Week 1 homework
Principles and practices 💼
1. First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Purification of enzymes for natural pigment synthesis facilitated by microalgal cell wall release
In recent decades, microalgae have emerged as promising platforms for the sustainable biosynthesis of various high-value compounds 1 2, however, during their purification, challenges can arise. This project aims to propose a method of metabolite purification from microalgal cultures by tapping into a largely overlooked resource of microalgal cells of various species, namely their cell wall. Purification of proteins of interest could be carried out by fusing them to elements of the microalgal cell wall and then harvesting the shedded cell walls following nitrogen starvation-induced sexual reproduction, as this method is inspired by the ecdysis of insects. Theoretically, this method of purification could be implemented for any fused proteins, but here, for a more tangible example stemming from an interest in the production of eco-friendly ink, the idea described below will focus on the synthesis of indigoidine synthetase, the primary enzyme implicated in the generation of the indigo dye.
The organism chosen for the purposes of this project is the model green microalga Chlamydomonas reinhardtii, as its physiology and metabolism have been extensively studied 3 4 5, while multiple tools have already been developed and established for its genetic manipulation 6 7 8. In more detail, the first step of this process should be to engineer C. reinhardtii 9 10 to overexpress pherophorin (microlgal cell wall protein)-indigoidine synthetase fusion proteins (Figure 1.1A). Subsequently, gamete generation can be induced by depleting the nitrogen in the growth medium, leading to mating of haploid microalgal cells followed by shedding of their cell walls (Figure 1.1B). The rejected cell wall components can then be isolated by sucrose-gradient centrifugation (Figure 1.1C), before being subjected to enzymatic processing both for polysaccharide degradation and for separation of indigoidine synthetase molecules from pherophorins (Figure 1.1D). Lastly, affinity chromatography can be employed for further purification of the synthesized indigoidine synthetase units (Figure 1.1E), which can afterwards be screened in an activity assay (Figure 1.1F) to monitor the enzymatic conversion of L-glutamate into indigoidine (indigo dye).
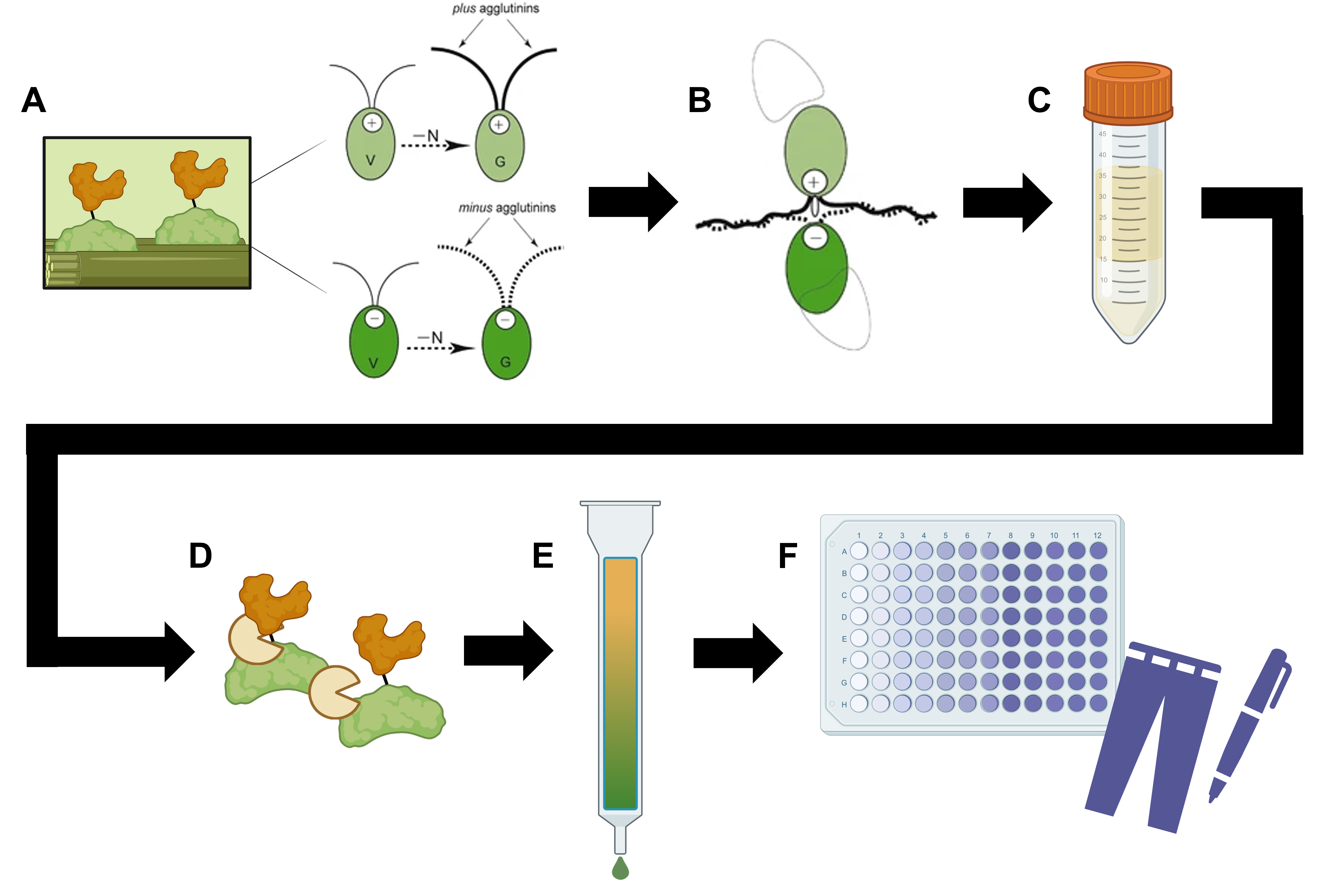 Figure 1.1 Schematic overview of cell wall release-based purification of indigoidine synthetase. Figure modified from Sekimoto, 2017 11 and partially created on BioRender.com.
Figure 1.1 Schematic overview of cell wall release-based purification of indigoidine synthetase. Figure modified from Sekimoto, 2017 11 and partially created on BioRender.com.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
The primary policy behind this project is sustainability, as microalgae require minimal resources to efficiently synthesize numerous valuable compounds utilized in human food and animal feed, in pharmaceuticals and cosmetics, and even in the energy sector with the production of biofuels through photosynthesis-driven carbon sequestration. Besides their role as promising green cell factories and a potential carbon sink, microalgae, and in turn projects revolving around them, contribute to sustainability by offering great spatial autonomy in terms of their cultivation, as they are not confined by the availability of arable land or freshwater-based irrigation. From a performance aspect, microalgae have been advocated for their high photosynthetic capacity too, enabling them to generate biomass more efficiently than most crops. Taking all the aforementioned benefits into consideration, another principle emerges, as microalgal cultivation gives equal opportunities for development both to established facilities, but most importantly, to low-income communities and developing countries, including small island nations. Culturing of microalgae merely requires exposing them to light and letting them fixate atmospheric inorganic carbon and has become even more affordable with the introduction of the plastic tubular photobioreactor 12, which also allows for the exploitation of otherwise unutilized vertical space.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
- Purpose: What is done now and what changes are you proposing?
- Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
- Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
- Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Option 1. Initiative to establish a teaching module on biosafety and biosecurity in high school Biology classes, with lectures from Biotechnology experts and academics of the local university or from iGEM teams that are active in the area. Raising awareness for biosafety and biosecurity issues as early as in high school years will ensure that future generations of researchers have the necessary stimuli to pursue good scientific practices. This initiative can also help high school students gain an overview both of the academic research being concucted at local universities, along with the regulatory frameworks defining that research, especially when that happens through the lens of an aspiring young scientist closer to their age, such as a university student member of a local iGEM team. In case of a smaller town or village without a university, lectures from invited experts coming from larger cities could be arranged, perhaps with the help and support of the Ministry of Education, which could even twin schools in rural areas with metropolitan universities.
Option 2. Enforcement of regular inspections from EU representatives to ensure that Biotechnology-focused academic, research, and industrial facilities adhere to imposed regulations. Surprisingly, there is no official EU institution, board, or committee primarily devoted to matters of biosafety and biosecurity, so pushing for the establishment of one should be a priority at this stage. In any case, laws that require regular inspections of biotechnological facilities even by experts appointed by the local government should be imposed. Close monitoring of academic and industrial facilities to verify that, for instance, HEPA filters are renewed and biohazardous waste is handled appropriately, is paramount and heavy fines should be issued in case of non-compliance.
Option 3. Increased funding of programs dedicated to designing and integrating novel kill-switches and other biocontainment mechanisms to expand the arsenal of available strategies both for a wider range of projects/conditions but also for a wider range of genetically modified organisms. This could be an action to promote innovative research in the field of biosecurity. By diverting resources or even creating new programs to fund research into novel biocontainment approaches at an EU (for example, the Marie-Skłodowska-Curie grants) or at a national level, more scientists could be encouraged to engage in discovering or devising cutting-edge kill-switch mechanisms 13 14 15 16 (Figure 1.2B), genetic safeguards or firewalls for preventing horizontal gene transfer, and auxotrophic strains 17 (Figure 1.2A). Hopefully, such an initiative could contribute to expanding the research and use of biocontainment not only to bacteria and yeast, for which the field of biosecurity is already established to a significant degree, but also to less widespread biotechnological hosts, such as microalgae, whose kill-switch and auxotrophy strategies are currently far less advanced.
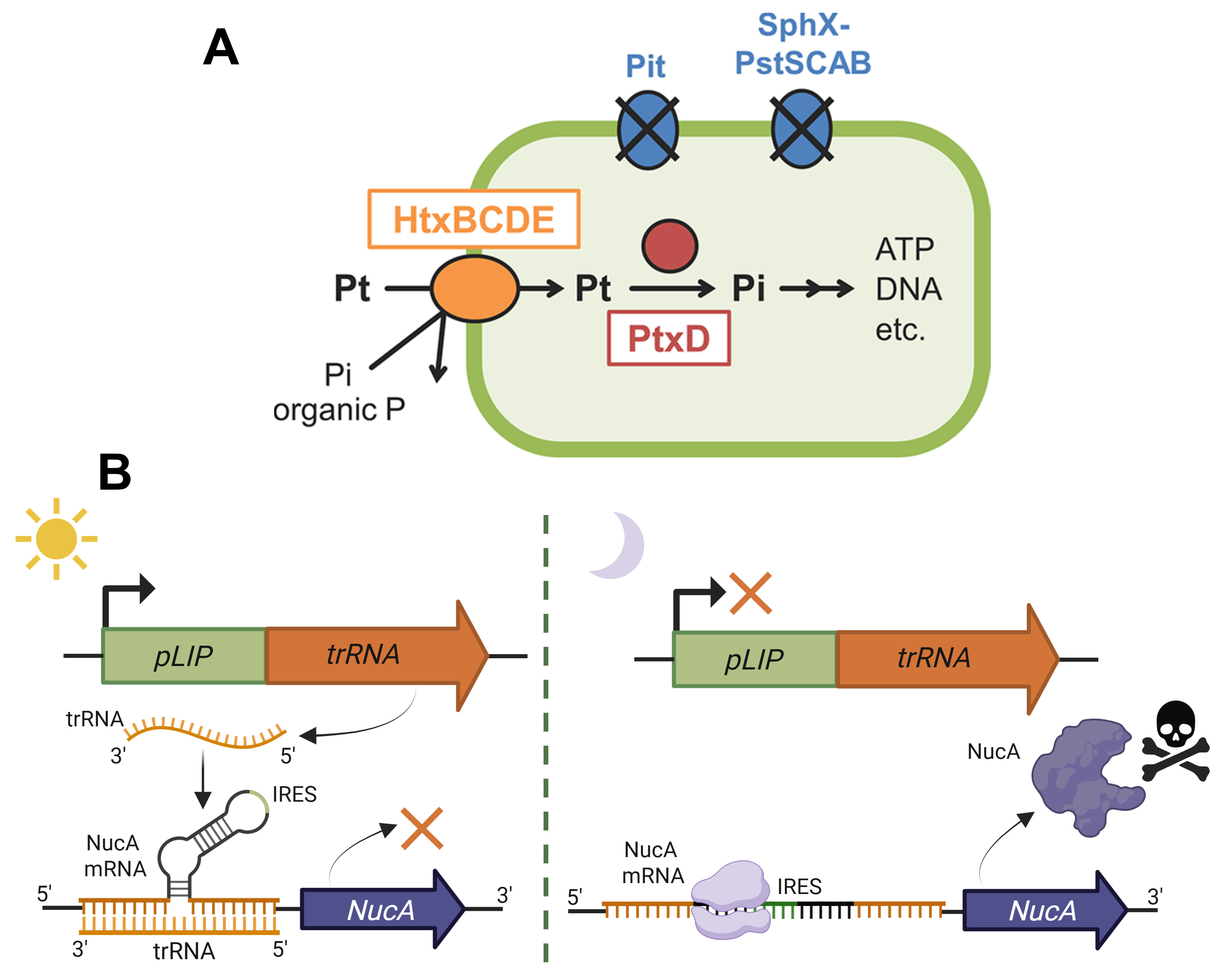 Figure 1.2 Proposed biocontainment strategies for genetically engineered microalgae. (A) A promising biocontainment method involves the development of auxotrophic strains, such as microalgae that can solely survive on a non-standard source of P, for example, phosphite 17. (B) Kill-switch mechanism employing a light-controlled riboregulator. The main premise behind this circuit is that genetically engineered microalgae are primarily cultured in well-illuminated ponds. In this case, the light-inducible pLIP promoter from Dunaliella sp. 13 drives the expression of a trigger RNA molecule (trRNA) that, through binding to a three-way junction riboregulator 14 15 at the 5’ UTR of NucA nuclease’s coding sequence, suppresses the gene’s expression. However, if the engineered microalgae accidentally end up in an underground aquifer or inside another organism after ingestion, the dark conditions will reveal an integrated internal ribosome entry site (IRES), enabling the nuclease’s expression 16 and, effectively, exterminating the microalgal cell. Figure from Motomura et al., 2018 17 and partially created on BioRender.com.
Figure 1.2 Proposed biocontainment strategies for genetically engineered microalgae. (A) A promising biocontainment method involves the development of auxotrophic strains, such as microalgae that can solely survive on a non-standard source of P, for example, phosphite 17. (B) Kill-switch mechanism employing a light-controlled riboregulator. The main premise behind this circuit is that genetically engineered microalgae are primarily cultured in well-illuminated ponds. In this case, the light-inducible pLIP promoter from Dunaliella sp. 13 drives the expression of a trigger RNA molecule (trRNA) that, through binding to a three-way junction riboregulator 14 15 at the 5’ UTR of NucA nuclease’s coding sequence, suppresses the gene’s expression. However, if the engineered microalgae accidentally end up in an underground aquifer or inside another organism after ingestion, the dark conditions will reveal an integrated internal ribosome entry site (IRES), enabling the nuclease’s expression 16 and, effectively, exterminating the microalgal cell. Figure from Motomura et al., 2018 17 and partially created on BioRender.com.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own.
Adhering to the suggested format, the scores of the proposed governance actions are presented below (Table 1.1).
Table 1.1 Scores of the proposed governance measures with respect to the rubric of policy goals.
| Does the option: | Option 1 | Option 2 | Option 3 |
|---|---|---|---|
| Enhance Biosecurity | |||
| • By preventing incidents | 1 | 2 | 1 |
| • By helping respond | 2 | 3 | 3 |
| Foster Lab Safety | |||
| • By preventing incident | 1 | 2 | 2 |
| • By helping respond | 1 | 3 | 2 |
| Protect the environment | |||
| • By preventing incidents | 2 | 2 | 1 |
| • By helping respond | 2 | 3 | 3 |
| Other considerations | |||
| • Minimizing costs and burdens to stakeholders | n/a | 1 | n/a |
| • Feasibility? | 1 | 2 | 2 |
| • Not impede research | 1 | 2 | 1 |
| • Promote constructive applications | 1 | 3 | 1 |
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix. Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
Among the three actions listed above, the least feasible one would be the enforcement of regular inspections by an EU board devoted to addressing biosafety and biosecurity issues. Since such an institution does not exist right now, its establishment would require time and a large bureaucratic, as well as administrative and legislative mobilization, implicating several bodies of the EU, including the European Council and the European Commission.
On the other hand, the other two initiatives, namely integrating modules about biosafety and biosecurity in high school curricula and increasing funding for research on biocontainment practices, appear more feasible and promising in the short term and should, therefore, be prioritized. The basic rationale behind this prioritization is that, judging from personal experience, the educational community, including secondary and higher education, has always been more open-minded and welcoming towards new initiatives compared to legislative bodies and political agents. As educational institutions, regardless of level, have always been united by ideals such as service to humanity and a strong drive towards innovation and intellectual progress, it is generally much easier and faster to put internal actions for institutional twinning and raising awareness about biosafety and biosecurity in motion, mostly concerning the first option. Another benefit of prioritizing the initiative presented in the first option would be providing the opportunity to promote other principles alongside biosafety and biosecurity to younger audiences too, for instance, sustainability and equity as previously mentioned. Regarding the third option, increasing funding for relevant strategies internally appears more feasible as well, since in many cases, research groups and laboratories are given substantial flexibility in terms of allocating resources to individual projects from national grants that do not specify a particular objective. In the long run, both options one and three could have a significantly positive influence in establising more fundamental and impactful governance policies in the future, as they incorporate a more “bottom-up” approach, where members of the general public that have become more aware of the aforementioned principles, school students, university students, and academics, can start building an initial framework. This will lay the foundation for more radical reforms, as more citizens become aware of biosafety, sustainability, and equity issues and push (through voting and other sociopolitical manifestations) for more generalized changes and a renewal of governance policies.
Preparation for week 2 lecture 🔎
Homework Questions from Prof. Jacobson
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
The primary polymerases for nuclear genome replication in human cells are DNA polymerases δ (delta) and ε (epsilon) 18 19 20. Purely based on their replicase activity, they display an error rate of 10-4 to 10-5 per base pair per replication cycle. Given that the entire haploid human genome spans approximately 3.3×109bp, it can be calculated that, in every replication cycle, (3.3×109bp) × 10-4 = 3.3×105bp can be erroneous in a worst-case scenario.
However, DNA polymerases δ and ε have evolved to have a 3’ to 5’ proofreading exonuclease activity, meaning that they can recognize and correct misplaced nucleotides based on strand complementarity by “going in reverse”, excising, and replacing them 18 19 20. It has been shown that, due to their proofreading capacity, those polymerases exhibit extremely low error rates, estimated at less than 10-9 per base pair per replication cycle 20, which, according to the calculation above, can be translated into approximately 3.3 errors for every replication. Except for the polymerases’ proofreading capacity, cells also possess mismatch repair (MMR) mechanisms that correct mistakes in DNA replication missed by the polymerases 20. In case all the previously mentioned systems fail, there are cell cycle checkpoints in place, which will not allow cellular division to proceed if mistakes in DNA replication persist.
Another factor that contributes to a lower rate of mutations in the “final product” of the genome, namely the proteins, is that protein-coding genes constitute about 1% of the entire genome, as they are “diluted” due to what has been called “junk DNA”. Because of this “dilution” effect, it seems far more likely that, even if an error occurs, it will be located in a non-coding region of the genome. Although this could influence the regulation of expression, it will not affect the final amino acid sequence of the protein of interest. To this end, namely the preservation of a protein’s primary structure, the genetic code has also evolved to be degenerate. The degeneracy of the genetic code, meaning the redundancy where multiple, distinct codons (nucleotide triplet combinations) encode for the same single amino acid, allows for increased flexibility in protein expression. Therefore, even if an error occurs within a coding region, the mutated codon can still be translated into the correct amino acid. Finally, as a fail-safe on the protein level, in case a replication error results in a mutated codon that corresponds to a different amino acid, the tertiary structure, as well as the functionality of the final protein, could still be preserved, if the altered amino acid demonstrates the same or very similar biochemical properties as the original one (for instance, aspartic acid and glutamic acid).
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice, what are some of the reasons that all of these different codes do not work to code for the protein of interest?
Each of the 20 standard amino acids can be represented by one to six different codons, with a three-codon representation in the genetic code on average. The coding sequence for an average human protein is 1,036bp long, meaning that an average human protein consists of approximately 1,036 / 3 = 345 amino acids. Based on this information, an average human protein could be translated from about 3345 = 4×10164 different codon combinations.
Although this is a valid theoretical assumption, in reality, every organism, including humans, does not show such flexibility in the coding sequences of its genome due to codon biases, as well as the genome’s GC content. Codon biases mostly relate to the actual availability of aminoacyl-tRNA synthetases and aminoacyl-tRNAs corresponding to every amino acid inside the cell, as aminoacyl-tRNAs are heavily involved in protein synthesis and their availability substantially affects the rate and the eventual success of the overall process. Specific codon usage databases (also known as “Kazusa tables”) have been composed based on studies of different organisms’ codon usages and biases 21 22. When synthesizing an artificial coding sequence for the expression of a human protein, the GC content of the genome needs to be taken into consideration too, as large variations can lead to silencing effects. Another factor that can drastically diminish the number of functional combinations for a protein’s coding sequence are the respective mRNA’s thermodynamic properties. Different combinations of codons, hence nucleotidic triplets, can facilitate the formation of secondary structures, such as hairpins, in a transcript, which can delay or even hinder translation. The selected codon sequence should, therefore, produce an mRNA with the appropriate thermodynamic profile for optimal protein expression. Finally, more limitations can be posed by the synthesis method in case the coding sequence has to be artificially assembled, as DNA molecules with low GC content and a low percentage of repeated sequences are preferred. Additionally, if the artificially synthesized gene also needs to contain introns that stabilize or enhance transcription, selection of a codon combination should ensure the presence of positions where introns can be inserted, for instance, GGs at determined intervals or spots.
Homework Questions from Dr. LeProust
1. What’s the most commonly used method for oligo synthesis currently?
Currently, the most established and widely used method for oligonucleotide synthesis is the solid-phase phosphoramidite (S-PP) method. This constitutes a type of chemical synthesis that utilizes a solid-phase material, typically controlled pore glass or microporous polystyrene, as a platform where oligonucleotides are added in a 3’ to 5’ direction. S-PP synthesis can be highly automated, enabling high-throughput production of oligonucleotides in 96- and 384-well plates.
The basic principle behind S-PP synthesis can be summarized as exposing biochemical groups that should react while simultaneously protecting groups that should not react. To this end, specifically modified nucleotides, called phosphoramidite monomers, which have their reactive groups “concealed”, need to be used. In particular, to prevent the reactive groups of the monomers from forming undesirable bonds during the process, their nitrogenous bases are protected by a benzoyl or isobutyryl moiety (Figure 1.3A, pink), their 3’-OH by a 2-cyanoethyl-diisopropylamino moiety (Figure 1.3A, purple, orange, and yellow), and their 5’-OH by a dimethoxytrityl (DMT) moiety (Figure 1.3A, green). In the case of RNA oligonucleotide synthesis, the highly reactive 2’-OH has to be “concealed” by a ter-butyldimethylsilyl moiety as well.
In more detail, the method consists of a cyclic four-step process that allows the assembly of the oligonucleotide chain by starting with a first monomer that has already been covalently attached to the synthesis platform and then elongating it one nucleotide at a time (Figure 1.3B):
- Step 1. Detritilation The 5′-DMT protecting group is removed by lowering the pH of the reaction, thus exposing the 5′-OH.
- Step 2. Coupling The incoming phosphoramidite monomer is added in a substantially high concentration and its phosphoramidite moiety “attacks” the now exposed 5′-OH (usually in the presence of an azole catalyst) to form a phosphite triester bond, ultimately linking the new monomer to the growing oligonucleotide. During this step, the diisopropylamino group is cleaved as well.
- Step 3. Capping Unreacted 5′-OH groups are capped through acetylation to avoid the formation of oligonucleotides with the wrong sequence.
- Step 4. Oxidation The unstable phosphite triester is oxidized into a stable phosphate triester bond.
Once the last cycle of the process is completed, the synthesized oligonucleotide is excised from the solid-phase platform by the implementation of alkaline conditions. This increase in pH will also cause the detachment of the 2-cyanoethyl groups, as well as of the nitrogenous bases’ protective moieties, from all the building blocks of the nucleotide, essentially rendering the newly synthesized oligonucleotide biochemically suitable for downstream applications 23 24 25.
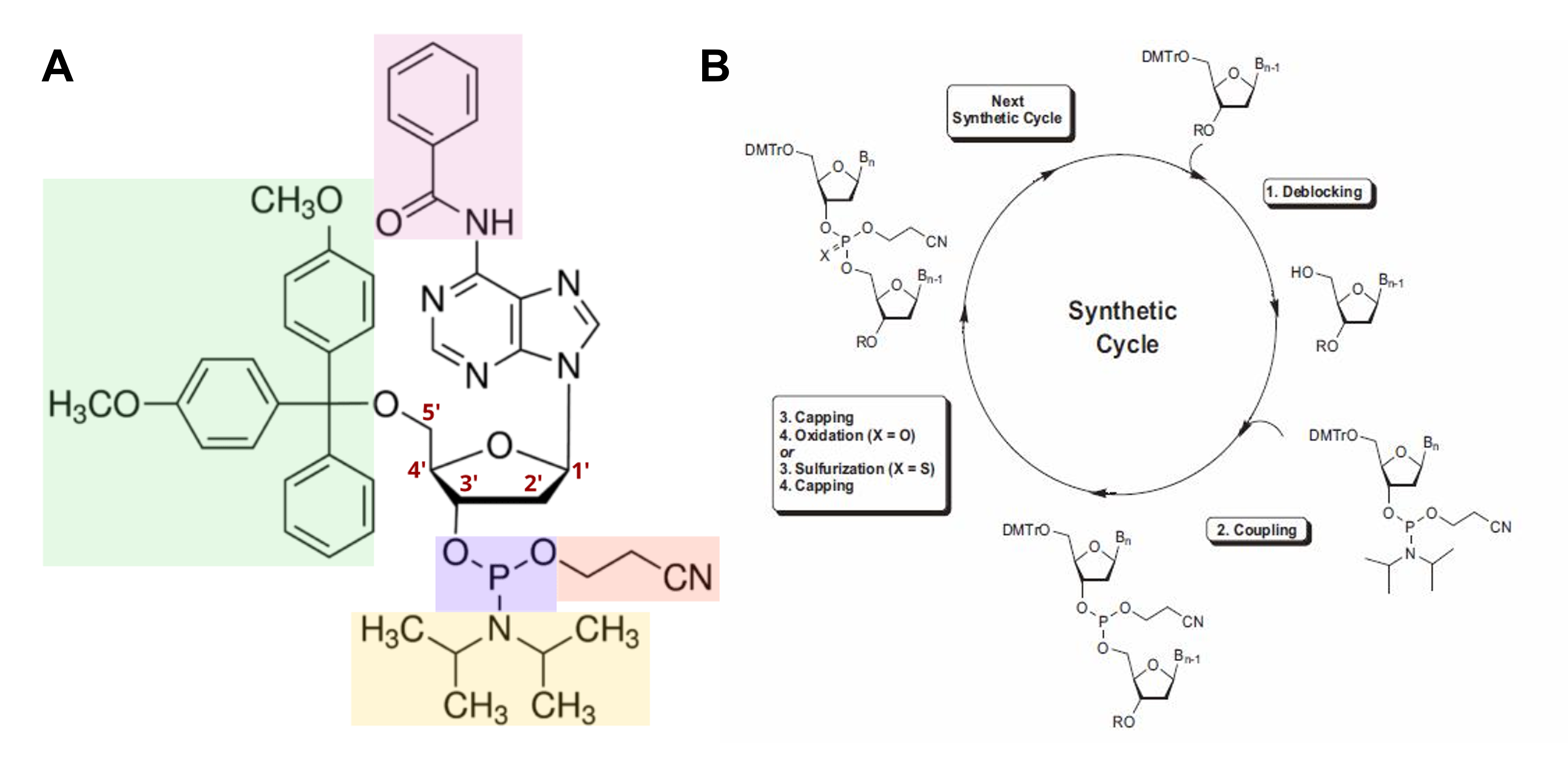 Figure 1.3 (A) Representation of a phosphoramidite monomer. (B) Schematic overview of solid-phase phosphoramidite synthesis. Figure from Twist Bioscience phosphoramidite chemistry and BOC Sciences S-PP cycle.
Figure 1.3 (A) Representation of a phosphoramidite monomer. (B) Schematic overview of solid-phase phosphoramidite synthesis. Figure from Twist Bioscience phosphoramidite chemistry and BOC Sciences S-PP cycle.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis? Why can 2000bp genes not be made via direct oligo synthesis?
Generating oligonucleotides via direct chemical synthesis, such as the S-PP method, presents challenges due to
cumulative inefficiencies in the building process. These inefficiencies ultimately lead to a drastic decrease in the yield and purity of the final product as, for several factors analyzed below, the latter drop exponentially with the increasing length of the desired oligonucleotide.
One such factor appears to be cumulative yield loss. While S-PP synthesis is highly efficient (>99% per step), this discrepancy from 100% increases exponentially as the chain grows. Given that the addition of every new phosphoramidite monomer probabilistically constitutes an independent event from previous additions and by assuming a coupling efficiency of 99%, the yield of a 100-mer is approximately 0.99100 = 36.6%, while the yield of a 200nt oligonucleotide amounts to an even lower percentage, 0.99200 = 13.4%. Accordingly, the yield of a 2,000nt sequence is practically 0, since 0.992,000 = 18.6×10-9%.
Another shortcoming stems from the accumulation of truncated sequences, which can occur at any stage of the assembly due to incomplete coupling. Those shorter oligonucleotides (also called “failure sequences”) accumulate as impurities in the pool of S-PP synthesized oligonucleotides. As the elongation advances, the length difference between the desired full-length product and the truncated oligonucleotide becomes negligible. For instance, the two nucleotides that differentiate a truncated 198-mer from the desired 200-mer correspond to a 1% discrepancy, however, in the case of a 1,198nt truncated oligonucleotide and the desired 2,000-mer, the discrepancy is 0.1%. Due to resolution constraints of high-performance liquid chromatography (HPLC) or polyacrylamide gel electrophoresis (PAGE), which are employed to separate the desired products from truncated entities, isolating the desired oligonucleotide (especially in the case of the 2,000-mer) proves to be extremely difficult, negatively impacting the purity of the final product.
Lastly, the physical properties of the support platform, as well as the biochemistry of the oligonucleotides themselves, can post limitations to the synthesis, which intensify with the increasing length of the desired oligonucleotide. More specifically, when CPG beads are used as a support matrix, their pores can become clogged as the oligonucleotide chain grows longer, ultimately hindering the diffusion of reagents to the reactive 5’-OH group and reducing coupling efficiency. Similarly, repeated cycles of detritilation under acidic conditions can induce depurination (chemical degradation and loss of purine bases). Depurination can accumulate over 200 cycles and even reach concerningly high levels at 2,000 cycles, leading to damaged, fragmented, or incorrect sequences.
Homework Questions from Prof. Church
3. Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:
For the generative optogenetics program, an approach could be to design and express a light-activated polymerase that has a “pinwheel” structure/shape and works the way a danish Christmas star is folded/woven (Figure 1.4A,B, and C). More specifically, the polymerase would have four arms (one for each of the standard DNA nucleotides) that are activated by a specific wavelength. For the light activation element, domains from different opsins can be incorporated into the molecule, which would cause conformational changes in the appropriate arm resulting in its bending towards the central cavity where the nascent oligonucleotide is anchored (Figure 1.4D). Recruitment of the right nucleotide could be facilitated by selective affinity through specific amino acid interactions, while each arm should retain the properties of a polymerase to also form phosphodiester bonds. For an initial DNA fragment, which is normally needed for enzymatic DNA amplification, a poly-A tail (such as the one mature eukaryotic trnascripts have attached in their 3’ end) could be employed, which can be cleaved after synthesis of the desired oligonucletide is completed. This approach can, theoretically, be easily automated and scaled up by utilizing a 96-well Light Plate Apparatus (LPA) or it can be enhanced with the addition of more arms to integrate non-standard nucleotides as well, such as pseudouridine and inosine, for different applications. 1
 Figure 1.4 (A) and (B) Photographs of folding/weaving a danish Christmas star, which can be seen finished in (C). (D) Schematic representation of the “pinwheel” polymerase, whose arms bend and add a particular nucleotide upon activation with a specific wavelength. Figure from How to make a danish Christmas star, the Bureau of Betterment, and partially created on BioRender.com.
Figure 1.4 (A) and (B) Photographs of folding/weaving a danish Christmas star, which can be seen finished in (C). (D) Schematic representation of the “pinwheel” polymerase, whose arms bend and add a particular nucleotide upon activation with a specific wavelength. Figure from How to make a danish Christmas star, the Bureau of Betterment, and partially created on BioRender.com.
Fabris M, Abbriano RM, Pernice M, et al. Emerging Technologies in Algal Biotechnology: Toward the Establishment of a Sustainable, Algae-Based Bioeconomy. Front Plant Sci. 2020;11:279. doi:10.3389/fpls.2020.00279 ↩︎ ↩︎
Brodie J, Chan CX, De Clerck O, et al. The Algal Revolution. Trends Plant Sci. 2017;22(8):726-738. doi:10.1016/j.tplants.2017.05.005 ↩︎
Johnson X, Alric J. Central carbon metabolism and electron transport in Chlamydomonas reinhardtii: metabolic constraints for carbon partitioning between oil and starch. Eukaryotic Cell. 2013;12(6):776-793. doi:10.1128/EC.00318-12 ↩︎
Rochaix JD. Chlamydomonas reinhardtii as the photosynthetic yeast. Annu Rev Genet. 1995;29:209-230. doi:10.1146/annurev.ge.29.120195.001233 ↩︎
Calatrava V, Tejada-Jimenez M, Sanz-Luque E, Fernandez E, Galvan A. Nitrogen metabolism in Chlamydomonas. In: The Chlamydomonas Sourcebook. Elsevier; 2023:99-128. doi:10.1016/B978-0-12-821430-5.00004-3 ↩︎
Ghribi M, Nouemssi SB, Meddeb-Mouelhi F, Desgagné-Penix I. Genome Editing by CRISPR-Cas: A Game Change in the Genetic Manipulation of Chlamydomonas. Life (Basel). 2020;10(11). doi:10.3390/life10110295 ↩︎
Perozeni F, Baier T. Current Nuclear Engineering Strategies in the Green Microalga Chlamydomonas reinhardtii. Life (Basel). 2023;13(7). doi:10.3390/life13071566 ↩︎
Einhaus A, Baier T, Kruse O. Molecular design of microalgae as sustainable cell factories. Trends Biotechnol. December 12, 2023. doi:10.1016/j.tibtech.2023.11.010 ↩︎
Baier T, Kros D, Feiner RC, Lauersen KJ, Müller KM, Kruse O. Engineered Fusion Proteins for Efficient Protein Secretion and Purification of a Human Growth Factor from the Green Microalga Chlamydomonas reinhardtii. ACS Synth Biol. 2018;7(11):2547-2557. doi:10.1021/acssynbio.8b00226 ↩︎
Torres-Tiji Y, Fields FJ, Yang Y, et al. Optimized production of a bioactive human recombinant protein from the microalgae Chlamydomonas reinhardtii grown at high density in a fed-batch bioreactor. Algal Research. 2022;66:102786. doi:10.1016/j.algal.2022.102786 ↩︎
Sekimoto H. Sexual reproduction and sex determination in green algae. J Plant Res. 2017;130(3):423-431. doi:10.1007/s10265-017-0908-6 ↩︎
Shahar B, Haim E, Kuc ME, Azerrad SP, Dudai N, Kurzbaum E. Simplified and cost-effective modulatory photobioreactor setup for upscaling microalgal culture for research and semi-industrial purposes. Algal Research. 2023;74:103200. doi:10.1016/j.algal.2023.103200 ↩︎
Park S, Lee Y, Lee JH, Jin E. Expression of the high light-inducible Dunaliella LIP promoter in Chlamydomonas reinhardtii. Planta. 2013;238(6):1147-1156. doi:10.1007/s00425-013-1955-4 ↩︎ ↩︎
Kim J, Zhou Y, Carlson PD, et al. De novo-designed translation-repressing riboregulators for multi-input cellular logic. Nat Chem Biol. 2019;15(12):1173-1182. doi:10.1038/s41589-019-0388-1 ↩︎ ↩︎
Zhao EM, Mao AS, de Puig H, et al. RNA-responsive elements for eukaryotic translational control. Nat Biotechnol. 2022;40(4):539-545. doi:10.1038/s41587-021-01068-2 ↩︎ ↩︎
Sebesta J, Xiong W, Guarnieri MT, Yu J. Biocontainment of Genetically Engineered Algae. Front Plant Sci. 2022;13. doi:10.3389/fpls.2022.839446 ↩︎ ↩︎
Motomura K, Sano K, Watanabe S, et al. Synthetic Phosphorus Metabolic Pathway for Biosafety and Contamination Management of Cyanobacterial Cultivation. ACS Synth Biol. 2018;7(9):2189-2198. doi:10.1021/acssynbio.8b00199 ↩︎ ↩︎ ↩︎
Thapa R. DNA Replication: Enzymes, Mechanism, Steps, Applications. November 2, 2023. Accessed February 8, 2026. https://microbenotes.com/dna-replication-steps/ ↩︎ ↩︎
Prindle MJ, Loeb LA. DNA polymerase delta in DNA replication and genome maintenance. Environ Mol Mutagen. 2012;53(9):666-682. doi:10.1002/em.21745 ↩︎ ↩︎
Bulock CR, Xing X, Shcherbakova PV. Mismatch repair and DNA polymerase δ proofreading prevent catastrophic accumulation of leading strand errors in cells expressing a cancer-associated DNA polymerase ϵ variant. Nucleic Acids Res. 2020;48(16):9124-9134. doi:10.1093/nar/gkaa633 ↩︎ ↩︎ ↩︎ ↩︎
Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28(1):292. doi:10.1093/nar/28.1.292 ↩︎
Codon Usage Database. Accessed February 8, 2026. https://www.kazusa.or.jp/codon/ ↩︎
McLaughlin L. What Is Oligonucleotide Synthesis? Phosphoramidite oligonucleotide synthesis. May 8, 2025. Accessed February 7, 2026. https://www.biotechnologyreviews.com/p/what-is-oligonucleotide-synthesis ↩︎
A Simple Guide to Phosphoramidite Chemistry and How it Fits in Twist Bioscience’s Commercial Engine. Accessed February 8, 2026. https://www.twistbioscience.com/blog/science/simple-guide-phosphoramidite-chemistry-and-how-it-fits-twist-biosciences-commercial ↩︎
the bumbling biochemist. Solid State Oligonucleotide Synthesis (Phosphoramidite Method). 2023. Accessed February 8, 2026. https://www.youtube.com/watch?v=t29CQywQpMY ↩︎