucleotide bases (ie NTPs): For transcription of DNA to form mRNA
Nucleic acid template: Encode protein/construct to be expressed
Buffer solution: Provide necessary salts, cofactors and buffers in an isosmotic system for optimal protein expression
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Cell-Free systems do not natively provide for substrate-level or oxidative phosphorylation required for ATP/GTP regeneration. ATP/GTP in turn is required for the endergonic processes involved in expression (eg GTP-driven ribosomal translation of primary transcript; ATP-driven tRNA aminoacetylation). I would include excess creatine phosphate to function as a Pi donor and creatine kinase to catalyze the phosphorylation of ATP/GTP in the cell-free system buffer solution.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free expression systems have higher yields of final product, are faster and less complex than eukaryotic cell-free expression systems, though the latter enable the production of larger, more complex proteins complexed with cofactors and coenzymes.
Consider the production of GFP in a prokaryotic system: simultaneous transcription and translation means multiple GFP polypeptides may be synthesized in parallel, while the autocatalytic folding of the protein removes the need for post-translational modification or energetically costly chaperonins.
Now consider, alternatively, the production of selectin in a eukaryotic system: given the glycoprotein nature of selectin, after translation yields the primary structure of selectin, it undergoes post-translational modification to have a glycosidic component linked chemically.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
After identifying the key components of the reaction mix and how they might impact expression (eg the relative concentrations of ions in the buffer, buffer pH, relative concentrations of GTP/ATP and components of the phosphorylative regeneration system), I would vary these conditions across a 96-well plate in triplicate and incubate the plate under constant conditions. At a fixed endpoint, I would quench the reaction and use spectrophotometry to determine the respective expression efficiency in each well, and thus the reaction conditions which produced the highest protein expression levels.
One challenge could be the imprecision of reaction mix formulation. I would use lab automation to reduce variation in reaction mix preparation and minimize error.
Another challenge could be a lack of standard lysate. I would prepare a large enough stock of lysate prior to experimentation.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Poor Quality of DNA template: Unoptimized codons and endonuclease contamination could reduce the efficiency of transcription and translation.
Troubleshooting: Using the same preparation and reaction mix, substitute a known optimized plasmid for the DNA template provided.
Insufficient ATP/GTP regeneration: ATP/GTP are rate-limiting due to the slow rate/insufficient regeneration
Troubleshooting: Repeat the experiment with an excess of creatine phosphatase and kinase
Insufficient cofactors: cofactors such as ${Mg}{2+}$ are rate-limiting due to a low concentration in reaction buffer which reduces the rate of formation of holoenzymes required for transcription and translation
Troubleshooting: Repeat the experiment with an excess of cofactors (eg. ${Mg}{2+}$) added to the reaction mix.
Homework question from Peter
Freeze-dried cell free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field - Architecture, Textiles/Fashion, or Robotics, and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
* Write a one-sentence summary pitch sentence describing your concept.
Imagine a smart layer you could add to windows to filter out UV rays in sunlight but which self-healed and wasn’t subject to photo-degradation.
* How will the idea work, in more detail? Write 3-4 sentences or more.
A cell-free system incorporated in a hydrogel will express photoprotective protein (eg. lycopene) under the regulation of a microfluidic logic chip. The logic chip could be hard-wired for a particular region’s climate, or incorporate a microprocessor to allow real-time updates from a photosensor. The lycopene itself will be degraded in the sunlight, but offer bespoke UV protection which compared to traditional UV films will be regenerable, allow vitamin D-promoting wavelengths and the natural warmth of sunlight through, and allow fine customization of which wavelengths of sunlight get filtered out.
* What societal challenge or market need will this address?
This would help minimize energy used for cooling as the world becomes more urbanized and meet the need for window conditioning in construction.
* How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Within the hydrogel film, the raw materials (eg rainwater) will be collected from the environment itself and stored in reservoirs which the microfluidic logic then transports to a well-protected reaction core housing the cell-free system proper.
Homework question from Ally
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
The risk of illness in long-duration human spaceflight is unconscionable; not being able to diagnose and consequently treat mystery illnesses only more so. I aim to provide a low-cost battery of diagnostics to rapidly identify pathogens, with strong translational potential for rural healthcare delivery on Earth.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Metabolomics.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Clearly state your hypothesis or research goal and explain the reasoning behind it (Maximum 150 words)
All pathogens carry metabolomic signatures which specifically identify strains, types and serotypes. My approach allows for a broad-range of pathogens to be identified without heavy, high-maintenance laboratory equipment.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I’ll use 3 samples of E. coli, T4 Phage and C. elegans to simulate bacterial, viral and nematodic pathogens in a BSL-2 environment. DI will be a -ve control. My experiment will involve the multi-repeat testing of a cell-free metabolomic screening system across serial dilutions of pathogen titers to establish diagnostic specificity and sensitivity.
z=32 based on consecutive m/z’s (875.4421, 848.9162); MW = 27982Da; Accuracy = 99.9%
No, its amplitude above background could mean it’s not reliably distinguished from background noise.
Waters Lab Part II
When a protein unfolds, several more ionizable sites on the polypeptide are exposed. As such, the denatured protein will exhibit several more m/z peaKs, and typically at higher m/z values than the native protein conformation.
The charge state is +0 based on the maximal abundance of the protein.
Theoretical Observed/measured on the Intact LC-MS: 28006
PPM Mass Error: 857ppm
Molecular weight (kDa): 28
Week 11 HW: Building Genomes
Part A
I enjoyed the sense of oneness and how it spoke to the power of science to transcend divisions of identity and geography
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
E. coli Lysate: Provides biological medium and machinery (eg ribosomes)
Salts/Buffer: Optimal pH, ionic atmosphere and osmolarity
Energy / Nucleotide System: Power transcription/translation
Translation Mix (Amino Acids): Allow for translation to produce mRNA from DNA
Additives: Nicotinamide in particular allows for the regeneration of NAD
Backfill: Maintain appropriate volume for manipulation of the biological systems
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
sfGFP: Maturation time - 13.6min
mRFP1: Maturation time - 60.0min
mKO2: Maturation time - 108.0min
mTurquoise2: Maturation time - 33.5min
mScarlet_I: Maturation time - 174.0min
Electra2: Lifetime - 3.4ns
Increasing the concentrations of AMP, UMP, CMP and GMP across all the proteins will enhance the total fluorescence over the incubation period by increasing the rate of reaction at the rate-limiting-step of translation.