Week 4 HW: Protein Design Part I
Part A
- Number of amino acids = 500g/(100 Da) = 5 mol = 3.0 x 10^24 molecules of amino acids (approx).
- Over the course of digestion, the polynucleotides are broken down into constituent nucleobases prior to absorption.
- Considering the degeneracy of codons, these 20 amino acids were sufficient to produce a diversity of proteins, and were evolutionarily conserved due to a lack of strong selection pressure against them (ie. they were “good enough”).
- .
- They could’ve formed abiotically given the presence of organic precursors such as carboxylic acids, amines and small molecule side groups, catalyzed by zeolites, and triggered by lightning and heat on the primordial earth or extraterrestrial asteroids respectively.
- Left-handed spirals.
- Yes, some examples are $\pi$ helices and $3_{10}$ helices.
- It’s more energetically stable given L-amino acids and D-saccharides predominate natural protein helices and the sugar-phosphate backbone of polynucleotides respectively.
- In aqueous environments, the hydrophobic portions of beta sheets are driven together by steric interactions while the hydrophilic groups facilitate relatively strong intermolecular hydrogen bonding.
- Beta sheets allow for strong intermolecular bonding, in hydrophobic zippers, that renders misfolded proteins more stable than their functional conformations and facilitates the addition of more proteins to the aggregate fibril - the biochemical basis of amyloid pathogenesis. Yes, you could make materials out of amyloid beta sheets but they’d only be stable under aqueous conditions and would be potential biotoxins.
- .
Part B
I chose to investigate an antifreeze protein from an arctic bacterium as a potential solution for winter snow-clearing.
The sequence, as obtained from UniProt is as follows:
- It is 280 residues long. Threonine is the most common AA residue, appearing a cumulative 33 times.
- There are 250 homologs from across the tree of life.
- It belongs to the ice-binding protein superfamily.
The structure was modelled in Apr 2014. The model is high-resolution, providing details down to 2.10Å. There are no non-protein components in the final protein structure. It belongs to the ice-binding protein superfamily
Cartoon View

Ribbon View 
Ball-and-Stick View 
Labelling by Secondary structure 
The protein has more sheets than helices, though the helices are substantially larger.
Labelling by residue hydropathy.
Blue represents hydrophilicity while red represents hydrophobicity 
Hydrophobic residues are encountered sparingly, on outward-oriented branches, while hydrophilic residues are found on the main beta sheets accessible to the protein’s surroundings.
Surface View 
There are a few pockets for water to bind to.
Part C
C1
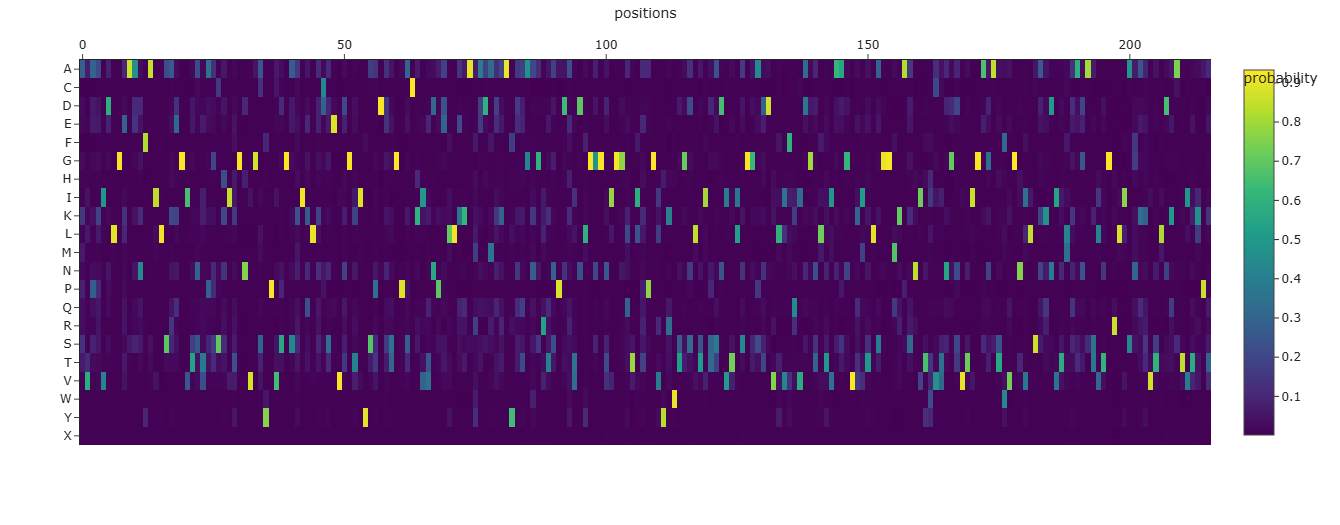
Deep Mutational Scans
Mutational Heat Map

- I think the bright bands which occur at regular intervals signify residues involved in linking helices together since they offer some flexibility in binding nucleation sites.
Latent Space Analysis
- Yes, they tend to be homologs
C2
Folding 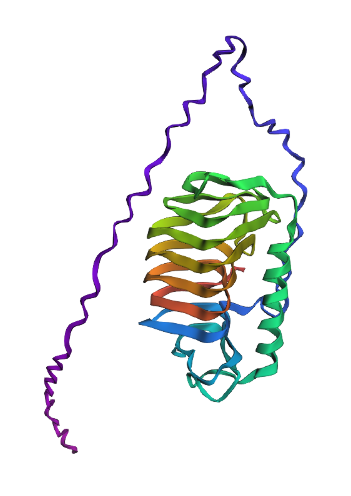
- It largely matches the original structure including the spiral linking several beta sheets and a large alpha helix. However, it fails to model the hydrophobic side branches of the protein separately and instead connects them using a polypeptide chain.
- No, it is quite sensitive to missense mutations. This comports with the earlier heatmap of mutational scans.
C3
Inverse Folding
Comparison of Structures
Original:
Modified Sequence: 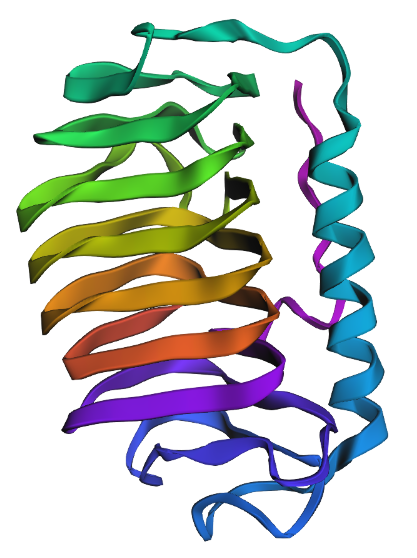
In comparison to the original, the predicted structure of the modified protein retains the broad helix and beta sheet spiral structure but its beta sheet spiral is misshapen and the hydrophobic branches are overlarge. Given the difference between experimentally-derived structure and computational folding prediction for the original protein, I would believe that this new sequence would fail to demonstrate ice-binding behavior in a biological environment.
D
Protein Engineering Goals
- Enhance the thermal stability of MS2-L peptide derived from the ssRNA Leviviridae phage
Protein Engineering Workflow
- Use Protein Language Models to conduct high-thoroughput in silico mutation of the protein
- Grade mutants’ thermal stability using Rosetta
- Carrying forward the top quintile of mutants as seeds for the next iteration, repeat the process till increments in stability are minimal