Project Description I want to develop a synthetic assymetrical-cell-division system to enable synthetic cell differentiation; as a toolkit for engineer multicellular organization, development, pattern formation and others. To futher detail, see the page of the final project
The project have two major subprojects:
A mitotic counter: a circuit capable of counting as states how many cell division the linage have sense since the system has been activated (firts division, second, third, and so on). The system uses the natural fluctuations of the cell’s cycle regulators to induce or activate distinct proteins. When the system is activated, a TF would be activated by an cell-cycle-dependent kinase (at the G1 phase), that would induce the expression of a recombinase, that would inverse the sequence of its own promoter, inducing the expression of an inactivated, second, TF. This TF2 would be activated via a second cell-cycle-dependent kinase, one that would be activated in the final of the cell cycle and not be expressed at the same time with the firts kinase. The phosphorylation upon TF2 would, for example, link two homodymers together, in an activated form (an option, but not necessarily how it would work). When the kinase 2 activates TF2, gene expression would not be available because of chromatin condesation upon mitosis. After the cell divides, at G1, TF2 could induce the activation of other genes, that, in consenquence and, using the architecture, enables the activation of other genes at the second cell division, and so on, making possible to count cell division in the cell lineage. The system also needs other things such as a degradation system, repression for past states, etc. Assymetrical component: ¿How can we engineer an assymetrical distribution of molecules to induce the assymetrical cell division? It should be transferable to other organisms. The proposed one is to used an synthetic phased segregated condensate that is capable of create one individual and stable condensate upon the cell. It should carry an mRNA that would express a TF. It should be formated upon the activation of the scaffolds and dissegregated upon the cell division, enabling the translation of the mRNA selected. The mRNA should be sequestred in the condensate, where its translation-initiation site would be blocked, and it would be protected of degradations by directed nucleases over that mRNA. All of this via linking the sites of union of the scaffolds with this important sites of the mRNA (nuclease recognition site, translation initiation site, rybozome binding site, etc). The scaffolds should be inactivated upon cell division via phosphorylation at the binding sites, liberating the mRNA. But how we can make a single condensate exist? The idea is to use the pyrenoid (a rubisco aggregate condensate that makes CO2 fixation more efficient) as an example for this, where, in a dynamics between phosphatases and kinases, the algae maintains the pyrenoid as a single condensate or multiple when needed. ¿So how all of this is going to work in order to make the assymetrical cell division system function?
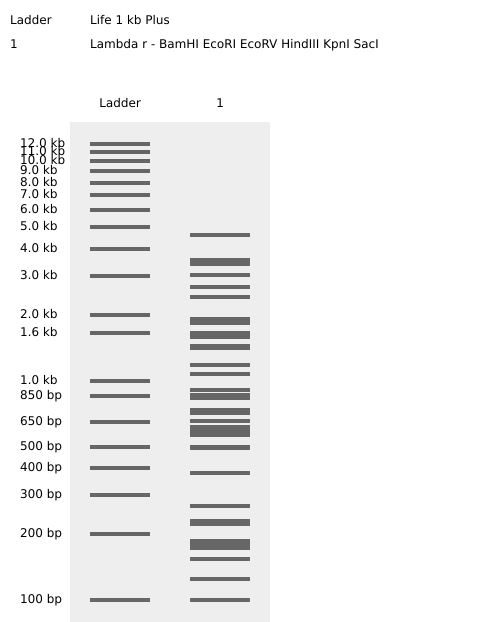
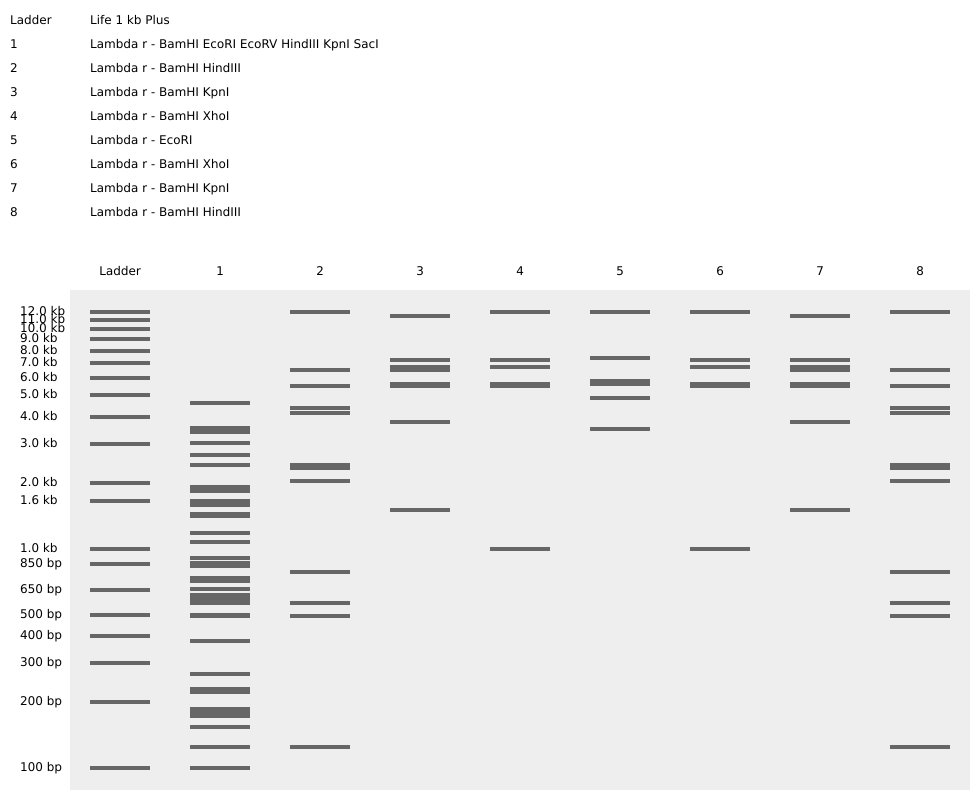
Homework Part 1: Benchling and in-silico gel art This is the virtual digestion of the Lambda genome with this restriction enzymes: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI and SalI.
Homework Part A. Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang: Why do humans eat beef but do not become a cow, eat fish but do not become fish? Because eating beef doesn´t make us incorporate the genetic information that make a cow, and even if we could, we don´t have machinery to process that information and make us a cow.
Homework Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM 1- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Assignment: DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The high Phusion High-fidelity polymerase, MgCl2, dNTPs, etc. The polymerase enable low error in the polymerization reaction.
What are some factors that determine primer annealing temperature during PCR? how much Hidrogen bombs are present (type of bases and how much).
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? It enable non-linear threshold computation over the concentration of the molecules.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal. An useful application could be the development of responses upon specific thresholds of morphogens, for long-space tissue patterning (and cell-fate determination).
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis enable rapid protein production without the need to wait for traditional transformation and growth. This fast prototyping enable fast test of multiple genes at the same time, genetic parts, variants, and other kind of libraries. Also, other variables, such as temperature, concentration of analytes, non-canonical AAs, etc, could be tested easily in CFE. One case could be when characterizing multiple parts in an organism, where making it in CFES would be much more time efficient. Other case could be when making proteins with non-canonical AAs, making it more easy in the CFES.
Waters Part I — Molecular Weight Based on the predicted amino acid sequence of eGFP and any known modifications, what is the calculated molecular weight? The calculated molecular weight of eGFP with the LE linker and His-tag is approximately 27.1 kDa.
Determine z for each adjacent pair of peaks using: Using adjacent peaks around 933.7 and 1000.4 m/z, the charge state is approximately +15.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I couldn´t contribuite to the pixel artwork sadly :(
Next year i become a TA without excuses.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction. E. coli Lysate: Provides the cellular machinery for transcription and translation, including ribosomes, tRNAs, enzymes, and metabolic proteins.
Lab questions Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively? The production of lycopene in E. coli is induced by the genes crtE, crtI, and crtB from Erwinia herbicola, which are carried in the pAC-LYC plasmid. Beta-carotene production requires the same pathway plus the additional crtY gene, present in the pAC-BETA plasmid.
I want to develop a synthetic assymetrical-cell-division system to enable synthetic cell differentiation; as a toolkit for engineer multicellular organization, development, pattern formation and others.
To futher detail, see the page of the final project
The project have two major subprojects:
A mitotic counter: a circuit capable of counting as states how many cell division the linage have sense since the system has been activated (firts division, second, third, and so on). The system uses the natural fluctuations of the cell’s cycle regulators to induce or activate distinct proteins. When the system is activated, a TF would be activated by an cell-cycle-dependent kinase (at the G1 phase), that would induce the expression of a recombinase, that would inverse the sequence of its own promoter, inducing the expression of an inactivated, second, TF. This TF2 would be activated via a second cell-cycle-dependent kinase, one that would be activated in the final of the cell cycle and not be expressed at the same time with the firts kinase. The phosphorylation upon TF2 would, for example, link two homodymers together, in an activated form (an option, but not necessarily how it would work). When the kinase 2 activates TF2, gene expression would not be available because of chromatin condesation upon mitosis. After the cell divides, at G1, TF2 could induce the activation of other genes, that, in consenquence and, using the architecture, enables the activation of other genes at the second cell division, and so on, making possible to count cell division in the cell lineage. The system also needs other things such as a degradation system, repression for past states, etc.
Assymetrical component: ¿How can we engineer an assymetrical distribution of molecules to induce the assymetrical cell division? It should be transferable to other organisms. The proposed one is to used an synthetic phased segregated condensate that is capable of create one individual and stable condensate upon the cell. It should carry an mRNA that would express a TF. It should be formated upon the activation of the scaffolds and dissegregated upon the cell division, enabling the translation of the mRNA selected. The mRNA should be sequestred in the condensate, where its translation-initiation site would be blocked, and it would be protected of degradations by directed nucleases over that mRNA. All of this via linking the sites of union of the scaffolds with this important sites of the mRNA (nuclease recognition site, translation initiation site, rybozome binding site, etc). The scaffolds should be inactivated upon cell division via phosphorylation at the binding sites, liberating the mRNA.
But how we can make a single condensate exist? The idea is to use the pyrenoid (a rubisco aggregate condensate that makes CO2 fixation more efficient) as an example for this, where, in a dynamics between phosphatases and kinases, the algae maintains the pyrenoid as a single condensate or multiple when needed.
¿So how all of this is going to work in order to make the assymetrical cell division system function?
When both systems are activated, the mitotic counter and the phase-condensate system, the single condensate is going to be segregated to any of the son’s cell, and, after the division have been made, the TF2 of the mitotic counter could express a kinase for the dissegregation of the condensate, enabling the translation of the mRNA. Also the system could make possible the use of logic gates, whether both components are present or no, x gene is activated (in the other cell that would have only the TF2 expressed, AND, NOT or OR gates based on this could enable the differentiation in this cell also, taking another fate that the father cells haved. Or even more complex differentiation dynamics). Anyways, the now different cell could express an auto-induced transcription regulator, that, as follows, would induce the differentiation of its own self, and his son’s cells (between other feedbacks).
Also the mRNA TF should repress the component of mRNA degradation.
¿And now what?
Those systems are toolkits for the multicellular engineering dicipline. To directly create organisms, we would need to have an autonomous cell differentiation system, a system that by its own is able to activate itself. The mitotic counter, also, for example could work as a signaling event upon certains cell divisions in the designated cell lineage, to activate cell-to-cell communications upon that moment, and only in that moment for example.
Possibilities are endless and only the imagination is the limit.
Governance Context
Autonomous multicellular systems capable of growth, differentiation, and spatial organization introduce additional biosafety and biosecurity considerations compared to single-cell engineering. Risks include unintended environmental persistence, uncontrolled proliferation, ecological interaction, and potential dual-use of self-assembling biological structures. Governance must therefore balance intrinsic biological containment, institutional oversight, and research feasibility.
Options considered
Option 1 — Mandatory Genetic Containment and Fail-Safe Design Intrinsic safeguards such as auxotrophy, kill-switches, replication limits, and environmental sensitivity.
Option 2 — Project Licensing, Design Review, and Traceability Registration, documentation, biosafety review, strain tracking, training, and incident reporting.
Option 3 — Voluntary Standards and Community Best Practices Non-binding guidelines and self-governance without formal enforcement.
Scoring scale: 1 (low contribution) to 5 (strong contribution).
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
5
4
2
• By helping respond
3
5
2
Foster Lab Safety
• By preventing incident
5
4
3
• By helping respond
3
5
3
Protect the environment
• By preventing incidents
5
4
1
• By helping respond
3
4
2
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
5
• Feasibility?
3
4
5
• Not impede research
2
4
5
• Promote constructive applications
4
5
3
Homework pre HW2:
Answers to Professor Jacobson
DNA replication is carried out by DNA polymerases, which exhibit a raw error rate of approximately 10⁻⁵ errors per base incorporated. However, proofreading activity and mismatch-repair pathways dramatically improve fidelity, reducing the effective error rate to roughly 10⁻⁹–10⁻¹⁰ per base.
Given that the human genome contains about 3 × 10⁹ base pairs, an uncorrected replication process would introduce tens of thousands of mutations per cell division. In practice, only a few or fewer mutations accumulate per division. This discrepancy is resolved through polymerase exonuclease proofreading and post-replication DNA repair systems that detect and correct mismatches before they become permanent mutations.
Because the genetic code is degenerate, most amino acids are encoded by multiple synonymous codons (on average roughly three per amino acid). Consequently, a typical human protein of several hundred amino acids could theoretically be encoded by an astronomically large number of different DNA sequences. In practice, however, only a small subset of these sequences functions efficiently. Constraints include codon bias and tRNA availability, unfavorable mRNA secondary structures, extreme GC content, unintended splice or regulatory signals, reduced translation efficiency, and technical limitations in DNA synthesis or cloning. These factors strongly restrict the number of sequences that yield robust protein expression.
Answers to Professor LeProust
The most widely used method for oligonucleotide synthesis is solid-phase phosphoramidite chemical synthesis. In this approach, DNA is assembled one nucleotide at a time on a solid support through automated cycles of nucleotide coupling, washing, and deprotection. This chemistry has remained the industry standard for decades because it is fast, scalable, and reliable for producing short DNA fragments.
Oligos longer than approximately 150–200 nucleotides are difficult to synthesize efficiently because each coupling step has slightly less than perfect efficiency. A small fraction of strands fails to extend at every cycle, and these losses accumulate exponentially as the number of cycles increases. As a result, the final mixture contains many truncated or error-containing products, while the yield of full-length DNA becomes very low. Additional chemical side reactions and base damage further degrade quality.
For this reason, a 2000 base-pair gene cannot be synthesized directly. The cumulative inefficiency across thousands of addition steps makes the probability of obtaining a correct full-length product essentially zero. Instead, long genes are constructed by synthesizing many shorter oligonucleotides and assembling them enzymatically using methods such as PCR assembly or Gibson assembly.
Answer to Professor Church
In most animals, including humans, nine amino acids cannot be synthesized de novo and must be obtained from the diet: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. Some sources include arginine as conditionally essential, particularly in growing organisms, yielding a list of ten.
Because lysine is already universally essential, engineering a synthetic “lysine contingency” does not create a fundamentally new biological vulnerability. Instead, it exploits an existing metabolic dependency. Organisms would already require external lysine, so forcing supplementation simply makes this requirement explicit rather than introducing a novel Achilles’ heel. Consequently, such a contingency is predictable and limited as a containment strategy rather than uniquely robust.
Week 2 HW: DNA read, write and edit
Homework
Part 1: Benchling and in-silico gel art
This is the virtual digestion of the Lambda genome with this restriction enzymes:
EcoRI,
HindIII,
BamHI,
KpnI,
EcoRV,
SacI and
SalI.
And here is some artwork:
Part 3: DNA design challenge
3.1- I have choose for the DNA nucleotidyllexotransferase, because it is used in the enzymatic DNA synthesis process, and been able to engineer it could conduct to synthesize DNA in vivo, thing that would make advance the field tremendously.
reverse translation of sp|P04053|TDT_HUMAN DNA nucleotidylexotransferase OS=Homo sapiens OX=9606 GN=DNTT PE=1 SV=3 to a 1527 base sequence of most likely codons.
3.3- Codon optimazed for Saccharomyces cerevisiae:
ATGGATCCACCAAGAGCATCACATTTGTCCCCAAGAAAAAAAAGACCTAGGCAAACTGGTGCATTAATGGCTTCCTCTCCACAGGACATTAAATTCCAAGACTTAGTCGTTTTTATCCTCGAAAAAAAGATGGGTACTACAAGAAGAGCTTTTCTCATGGAATTGGCTAGACGTAAAGGTTTCAGAGTTGAAAATGAACTCAGTGATTCTGTTACACATATTGTTGCTGAAAATAATTCTGGTTCAGATGTTTTGGAATGGTTGCAAGCTCAAAAAGTTCAAGTCTCTTCTCAACCAGAATTGTTAGATGTCAGTTGGTTAATAGAATGCATTAGAGCTGGTAAGCCAGTTGAAATGACTGGCAAACATCAATTAGTAGTAAGAAGAGATTACTCAGATTCAACAAATCCAGGTCCACCAAAAACTCCACCAATAGCTGTTCAAAAAATTTCTCAATACGCTTGTCAACGTAGGACTACTTTAAATAATTGTAATCAAATTTTTACAGATGCTTTTGACATCTTGGCTGAAAACTGTGAATTTAGAGAAAATGAAGATAGCTGCGTTACGTTTATGAGAGCTGCCTCAGTTTTAAAAAGCTTACCTTTTACTATAATTTCTATGAAAGATACCGAAGGAATTCCATGTTTGGGTTCAAAAGTTAAGGGTATTATAGAAGAAATAATTGAAGATGGTGAATCATCAGAAGTTAAAGCAGTGTTGAATGATGAAAGATACCAATCTTTTAAATTATTTACTTCAGTCTTTGGTGTTGGTTTAAAAACCTCCGAAAAATGGTTTAGAATGGGCTTCAGAACCTTGAGTAAGGTGAGATCTGATAAATCATTGAAATTTACGAGAATGCAAAAAGCTGGTTTTTTGTACTACGAGGATTTGGTCAGCTGTGTTACTAGAGCTGAAGCTGAAGCTGTTTCTGTTCTGGTTAAAGAAGCAGTTTGGGCATTTTTGCCTGATGCTTTCGTTACAATGACAGGTGGTTTTAGAAGGGGTAAAAAAATGGGTCATGATGTAGATTTTTTGATTACTTCCCCTGGTTCTACTGAAGATGAAGAACAATTGCTACAAAAAGTGATGAACTTATGGGAGAAAAAAGGTTTGTTGTTGTACTATGATTTAGTTGAGTCTACTTTTGAAAAGTTGAGATTACCATCTCGCAAAGTTGATGCTTTAGATCATTTCCAAAAATGTTTTTTGATCTTTAAGTTGCCAAGACAAAGAGTAGATTCCGATCAGAGTTCCTGGCAAGAAGGTAAGACTTGGAAAGCCATTAGAGTCGACTTGGTTCTGTGTCCATACGAAAGAAGAGCTTTTGCATTATTAGGTTGGACAGGTTCCAGACAATTTGAAAGAGATTTGAGAAGATATGCCACACATGAGAGAAAGATGATTTTAGATAACCATGCATTATATGATAAAACTAAAAGAATCTTCTTGAAAGCCGAATCAGAGGAAGAAATTTTTGCCCATCTAGGTTTGGATTACATTGAACCATGGGAGAGAAATGCT
Avoid cleavage sites of restriction enzymes:
BbsI BsaI
3.4- I can produce it in S. cerevisiae via putting the fragment into a vector with the corresponding parts (RBS, promoter, terminator, etc). I can add tags in the 3´ sequence if i wanted to purify it with a desire method. Also cell-free methods could be use to produce it.
Part 4: prepare a Twist DNA Synthesis Order
Part 5: DNA Read/Write/Edit
Part 5.1: DNA Read
5.1- What DNA would you want to sequence (e.g., read) and why?
For the develop of my personal project, i would need to sequence a lot of the plasmids and designs that i will design in it. Also, genomic DNA sequencing to see the integration of the sequences in the corresponding site might be needed.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
TGS. Nanopore, for example.
To plasmid and genomic site-especific integration view i would amplify the concentration of the sequences by PCR they, and i would fragmentate (e.g via sonification) the sequences. Then, i would design primers to bind to specific regions (e.g adeine tails) and add adaptors to the sequencing machine and barcodes to aling the fragments, and PCR the sequences. What is next is to prepare the material for the sequencing machine (dilute, pipete). And for transcriptome reading the use of a reverse transcriptase in the firts PCR is needed.
For nanopore sequencing, after the preparation of the library, the sample is loaded onto the flow cell, where a flow of ions create a current that passes throught pores. A protein motor guide that pass of the dna throught the pore, where each base disrupt the voltage and the machine is able to interpret it. The raw electrical signals are stored as FAST5 and by algorithms is converted into FASTQ.
5.2 DNA write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would to synthesize the components of the system (e.g transcription factors, kinases, phosphatases) and their respective vectors and cassettes components (promoters, RBSs, terminators). Maybe, as the design process uses tools of protein design, some components may need to be prepared in libraries where certain AA sequences are changign for a more desired output. And, for a more broad characterization, libraries where the components of the cassettes are different.
For now, the components are TF1,TF2, peptidase 1, kinase 1 , cas 13 and their gRNA, condensate andamio, condesate andamio 2, TF3, TF 4 and TF5. All of this components are general because some would be engineered (modified domains, design of motives, etc).
5.3 DNA edit
(i) What DNA would you want to edit and why?
For my system to work i would need to integrate the components in the genome with, maybe, some recombinase-use engineering.
Week 3 HW: Lab automation
Homework
I create the following code by drawing at the opentrons art platform, coping the coordenates and asking chatgpt to generate the code below the code provided by the original opentrons HTGAA google colabs.
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
A massively parallel reporter assay library to screen short synthetic promoters in mammalian cells
The paper uses massive parallel reporter assays (MPRAs) as a high-throughput method to cuantify the barcoded mRNA produced by a series of synthetic promoters, and how the regulatory dynamics change over different compounds. They tested 6144 promoters in conventional and specific mammalian cell lines
The results were obtein by doing NGS and counting the reads of the mRNAs presents in the cells
Write a description about what you intend to do with automation tools for your final project
For my final project i can use cell-free protein synthesis to screen the post-translational modifications with fluorescence based arrays, in a cloud laboratory.
Im going to express the cdks-cyclins complexes, a peptidase, and the correspond TF that would have the post-translational modifications.
If im going to use a peptidase that depends on those cdks/cyclins complexes i would be easy, but it firts needs to exist, and not have other activations, otherwise the system would be active at a different time, and i find this very difficult. Another option could be design it
If i design the peptidase, firts i should test it in the same type of array.
I can build libraries of different changes in the domains and test the perfomance in a well plate.
A possible automated workflow would be:
Echo transfer DNA templates encoding CDK-cyclin complexes, transcription factor variants, protease/peptidase candidates, and fluorescent reporter constructs into specified wells of a 96-well or 384-well plate.
Echo transfer any required cofactors, kinase reagents, ATP supplementation conditions, or small molecules into selected wells depending on the test condition.
Bravo dispense or stamp cell-free protein synthesis master mix into all wells.
Multiflo add lysate or additional common reagents to initiate expression.
PlateLoc seal the plate to prevent evaporation.
Inheco incubate the plate under defined temperature conditions to allow protein synthesis and post-translational regulation to occur.
XPeel remove the seal after incubation.
PHERAstar measure fluorescence or time-course reporter output to compare activation, background signal, and dynamic range across designs.
Answer any NINE of the following questions from Shuguang Zhang:
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because eating beef doesn´t make us incorporate the genetic information that make a cow, and even if we could, we don´t have machinery to process that information and make us a cow.
Why are there only 20 natural amino acids?
I think because they were the optimal to being building blocks and have catalytic function. Also maybe have to be with the abundance of those in early life and that maybe suited interactions with RNAs at that time.
Can you make other non-natural amino acids? Design some new amino acids.
My naive try is to mix diketopyrrolopyrrole as the functional group
(Quiting the H close to the N and making there a covalent bound).
Maybe it could work as organic photovoltaics for protein electrical processing
Another could use cyclopentadienyl as the functioanl group to bind rare earth minerals
Or pentamethylcyclopentadiene
Where did amino acids come from before enzymes that make them, and before life started?
From abiotic reactions in special enviroments, with rich carbon, nitrogen and oxygen sources.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
left handedness.
Can you discover additional helices in proteins?
I found that proteins contain alternative helical structures—most notably
-helices,
-helices, and polyproline helices.
Why are most molecular helices right-handed?
I think the repulsion between the CO from the second and third AA, when curving to the left is going to be too strong (they are going to be facing each other). Also, the H-bounds formed in the right handed wouldn´t be so stable in the left handed.
Why do β-sheets tend to aggregate?
For the repetitiveness of the motif that form the initial bounds of the B-sheets that are exposed on the other side of the firts B-sheet.
What is the driving force for β-sheet aggregation?
The hydrophobic effect, because it drives a hiddeness of the functional hydrophobic groups from aqueos solvent.
Why do many amyloid diseases form β-sheets?
I think because, being b-sheets a common motif, if there are problems in the folding process, they could thend to aggregate. And if we think about the a-helix, it could be very difficult to aggregate there, so if b-sheets are termodynamic stable, it would be easy to misfolded protein to aggregate in conjuction if the hydrophobic motif enable them.
Can you use amyloid β-sheets as materials?
If they are well designed, yeah why not.
Design a β-sheet motif that forms a well-ordered structure.
What comes to my mind is a toroid-like estructure.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
I selected TdT, because it polymerases DNA without any template, and i find it cool.
Identify the amino acid sequence of your protein.
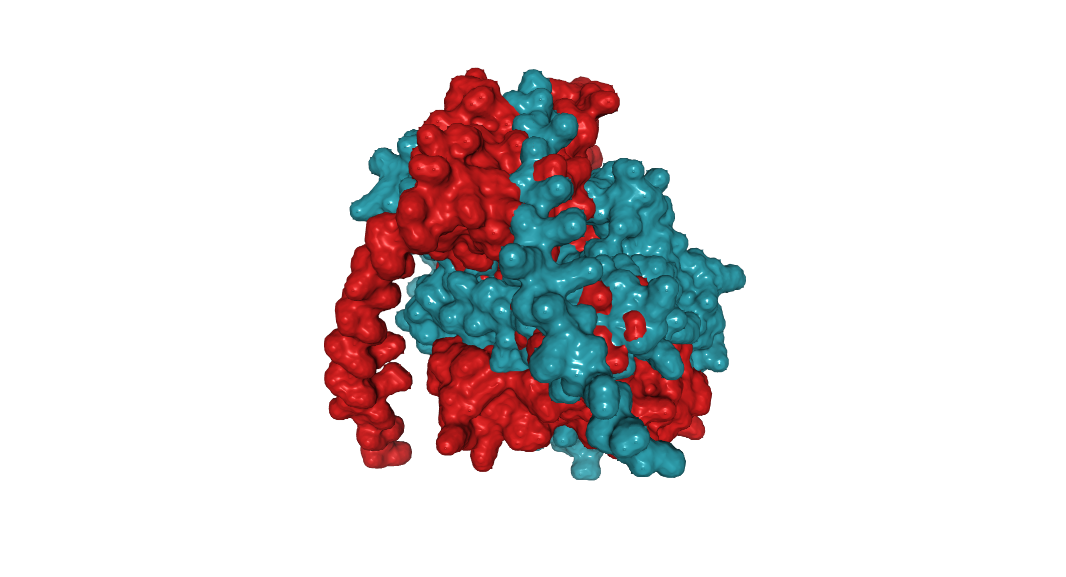
MDPPRASHLSPRKKRPRQTGALMASSPQDIKFQDLVVFILEKKMGTTRRAFLMELARRKG FRVENELSDSVTHIVAENNSGSDVLEWLQAQKVQVSSQPELLDVSWLIECIRAGKPVEMT GKHQLVVRRDYSDSTNPGPPKTPPIAVQKISQYACQRRTTLNNCNQIFTDAFDILAENCE FRENEDSCVTFMRAASVLKSLPFTIISMKDTEGIPCLGSKVKGIIEEIIEDGESSEVKAV LNDERYQSFKLFTSVFGVGLKTSEKWFRMGFRTLSKVRSDKSLKFTRMQKAGFLYYEDLV SCVTRAEAEAVSVLVKEAVWAFLPDAFVTMTGGFRRGKKMGHDVDFLITSPGSTEDEEQL LQKVMNLWEKKGLLLYYDLVESTFEKLRLPSRKVDALDHFQKCFLIFKLPRQRVDSDQSS WQEGKTWKAIRVDLVLCPYERRAFALLGWTGSRQFERDLRRYATHERKMILDNHALYDKT KRIFLKAESEEEIFAHLGLDYIEPWERNA
It have 509 AA. Lysine is the most frequent amino acid with 50 counts (9.82%). Its homologous to 250 proteins (using uniprot blast tool). Belongs to the DNA polymerase type-X family.
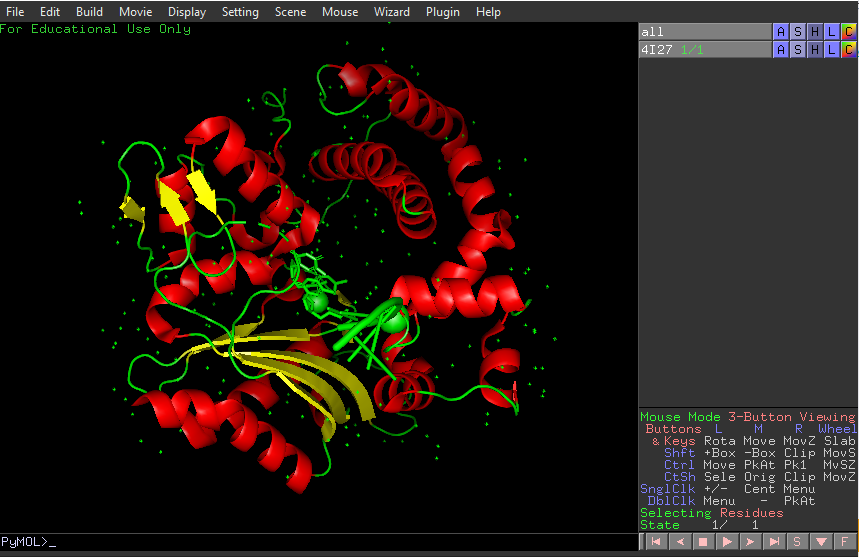
Identify the structure page of your protein in RCSB.
4I27 | pdb_00004i27.
It was released in 2013-07-24. It has a good quality resolution (2.60 Å). In the structure is involved 2’,3’-DIDEOXY-THYMIDINE-5’-TRIPHOSPHATE, magnesium ion and sodium ion. Using the SCOP 2 database, it belong to the DNA nucleotidylexotransferase structural family.
It have more helices thant sheets.
It have a hole where DNA binds to it.
Part C: Using ML-Based Protein Design Tools
Deep Mutational Scans
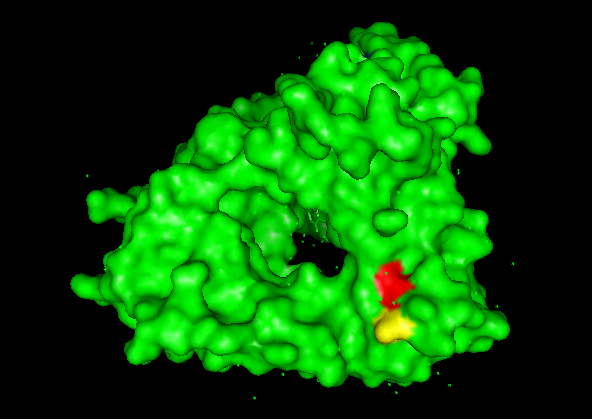
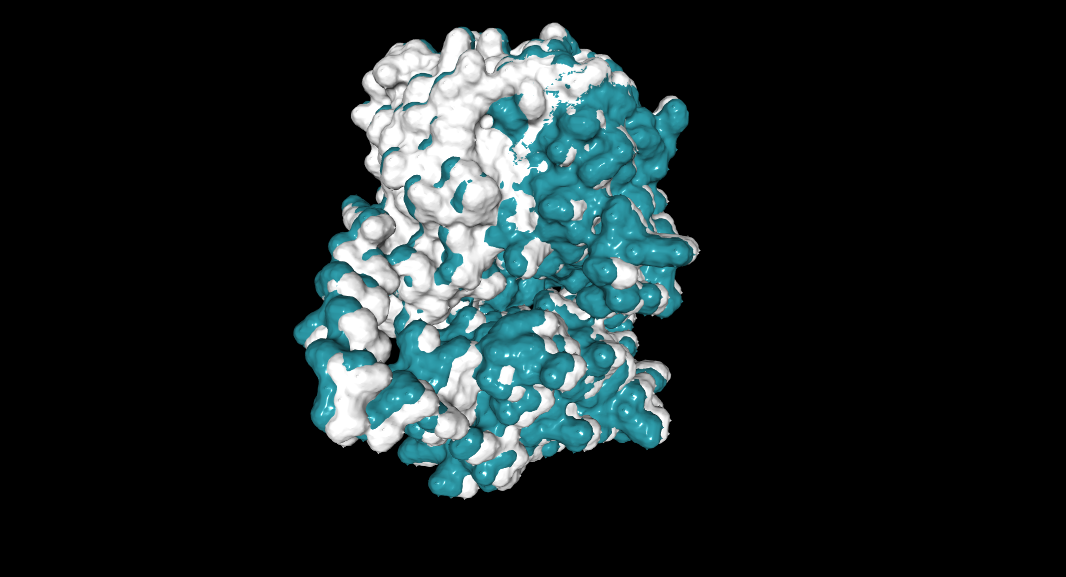
A series of mutations that stands out is in the T from the 273 AA to D and E (+2 score). Another close one is in the L in the 274 to D and E (+3 score). Another one is 264 E to L (4.76 score).
This mutations are close to the binding pocket of the DNA, affecting maybe the catalysis of the TdT or the affinity for the DNA.
In red L 274, in yellow T 273.
In magenta E 264, in cyan single strand DNA.
Latent Space Analysis
The neighborhoods approximate similar proteins! I could find neighborhoods of polymerases, transcription factors, etc etc. But i couldn´t find my protein.
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I couldn`t see it with py3Dmol, so i open it with NGLviewer
ptm: 0.756 plddt: 89.294
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
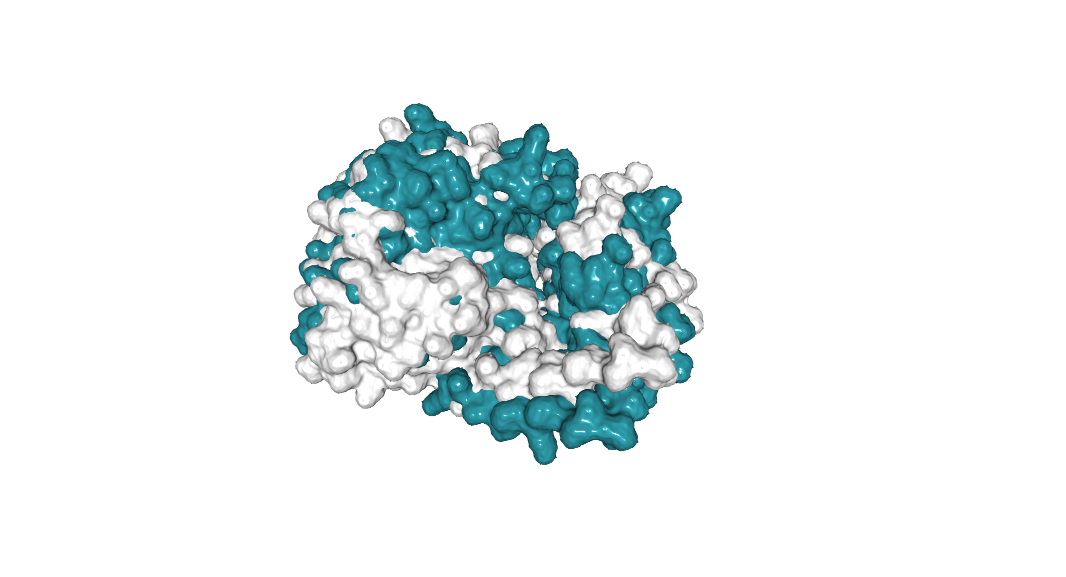
264 E to L (Non-mutated: cyan, mutated: white.):
ptm: 0.756 plddt: 89.287.
The structure is maintain. This mutation could improve the binding to DNA or other ligands.
Here i tried a desestabilitation mutation predicted by the deep mutational scan. 256 F to P:
ptm: 0.755 plddt: 89.634
It have the form of the non mutated version, but i thing it could work worse than the original protein. The binding pocket changes significantly.
Protein Generation. Inverse-Folding a protein
Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN.
New Sequence:PPPPPKVIRPPRPPPPPPPSPPPPPPPSLQKFKDIVVYVLEDNLGEKKRKELKEKLRAAGFTVSDKLNNDVTHIVAYNLTGTEVLDLIKASGIKLDNTPKLLKISWAEDCIEAGKPVEITPEYILPVEPPPKDKSIPPPPPPPPPPREPLSPYACKRRCSLIDYNKKFVDTFNILAEYYRFLNNSEKADKYNRAAAQLKSLPFEIKSMEDLEGIPHIYPEIRKIIEEILKNGYSTEVEKILNDPYFKTKKLFTSIYGFGLATADKYYKAGYTSIEEVKSDKSIKFSEEQKAGLKYLKDLTRPITREEALRIHEIIKEAVHAFLPDAIVELVGSFARGAETSRDVDFLISSPTWKGDQTLLEKVIEYLKEKGLLLYYKLTPSTYDPNALPSTDVNAPSPFQRVDMIFKLPLEEEEEQLGDRPPGKKWRAVKVDLTLVPYDRFAYARLYFTASPQFRRDLIEYARDERGMLLSSTSLYDLKKKEFISASSVEEIYAALGLPYIPPEELNC
I went and use ESMFold to see how the New Sequence folded. It was interesting.
ptm: 0.761 plddt: 93.088
Its different, but no at the same time, idk..
Then i wanted to try to change only the AA of the binding site. I use pymol to select the AA 4 A around the ligand, and asked chatgpt to write me a code to make this possible.
The code is:
after print(f"Length of chain {chain} is {l}"), put:
positions_to_design = [253, 255, 256, 257, 258, 259, 260, 261, 262,
288, 332, 333, 336, 338, 340, 341, 342, 343,
345, 381, 397, 398, 405, 432, 434, 449, 450,
452, 457, 461]
fixed_positions_dict = {}
name = pdb_dict_list[0]['name']
fixed_positions_dict[name] = {}
for chain in designed_chain_list:
chain_length = len(pdb_dict_list[0][f"seq_chain_{chain}"])
fixed_positions = [i for i in range(1, chain_length + 1) if i not in positions_to_design]
fixed_positions_dict[name][chain] = fixed_positions
print("fixed_positions_dict:")
print(fixed_positions_dict)
New Sequence:MDPPRASHLSPRKKRPRQTGALMASSPQDIKFQDLVVFILEKKMGTTRRAFLMELARRKGFRVENELSDSVTHIVAENNSGSDVLEWLQAQKVQVSSQPELLDVSWLIECIRAGKPVEMTGKHQLVVRRDYSDSTNPGPPKTPPIAVQKISQYACQRRTTLNNCNQIFTDAFDILAENCEFRENEDSCVTFMRAASVLKSLPFTIISMKDTEGIPCLGSKVKGIIEEIIEDGESSEVKAVLNDERYQSFKLFTSVYGVGLKTSEKWFRMGFRTLSKVRSDKSLKFTRTQKAGFLYYEDLVSCVTRAEAEAVSVLVKEAVWAFLPDAFVTMTGSFRRGAKESRDVDFLITSPGSTEDEEQLQKVMNLWEKKGLLLYYDLVESTFEKLRLPSRKVDALSPFQKCFLIFKLPRQRVDSDQSSWQEGKTWKAIRVDLTLCPYERRAFALLGWTGSRQFERDLRRYATHERKMILDNHALYDKTKRIFLKAESEEEIFAHLGLDYIEPWERNA
This is a library with only the binding site changed. Some will perform better, some worse. If i want to optimaze the structure, i can do the inverse.
Part D. Group Brainstorm on Bacteriophage Engineering
I think the no-dependency for DnaJ can be solved computationaly. Software like proteinMPNN to do inverse folding of the sequence or other sequences, EvolvePro to do in silico mutagenesis to explore variants, fooldseek for search of other natural ocurring sequences with the same structure, ESMFold to corroborate the 3D structure, etc.
An idea that i have is to anchor a synthetic domain into the L protein to help the conformational change. So that one L protein could help another without the need of Dnaj. The interactions may be difficult to stablish, but this could help the acumulation of phages upon oligomeration.
The problem is size of the MS2 genome. This could make it more bigger, more inefficient translation and replication, and less RNA accumulated in the capsid.
Other is use the Loajd mutant but with a low translation efficienty, changing nucleotides involve in the translation of the peptide, to help acumulate enough Loadj upon for the lysis. Even replication rate could be manipulated.
About the firts idea, the pipeline could be:
Know the residues in Dnaj that make the interaction with L protein, and their position in the tridimentional structure. Then, use RFdiffusion or other software to stablish those residues as constrains and make a backbone for that domain (small if possible), or maybe using the entire domain in the Dnaj. Check folding, stability, etc. Anchor that domain with a linker into the L protein. If the interactions between the domain an the N-terminal domain doesn`t occur in the same protein would be good. A good design could help to this. Then test for interactions. How the domain interacts with N-terminous domain vs Dnaj, different possittion to put it, optimazing, etc. Alphafold-Multimer and Boltz-1 to this, proteinMPNN to make the inverse folding, ESMFold to check 3D structure, EvolvePro and ESM-2 for mutational scoring for optimazing and for knowing the residues involve in the interactions use Alphafold-Multimer and Boltz-1 to get the pdb and a visualization system to see the residues.
A pitfall could be not getting enough data of the interactions between the Dnaj and the L protein, or the designed peptides don´t work well because of the limits of the tools.
For the second idea it could be identify the translational motif that interact with the ribosome or other translational structure, find the sequences and see if there are data about more or less efficient translation rate acording to the sequence, and design the new sequence.
Week 5 HW: Protein design part II
Homework
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
1- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
2- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card.
3- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence:
Control: FLYRWLPSRRGG
(X were A in all the peptides)
Part 2: Evaluate Binders with AlphaFold3
1- Navigate to the AlphaFold Server: alphafoldserver.com.
2- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
3- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Results:
P0: ipTM = 0.22 pTM = 0.8. It appears to bind to the dimer interface, surface-bound.
P1: ipTM = 0.2 pTM = 0.83. Binds between the N-terminus and the B-barrel, partially buried. (ipTM scores 0.3 in other run that i make accidentally)
P2: ipTM = 0.48 pTM = 0.89. Binds between the B-barrel and the dimer interface, partially buried?
4- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The ipTM values where generally shorts, but the P2 is the most notorious of the generated peptides. This also exceeds the know binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
1- Paste the peptide sequence.
2- Paste the A4V mutant SOD1 sequence in the target field.
3- Check the boxes
(For the P0, as example)
💧 Solubility,Soluble 1.000
🔬 Permeability (Penetrance), Non-permeable 0.043
🩸 Hemolysis, Non-hemolytic 0.067
👯 Non-Fouling, Fouling 0.360
⏱️ Half-Life 1.167 h
🔗 Binding Affinity, Weak binding 5.364
⚖️ Molecular Weight 1431.6
⚡ Net Charge (pH 7) -1.14
🎯 Isoelectric Point 5.55
💦 Hydrophobicity (GRAVY) -0.50
P0: 5.364,pKd/pKi
P1: 6.013,pKd/pKi
P2: 5.406,pKd/pKi
P3: 5.446,pKd/pKi
Control: 5.555,pKd/pKi
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Well, not necesarily. P2 wasn´t the best, as in comparison with P1. There is no correlation per se between AlphaFold 3 and PeptiVerse. No, there is no one with high hemolysis and poorly soluble. Im going to continue with P1 as has the best binding predicted, the other components seems to be okay, and in AlphaFold 3 the ipTM wasn´t so bad.
Part 4: Generate Optimized Peptides with moPPIt
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
ipTM were very low. Some were far away from the 1-10 region selected. 1, 4 and 5 were closest to the N-terminus, all surface bounded.
Part C: Final Project: L-Protein Mutants
Im going for the Option 2: Mutagenesis using Af2-Multimer.
Going to optimize the structure of the N-terminus domain while optimizing worst binding with Dnaj.
Useful data:
L protein sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
989 50 K L 2.561464
574 29 C R 2.395427
769 39 Y L 2.241778
575 29 C S 2.043150
173 9 S Q 2.014323
573 29 C Q 1.997049
572 29 C P 1.971028
569 29 C L 1.960646
987 50 K I 1.928798
1049 53 N L 1.864930
1209 61 E L 1.818096
1029 52 T L 1.813965
984 50 K F 1.802066
576 29 C T 1.797247
568 29 C K 1.795877
93 5 F Q 1.795244
94 5 F R 1.659716
560 29 C A 1.648655
534 27 Y R 1.628060
434 22 F R 1.602028
92 5 F P 1.596889
997 50 K V 1.594573
995 50 K S 1.574555
96 5 F T 1.559023
95 5 F S 1.556416
889 45 A L 1.539248
775 39 Y S 1.517457
535 27 Y S 1.497052
789 40 V L 1.477630
529 27 Y L 1.474638
435 22 F S 1.423357
563 29 C E 1.383282
760 39 Y A 1.364997
571 29 C N 1.362601
980 50 K A 1.357792
567 29 C I 1.344121
89 5 F L 1.332615
334 17 N R 1.323652
767 39 Y I 1.320101
776 39 Y T 1.302803
514 26 D R 1.268762
566 29 C H 1.246106
764 39 Y F 1.245850
777 39 Y V 1.244389
454 23 K R 1.236555
494 25 E R 1.229349
474 24 H R 1.227778
996 50 K T 1.222128
533 27 Y Q 1.218850
536 27 Y T 1.215567
Amino Acid Position Score
0 L 50 2.561468
1 L 39 2.241780
2 I 50 1.928801
3 L 53 1.864932
4 L 52 1.813968
5 F 50 1.802069
6 V 50 1.594576
7 S 50 1.574557
8 L 45 1.539248
9 S 39 1.517457
10 L 40 1.477630
11 A 39 1.364999
12 A 50 1.357795
13 I 39 1.320103
14 T 39 1.302804
15 F 39 1.245851
16 V 39 1.244390
17 T 50 1.222131
18 L 54 1.120860
19 R 39 1.064191
Position Wild_Type_AA Mutation_AA LLR_Score
989 50 K L 2.561468
574 29 C R 2.395427
769 39 Y L 2.241780
575 29 C S 2.043150
173 9 S Q 2.014325
573 29 C Q 1.997049
572 29 C P 1.971029
569 29 C L 1.960646
987 50 K I 1.928801
1049 53 N L 1.864932
996 50 K T 1.222131
533 27 Y Q 1.218851
536 27 Y T 1.215567
I select this mutations based on the scores predicted by the AIs and the N-terminus region utilize to model: C29R, C29S, S9Q, C29Q, C29P, C29L, C29T, C29K, F5Q, F5R, C29A, Y27R, F22R, F5P, F5T, F5S, Y27S, Y27L, F22S, C29E, C29N, C29I, F5L, N17R, D26R, C29H, K23R, E25R, H24R, Y27Q, Y27T
I make a code with chatgpt to
from the mutations (using only 1-30 AA of the L protein)
→ ESM2 LLR
→ choose top candidates stables
→ ColabFold peptide only
→ ColabFold peptide + chaperon
→ score final by:
mono_pLDDT high
mono_pTM high
complex_ipTM low
contact peptide–chaperon low
final_score high
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The high Phusion High-fidelity polymerase, MgCl2, dNTPs, etc. The polymerase enable low error in the polymerization reaction.
What are some factors that determine primer annealing temperature during PCR?
how much Hidrogen bombs are present (type of bases and how much).
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Restriction enzyme linearization could be more sequence dependent, and also not super escalable. In comparison, PCR linearization is more independent in those aspects, but have the downsides of primer design.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
By amplifying with exacts overlaps fragments that flank the desire product.
How does the plasmid DNA enter the E. coli cells during transformation?
By making the cells competetent by (normally) two ways: electroporation and heatshock, inducing pores into the membrane that enables the DNA to entry into the cells.
Describe another assembly method in detail (such as Golden Gate Assembly). Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online). Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly is a method of cloning that uses IIs restriction enzymes. Thesee enzymes cut not in but but close the recognition site, allowing to make specifics overhangs to assembly the parts. There entry vectors and overhangs to the parts, containing the correspond recognition sites and future overhangs.
This method allow to cloning up to 50 parts in one reaction, useful for multigene and from library-parts assembly. There is a common notation to the overhangs based on the type of parts, enabling MoClo (modular cloning) (leves 0: parts of a gene assembly; level 1: multigene assembly; level 2: multi-multigene assembly).
THe reaction usually is the following: (5 minutes 40 C, 5 minutes 16 C)x50, 5 minutes 50 C.
The entry vectors usually have a sequence of Lacz for blue-white colony selection, where the whites have final insert.
For the Asimov (Kernel) i didn´t have the acces to the API to do the homework
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
It enable non-linear threshold computation over the concentration of the molecules.
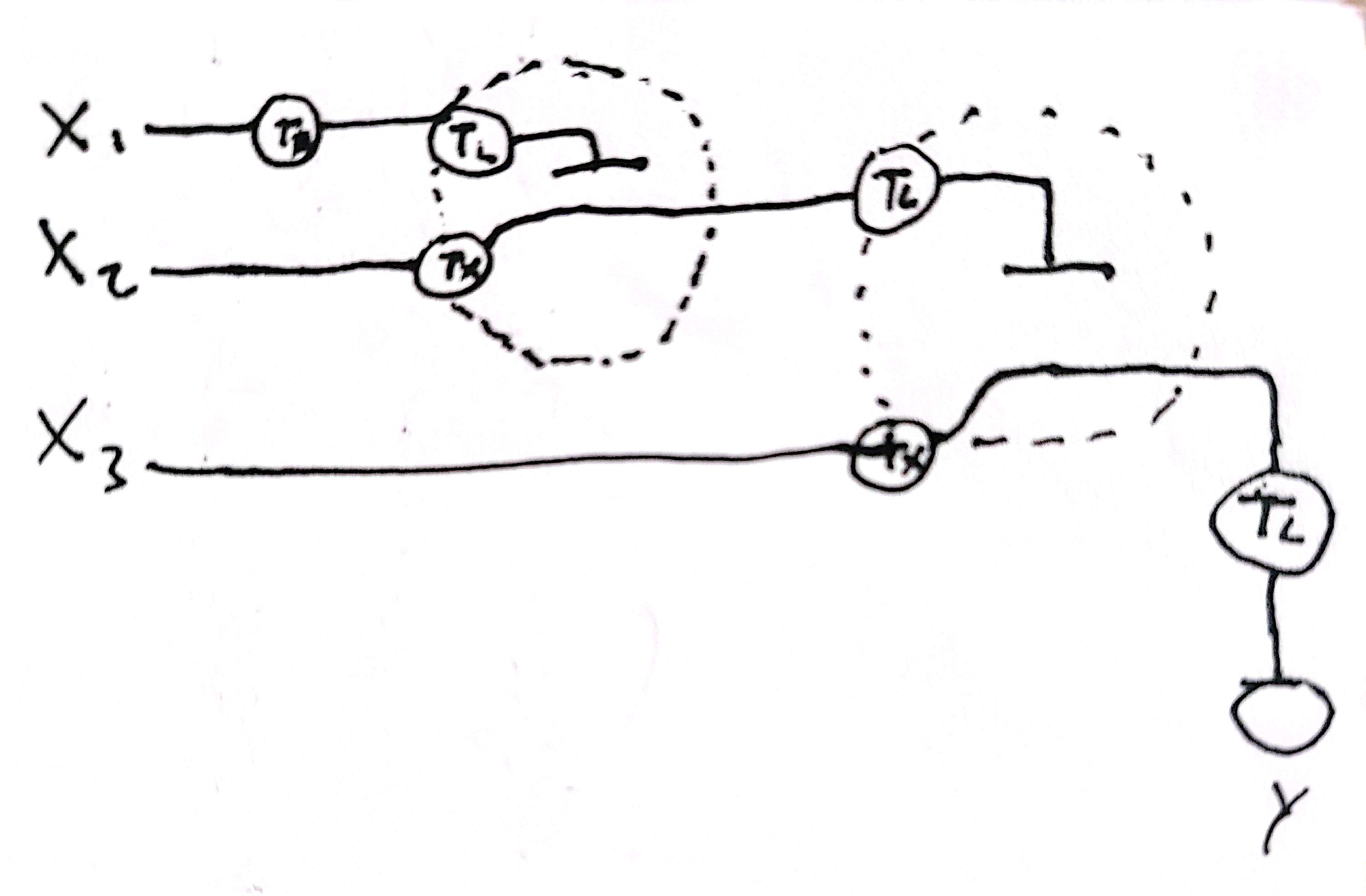
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
An useful application could be the development of responses upon specific thresholds of morphogens, for long-space tissue patterning (and cell-fate determination).
Where A, B and C are threshold (A being the most with high concentration requeriment). A should repress B and C, and B should repress C.
A limitation could be the specifity of the range of operation of the cassettes (crosstalk between the thresholds response modules).
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Pleurotus ostreatus for construction (concret crack healing) by Konwarh et al, 2020. Pleurotus ostreatus for concrete crack healing offers self-repair capability, lower maintenance costs, and greater sustainability compared to traditional sealants and repair materials. However, it still has limitations in long-term durability, mechanical reliability, and large-scale implementation; fungi skin wound healing by Ruggeri et al, 2023. Are biocompatible, biodegradable, and may promote tissue regeneration better than some synthetic dressings. Their disadvantages include sterilization challenges, regulatory barriers, and inconsistent material properties; and Ganoderma sessile as a block for architectural design by Attias et al, 2020. Ganoderma sessile as an architectural block is lightweight, sustainable, biodegradable, and provides good insulation compared to conventional building materials. However, it has lower structural strength, moisture sensitivity, and limited durability for demanding construction applications.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
One idea that i have in the past was to engineer fungi to produce melanin and other UV/radiation absorbent materials to serve as building blocks for the astronauts. Fungi offers the advantages of euakariots (PT modifications, multicelullarity, specialized organelles, etc) + the advantages of bacteria (rapid growth, industrial protein production, fast prototyping, etc).
Week 9 HW: Cell-Free Systems
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis enable rapid protein production without the need to wait for traditional transformation and growth. This fast prototyping enable fast test of multiple genes at the same time, genetic parts, variants, and other kind of libraries. Also, other variables, such as temperature, concentration of analytes, non-canonical AAs, etc, could be tested easily in CFE.
One case could be when characterizing multiple parts in an organism, where making it in CFES would be much more time efficient. Other case could be when making proteins with non-canonical AAs, making it more easy in the CFES.
Describe the main components of a cell-free expression system and explain the role of each component.
Lysate (provide cellular machinary), phosphates (e.g potassium phosphate monobasic and potassium phosphate dibasic, wich maintains a constant pH, and lowers the phosphatase activity of the lysate), AAs (for efficient protein production), ribonucleosides (nucleotide and energy metabolite precursors), magnesium glutamate. potassium glutamate (both for ions), PEG 8000 (for molecular crwowding) and the genetic component to incorporate.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
For short-term the best could be the incorporation of phosphoenolpyruvate (PEP) and pyruvate Kinase (PK), wich transfer one phosphate from PEP to ADP. For medium term could be adding the enzymes for the glycolitic pathway, turning glucose-1-phosphate into pyruvate, generating ATP through glycolysis. For long term, the option of the use of photosynthetic and CO2 fixation pathways, or analogous for autonomous energy production to maintain large productions of proteins, would be crucial.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic CFES produce high yield of proteins, but doesn`t worf for folding comple eukaryotic proteins and for PTM. Eukaryotic CFES, in the other hand, enable this, but have lower yields, and higher costs. In prokaryotics CFES i could produce T5 exonuclease, and in eukaryotic CFES, monoclonal antibodys, wich need to have PTMs.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
One option could be the use of lipid vesicles plus the machinary to drive the protein there, but this may require a lot of genes, wich could make all more difficult. Or maybe not. maybe 3-4 proteins that 1. bind to the liposome membrane, drive the ribosome or protein there, and enable integration of the protein there. Other option could be the use of equivalent proteins that are engineer to be soluble in water, while maintaining the same functionalites that the hydrophobic counterpart. One idea also could be the use biomolecular condensates that have an hydrphobic interior and could help fold the protein there.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Poor DNA/mRNA quantity and quality, and a troubleshoot could be checking the purity in the spectrophotometer, run agarose gel, and making enzymes digestions. Inapropiate folding, could be treated by reducing the incubation temperature, making the folding more slow. Insolubility is another problem that should be checked maybe computationally, creating soluble versions. And rapid depletion of energy substrates. For this, switching from a bacth mode to an continuous exchange cell free dialysis system could make it work more time.
Based on the predicted amino acid sequence of eGFP and any known modifications, what is the calculated molecular weight?
The calculated molecular weight of eGFP with the LE linker and His-tag is approximately 27.1 kDa.
Determine z for each adjacent pair of peaks using:
Using adjacent peaks around 933.7 and 1000.4 m/z, the charge state is approximately +15.
Determine the MW of the protein using the relationship between m/z, MW, and z.
The deconvoluted molecular weight is approximately 27.1 kDa.
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1.
The measured MW was very close to the theoretical MW, with only a few ppm error, indicating high accuracy.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No. The isotopic peaks are not sufficiently resolved in the intact denatured spectrum, so the exact charge state cannot be directly determined.
Waters Part II — Secondary/Tertiary structure
Based on learnings in the lab, please explain the difference between native and denatured protein conformations.
Native proteins remain folded and compact, exposing fewer protonation sites, so they show lower charge states. Denatured proteins unfold, exposing more residues and producing higher charge states.
What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
The denatured spectrum shows many high charge states at lower m/z values, while the native spectrum shows fewer and lower charge states at higher m/z values.
Zooming into the native mass spectrum of eGFP, can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
Yes. The peak is approximately charge state +10. The isotope spacing indicates the charge state.
Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP?
There are 20 Lysines (K) and 6 Arginines (R).
How many peptides will be generated from tryptic digestion of eGFP?
Approximately 27 peptides are expected after tryptic digestion.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
The PeptideMass tool predicts about 27 peptides.
Based on the LC-MS data for the Peptide Map data generated in lab how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes?
Approximately 18–20 major chromatographic peaks are observed.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
There are fewer observed chromatographic peaks than predicted peptides.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b.
The main peptide peak is observed at m/z 525.767.
What is the charge (z) of the most abundant charge state of the peptide?
The most abundant charge state is +2.
Calculate the mass of the singly charged form of the peptide (M) based on its m/z and z.
The peptide mass is approximately 1049.5 Da.
Identify the peptide based on comparison to expected masses in the PeptideMass tool.
The peptide likely corresponds to a predicted tryptic GFP peptide near 1050 Da.
What is mass accuracy of measurement? Please calculate the error in ppm.
The measurement error is very low, approximately a few ppm.
What is the percentage of the sequence that is confirmed by peptide mapping?
Approximately 80–90% of the eGFP sequence is confirmed by peptide mapping.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I couldn´t contribuite to the pixel artwork sadly :(
Next year i become a TA without excuses.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate: Provides the cellular machinery for transcription and translation, including ribosomes, tRNAs, enzymes, and metabolic proteins.
BL21 (DE3) Star Lysate: A lysate strain optimized for protein expression. It includes T7 RNA polymerase, which transcribes genes controlled by a T7 promoter.
Potassium Glutamate: Maintains ionic strength and mimics the intracellular salt environment needed for efficient translation.
HEPES-KOH pH 7.5: Buffers the reaction to keep the pH stable near physiological conditions.
Magnesium Glutamate: Provides Mg²⁺, essential for ribosome function, RNA stability, and enzyme activity.
Potassium phosphate monobasic: Helps buffer the reaction and contributes phosphate for energy metabolism.
Potassium phosphate dibasic: Works with monobasic phosphate to maintain pH and phosphate balance.
Ribose: Carbon precursor used to regenerate nucleotides and sustain long-term transcription.
Glucose: Energy source that feeds metabolism in the lysate to support ATP regeneration.
AMP: Precursor for ATP and RNA synthesis.
CMP: Precursor for CTP and RNA synthesis.
GMP: Precursor for GTP and RNA synthesis.
UMP: Precursor for UTP and RNA synthesis.
Guanine: Purine base that can be salvaged by lysate enzymes to make GMP/GTP.
17 Amino Acid Mix: Supplies most amino acids needed for protein translation.
Tyrosine: Added separately because it has lower solubility or different preparation needs.
Cysteine: Added separately because it is chemically sensitive and can oxidize.
Nicotinamide: Supports NAD-related metabolism and helps sustain long-duration energy regeneration.
Nuclease Free Water: Used to bring the reaction to the final volume without adding nucleases that could degrade DNA or RNA.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above.
The 1-hour PEP-NTP mix uses ready-made NTPs and PEP for fast, immediate transcription and energy supply. The 20-hour NMP-Ribose-Glucose mix uses NMPs, ribose, glucose, phosphate, and guanine so the lysate can regenerate nucleotides and energy more slowly and sustainably over long incubation. It is designed for longer protein production rather than maximum short-term speed.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Guanine can be converted by salvage-pathway enzymes in the E. coli lysate into GMP, then further phosphorylated to GDP and GTP. That regenerated GTP can then be used by T7 RNA polymerase during transcription.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems.
sfGFP: Folds very efficiently and matures well, so it usually gives strong fluorescence in cell-free reactions.
mRFP1: Often matures more slowly and can be less bright than newer red fluorescent proteins, reducing signal during shorter incubations.
mKO2: Its orange fluorescence depends on proper folding and chromophore maturation, which can be affected by oxygen and reaction conditions.
mTurquoise2: A cyan fluorescent protein with good brightness, but readout depends strongly on correct excitation/emission settings and folding.
mScarlet_I: Very bright red fluorescent protein, but full fluorescence still requires proper folding and oxygen-dependent chromophore maturation.
Electra2: Fluorescence output depends on efficient expression, folding, and maturation; its signal may be affected by pH, oxygen, and reaction duration.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve fluorescence over a 36-hour incubation.
For mScarlet_I, increasing glucose/ribose-based energy regeneration and maintaining HEPES/phosphate buffering should improve long-term translation and chromophore maturation over 36 hours. Expected effect: more properly folded and matured red fluorescent protein, giving higher final fluorescence.
For the following parts i sadly wasnt`t able to continue because i couldn´t send the master mix.
Week 12 HW: Building Genomes
Lab questions
Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively?
The production of lycopene in E. coli is induced by the genes crtE, crtI, and crtB from Erwinia herbicola, which are carried in the pAC-LYC plasmid. Beta-carotene production requires the same pathway plus the additional crtY gene, present in the pAC-BETA plasmid.
Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene?
The antibiotic resistance gene allows researchers to selectively grow only the bacteria that successfully received the plasmid. Since the media contains chloramphenicol, only transformed cells carrying the resistance gene survive and continue producing the desired pigments.
What outcomes might we expect to see when we vary the media, presence of fructose, and temperature conditions of the overnight cultures?
Changing media composition, carbon source, and temperature can affect both bacterial growth and metabolic flux toward carotenoid production. Richer media such as 2YT may increase biomass, fructose may improve recombinant gene expression and pigment synthesis, and lower temperatures like 30°C may improve folding and reduce metabolic stress, while 37°C may increase growth rate but sometimes reduce pigment accumulation.
Generally describe what “OD600” measures and how it can be interpreted in this experiment.
OD600 measures the turbidity of the bacterial culture at 600 nm, which reflects cell density because suspended cells scatter light. In this experiment, OD600 is used to estimate bacterial growth so pigment production can be normalized relative to the number of cells present.
What are other experimental setups where we may be able to use acetone to separate cellular matter from a compound we intend to measure?
Acetone can be used in experiments involving extraction of hydrophobic compounds such as pigments, lipids, chlorophylls, or carotenoids from cells or tissues. It is also useful for precipitating proteins while keeping smaller metabolites or pigments dissolved in solution for spectrophotometric analysis.
Why might we want to engineer E. coli to produce lycopene and beta-carotene pigments when Erwinia herbicola naturally produces them?
E. coli is easier to culture, genetically manipulate, and scale industrially than many native producers. Engineering E. coli allows researchers to optimize production conditions, tune metabolic pathways, and potentially produce carotenoids more efficiently and at lower cost for biotechnology or food applications.
What are the enzymes of the carotene pathway?
The carotene pathway described in the lab includes the enzymes encoded by crtE, crtB, crtI, and crtY. These enzymes convert metabolic intermediates such as farnesyl diphosphate into lycopene and finally into beta-carotene.
Within this pathway, which is the rate determining step? Which enzyme is responsible for this step?
A commonly limiting step in carotenoid biosynthesis is the desaturation process converting phytoene into lycopene, catalyzed by CrtI (phytoene desaturase). This step involves multiple sequential desaturation reactions and often strongly influences pathway flux.
The first thing to do is to decide what organism you are going to use for this (E. coli or S. cerevisiae) for production. Which would you choose and why?
I would choose E. coli because it grows rapidly, is inexpensive to culture, and has highly developed genetic engineering tools. It is especially useful for fast and scalable bacterial bioproduction, although S. cerevisiae may be preferable for some eukaryotic proteins or pathways requiring compartmentalization.
What is the function of a promoter?
A promoter is a DNA sequence that recruits RNA polymerase and regulatory proteins to initiate transcription of a gene. It determines when, where, and how strongly a gene is expressed.
What types of promoters do we have?
Promoters can be constitutive, inducible, repressible, strong, weak, tissue-specific, or environmentally responsive. Some promoters are always active, while others respond to signals such as metabolites, temperature, or chemicals.
If we wanted to turn off the transcription of a gene in response to a metabolite, what type of promoter would be most useful? What if we wanted this to increase in the presence of the metabolite?
To turn transcription off in response to a metabolite, a repressible promoter would be useful. To increase transcription in response to the metabolite, an inducible promoter would be more appropriate.
Now choose one of the genes of the metabolic pathway and choose one enzyme to make an expression construct. What promoter could you use for this? Why did you choose it?
For expression of crtI, I would use the T7 promoter in E. coli. The T7 system is widely used because it produces very high transcription levels when T7 RNA polymerase is present, which could increase carotenoid production.
What is the origin of replication?
The origin of replication (ori) is the DNA sequence where plasmid replication begins inside the host cell. It controls plasmid maintenance and copy number.
What types of origin of replication do we have?
Origins of replication can be high-copy or low-copy, broad-host-range or host-specific. Different origins determine how many plasmid copies exist per cell and which organisms can replicate the plasmid.
(Extra) What are compatibility groups?
Compatibility groups classify plasmids according to whether they can coexist in the same cell. Plasmids with similar replication control systems are usually incompatible because they interfere with each other’s replication.
Now for the previously chosen promoter and gene what will be the best origin of replication?
For strong crtI expression with a T7 promoter, a medium- or high-copy-number origin such as ColE1-derived origins would be useful because it increases plasmid abundance and potentially increases enzyme production.
Elaborate further on other bioparts like RBS, terminators, operators you would use for a correct design and further bioproduction.
A strong ribosome binding site (RBS) would improve translation efficiency of the carotenoid enzymes. Terminators would ensure proper transcription termination and prevent readthrough into other genes. Operators could be added to allow metabolic regulation by repressors or activators, helping reduce metabolic burden and optimize pathway balance.
What are aptamers and riboswitches and how can they be used for metabolic tuning or engineering in prokaryotes?
Aptamers are nucleic acid sequences that bind specific molecules, while riboswitches are regulatory RNA elements that change gene expression after binding metabolites. In metabolic engineering, they can dynamically regulate enzyme expression depending on intracellular metabolite concentration, improving pathway balance and reducing toxicity.
What approach can be used to join all these parts together?
Methods such as Gibson Assembly, Golden Gate Assembly, or restriction enzyme cloning can be used to combine promoters, genes, RBSs, and terminators into a plasmid. Sequence analysis is important to avoid problematic restriction sites and ensure compatible overhangs or homologous regions for assembly.
Week 13 HW: Biodesign & Engineered Living Materials