Week 2 homework
DNA read, write, and edit 🧬
Part 1: Benchling and in-silico gel art
The genome of the λ-phage was imported and virtually digested with the following restriction endonucleases: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI before being visualized on Benchling’s agarose gel simulator (Figure 2.1).
 Figure 2.1 Virtual digest of λ-phage’s DNA treated with seven different restriction enzymes (as indicated by the gel lane legend on the top left). Figure created on Benchling.com.
Figure 2.1 Virtual digest of λ-phage’s DNA treated with seven different restriction enzymes (as indicated by the gel lane legend on the top left). Figure created on Benchling.com.
Part 3: DNA design challenge
3.1. Choose your protein. In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, Google), obtain the protein sequence for the protein you chose.
The entire medical establishment relies heavily on a sea creature so ancient and resilient that it has barely changed since its ancestral form first appeared in the tree of life when Planet Earth had its own ring system (like Saturn and Uranus), approximately 450 million years ago. Besides a hardy armour-like exoskeleton and a bizarre body plan, horseshoe crabs bring to the table a primitive, yet extremely functional, immune system tightly-packed in their blue-colored blood. A key component of their immune response are the granular amoebocytes found in their blood (Figure 2.2B), which, upon contact with a bacterial endotoxin, initiate a coagulation cascade that protects the horseshoe crab by sequestering and neutralizing the harmful agent (Figure 2.2C). This very property of horseshoe crab immunity has been harnessed by numerous medical corporations since the 1960s-1970s as an effective method to safely screen vaccines, other injectable pharmaceuticals, as well as implantable biomedical devices, for the presence of bacteria-derived toxins in a procedure called “Limulus amoebocyte lysate (LAL) assay” 1 (Figure 2.2A).
 Figure 2.2 (A) Photograph from a horseshoe crab blood harvesting facility. (B) The blue blood of horseshoe crabs contains granular amoebocytes (depicted in brown), which, upon contact with a bacterial endotoxin (mostly a lipopolysaccharide residue), trigger a coagulation cascade that sequesters the foreign compound. This property has been utilized by the medical establishment for decades to test pharmaceuticals for the presence of bacterial endotoxins (C). Figure from an NPR’s report on horseshoe crab blood harvesting and partially created on BioRender.com.
Figure 2.2 (A) Photograph from a horseshoe crab blood harvesting facility. (B) The blue blood of horseshoe crabs contains granular amoebocytes (depicted in brown), which, upon contact with a bacterial endotoxin (mostly a lipopolysaccharide residue), trigger a coagulation cascade that sequesters the foreign compound. This property has been utilized by the medical establishment for decades to test pharmaceuticals for the presence of bacterial endotoxins (C). Figure from an NPR’s report on horseshoe crab blood harvesting and partially created on BioRender.com.
Based on this premise, an interesting idea to pursue would be to perform whole-cell engineering of bacteria or, preferably, yeast cells to render them functionally similar to the granular amoebocytes contained in horseshoe crab blood. This endeavor, primarily inspired by an old iGEM project aiming to convert Escherichia coli bacteria into red blood cells 2, could contribute to the conservation of the fragile ecosystem to which horseshoe crabs belong, but also drastically confine the invasive, time-consuming, and expensive practice of harvesting horseshoe crab haemolymph 3.
To this end, a critical step is to transform the cells-to-be-engineered with the gene coding for factor C, namely the main protein that initiates the immune response and triggers the coagulation pathway 4 5. The amino acid sequence for the factor C protein of the mangrove horseshoe crab Carcinoscorpius rotundicauda (CrFactor C) was retrieved from UniProt under the accession number “Q26422” 4 6:
>sp|Q26422|LFC_CARRO Limulus clotting factor C OS=Carcinoscorpius rotundicauda OX=6848 PE=2 SV=1 MVLASFLVSGLVLGLLAQKMRPVQSKGVDLGLCDETRFECKCGDPGYVFNIPVKQCTYFY RWRPYCKPCDDLEAKDICPKYKRCQECKAGLDSCVTCPPNKYGTWCSGECQCKNGGICDQ RTGACACRDRYEGVHCEILKGCPLLPSDSQVQEVRNPPDNPQTIDYSCSPGFKLKGMARI SCLPNGQWSNFPPKCIRECAMVSSPEHGKVNALSGDMIEGATLRFSCDSPYYLIGQETLT CQGNGQWNGQIPQCKNLVFCPDLDPVNHAEHKVKIGVEQKYGQFPQGTEVTYTCSGNYFL MGFDTLKCNPDGSWSGSQPSCVKVADREVDCDSKAVDFLDDVGEPVRIHCPAGCSLTAGT VWGTAIYHELSSVCRAAIHAGKLPNSGGAVHVVNNGPYSDFLGSDLNGIKSEELKSLARS FRFDYVRSSTAGKSGCPDGWFEVDENCVYVTSKQRAWERAQGVCTNMAARLAVLDKDVIP NSLTETLRGKGLTTTWIGLHRLDAEKPFIWELMDRSNVVLNDNLTFWASGEPGNETNCVY MDIQDQLQSVWKTKSCFQPSSFACMMDLSDRNKAKCDDPGSLENGHATLHGQSIDGFYAG SSIRYSCEVLHYLSGTETVTCTTNGTWSAPKPRCIKVITCQNPPVPSYGSVEIKPPSRTN SISRVGSPFLRLPRLPLPLARAAKPPPKPRSSQPSTVDLASKVKLPEGHYRVGSRAIYTC ESRYYELLGSQGRRCDSNGNWSGRPASCIPVCGRSDSPRSPFIWNGNSTEIGQWPWQAGI SRWLADHNMWFLQCGGSLLNEKWIVTAAHCVTYSATAEIIDPNQFKMYLGKYYRDDSRDD DYVQVREALEIHVNPNYDPGNLNFDIALIQLKTPVTLTTRVQPICLPTDITTREHLKEGT LAVVTGWGLNENNTYSETIQQAVLPVVAASTCEEGYKEADLPLTVTENMFCAGYKKGRYD ACSGDSGGPLVFADDSRTERRWVLEGIVSWGSPSGCGKANQYGGFTKVNVFLSWIRQFI
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence. The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (Google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
The nucleotide coding sequence for CrFactor C was extracted from the respective NCBI entry 7:
>S77063.1 factor C=endotoxin-sensitive intracellular serine protease zymogen {clone CrFC21} [Carcinoscorpius rotundicauda=Singapore horseshoe crabs, blood, amoebocytes, CDS, 3060 nt] ATGGTCTTAGCGTCGTTTTTGGTGTCTGGTTTAGTTCTAGGGCTACTAGCCCAAAAAATGCGCCCAGTTCAGTCCAAAGGAGTAGATCTAGGCTTGTGTGATGAAACGAGGTTCGAGTGTAAGTGTGGCGATCCAGGCTATGTGTTCAACATTCCAGTGAAACAATGTACATACTTTTATCGATGGAGGCCGTATTGTAAACCATGTGATGACCTGGAGGCTAAGGATATTTGTCCAAAGTACAAACGATGTCAAGAGTGTAAGGCTGGTCTTGATAGTTGTGTTACTTGTCCACCTAACAAATATGGTACTTGGTGTAGCGGTGAATGTCAGTGTAAGAATGGAGGTATCTGTGACCAGAGGACAGGAGCTTGTGCATGTCGTGACAGATATGAAGGGGTGCACTGTGAAATTCTCAAAGGTTGTCCTCTTCTTCCATCGGATTCTCAGGTTCAGGAAGTCAGAAATCCACCAGATAATCCCCAAACTATTGACTACAGCTGTTCACCAGGGTTCAAGCTTAAGGGTATGGCACGAATTAGCTGTCTCCCAAATGGACAGTGGAGTAACTTTCCACCCAAATGTATTCGAGAATGTGCCATGGTTTCATCTCCAGAACATGGGAAAGTGAATGCTCTTAGTGGTGATATGATAGAAGGGGCTACTTTACGGTTCTCATGTGATAGTCCCTACTACTTGATTGGTCAAGAAACATTAACCTGTCAGGGTAATGGTCAGTGGAATGGACAGATACCACAATGTAAGAACTTGGTCTTCTGTCCTGACCTGGATCCTGTAAACCATGCTGAACACAAGGTTAAAATTGGTGTGGAACAAAAATATGGTCAGTTTCCTCAAGGCACTGAAGTGACCTATACGTGTTCGGGTAACTACTTCTTGATGGGTTTTGACACCTTAAAATGTAACCCTGATGGGTCTTGGTCAGGATCACAGCCATCCTGTGTTAAAGTGGCAGACAGAGAGGTCGACTGTGACAGTAAAGCTGTAGACTTCTTGGATGATGTTGGTGAACCTGTCAGGATCCACTGTCCTGCTGGCTGTTCTTTGACAGCTGGTACTGTGTGGGGTACAGCCATATACCATGAACTTTCCTCAGTGTGTCGTGCAGCCATCCATGCTGGCAAGCTTCCAAACTCTGGAGGAGCGGTGCATGTTGTGAACAATGGCCCCTACTCGGACTTTCTGGGTAGTGACCTGAATGGGATAAAATCGGAAGAGTTGAAGTCTCTTGCCCGGAGTTTCCGATTCGATTATGTCCGTTCCTCCACAGCAGGTAAATCAGGATGTCCTGATGGATGGTTTGAGGTAGACGAGAACTGTGTGTACGTTACATCAAAACAGAGAGCCTGGGAAAGAGCTCAAGGTGTGTGTACCAATATGGCTGCTCGTCTTGCTGTGCTGGACAAAGATGTAATTCCAAATTCGTTGACTGAGACTCTACGAGGGAAAGGGTTAACAACCACGTGGATAGGATTGCACAGACTAGATGCTGAGAAGCCCTTTATTTGGGAGTTAATGGATCGTAGTAATGTGGTTCTGAATGATAACCTAACATTCTGGGCCTCTGGCGAACCTGGAAATGAAACTAACTGTGTATATATGGACATCCAAGATCAGTTGCAGTCTGTGTGGAAAACCAAGTCATGTTTTCAGCCCTCAAGTTTTGCTTGCATGATGGATCTGTCAGACAGAAATAAAGCCAAATGCGATGATCCTGGATCACTGGAAAATGGACACGCCACACTTCATGGACAAAGTATTGATGGGTTCTATGCTGGTTCTTCTATAAGGTACAGCTGTGAGGTTCTCCACTACCTCAGTGGAACTGAAACCGTAACTTGTACAACAAATGGCACATGGAGTGCTCCTAAACCTCGATGTATCAAAGTCATCACCTGCCAAAACCCCCCTGTACCATCATATGGTTCTGTGGAAATCAAACCCCCAAGTCGGACAAACTCGATAAGTCGTGTTGGGTCACCTTTCTTGAGGTTGCCACGGTTACCCCTCCCATTAGCTAGAGCAGCCAAACCTCCTCCAAAACCTAGATCCTCACAACCCTCTACTGTGGACTTGGCTTCTAAAGTTAAACTACCTGAAGGTCATTACCGGGTAGGGTCTCGAGCCATCTACACGTGCGAGTCGAGATACTACGAACTACTTGGATCTCAAGGCAGAAGATGTGACTCTAATGGAAACTGGAGTGGTCGGCCAGCGAGCTGTATTCCAGTTTGTGGACGGTCAGACTCTCCTCGTTCTCCTTTTATCTGGAATGGGAATTCTACAGAAATAGGTCAGTGGCCGTGGCAGGCAGGAATCTCTAGATGGCTTGCAGACCACAATATGTGGTTTCTCCAGTGTGGAGGATCTCTATTGAATGAGAAATGGATCGTCACTGCTGCCCACTGTGTCACCTACTCTGCTACTGCTGAGATTATTGACCCCAATCAGTTTAAAATGTATCTGGGCAAGTACTACCGTGATGACAGTAGAGACGATGACTATGTACAAGTAAGAGAGGCTCTTGAGATCCACGTGAATCCTAACTACGACCCCGGCAATCTCAACTTTGACATAGCCCTAATTCAACTGAAAACTCCTGTTACTTTGACAACACGAGTCCAACCAATCTGTCTGCCTACTGACATCACAACAAGAGAACACTTGAAGGAGGGAACATTAGCAGTGGTGACAGGTTGGGGTTTGAATGAAAACAACACCTATTCAGAGACGATTCAACAAGCTGTGCTACCTGTTGTTGCAGCCAGCACCTGTGAAGAGGGGTACAAGGAAGCAGACTTACCACTGACAGTAACAGAGAACATGTTCTGTGCAGGTTACAAGAAGGGACGTTATGATGCCTGCAGTGGGGACAGTGGAGGACCTTTAGTGTTTGCTGATGATTCCCGTACCGAAAGGCGGTGGGTCTTGGAAGGGATTGTCAGCTGGGGCAGTCCCAGTGGATGTGGCAAGGCGAACCAGTACGGGGGCTTCACTAAAGTTAACGTTTTCCTGTCATGGATTAGGCAGTTCATTTGA
3.3. Codon optimization. Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize Google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Different organisms display different codon biases regarding protein translation 8 9. Codon bias and codon usage are predominantly determined by the relative abundance of aminoacyl-tRNAs and aminoacyl-tRNA synthetases inside a cell, as they both constitute crucial components of the translational machinery, carrying the proteinogenic amino acids and loading the aminoacyl-tRNAs with the proper amino acid respectively. This codon bias has to be taken into account when transforming an organism with a gene from another organism to modify the coding sequence accordingly, enabled by the degeneracy of the genetic code, and render it compatible with the host cell’s translational machinery, thus ensuring smooth heterologous expression of the protein of interest. If the gene of interest is not codon optimized for the expression host, it is likely that the protein will be synthesized at very low levels or not at all.
Similarly, when choosing an expression host for synthesizing the protein of interest, several parameters have to be considered as well. In this specific case, factor C is a protein derived from a eukaryote, has a complex molecular structure involving disulfide bonds, and is glycosylated at several amino acid positions. For the expression of a protein with those characteristics, a putative host should also be a eukaryote, as eukaryotic cells harbor the necessary biochemical pathways for protein post-translational modifications, such as glycosylation, and, additionally, should have an oxidizing intracellular environment to facilitate the formation of disulfide bridges. A promising candidate that fulfils all those criteria is the methylotrophic yeast Pichia pastoris, for which the original coding sequence for factor C has been codon optimized employing Benchling’s codon optimization tool:
>factor C=endotoxin-sensitive intracellular serine protease zymogen {clone CrFC21} [codon optimized for Pichia pastoris, CDS, 3060 nt] ATGGTCTTAGCGTCGTTTTTGGTTTCTGGTTTAGTTCTAGGGCTACTAGCCCAAAAAATGCGCCCAGTTCAGTCCAAAGGAGTAGATCTAGGCTTGTGTGATGAAACGAGGTTCGAGTGTAAGTGTGGCGATCCAGGCTATGTTTTCAACATTCCAGTCAAACAATGCACATACTTTTATCGATGGAGGCCGTATTGTAAACCATGTGATGACCTGGAGGCTAAGGATATTTGTCCAAAGTACAAGCGATGTCAAGAGTGTAAGGCTGGTCTTGATAGTTGTGTTACTTGTCCACCTAACAAGTATGGTACTTGGTGTAGCGGTGAATGTCAGTGCAAGAACGGAGGTATCTGTGACCAGAGGACAGGAGCTTGTGCATGTCGTGACAGATATGAAGGGGTGCACTGCGAAATTCTCAAAGGTTGTCCTCTTCTTCCATCGGATTCTCAGGTTCAAGAAGTCAGAAATCCACCAGATAATCCCCAAACTATTGACTACAGCTGCTCACCAGGGTTCAAGCTTAAGGGTATGGCACGAATTAGCTGCCTCCCAAATGGACAGTGGAGTAACTTTCCACCAAAATGTATTAGAGAATGTGCCATGGTTTCATCTCCAGAACATGGTAAAGTTAATGCTCTTTCCGGTGATATGATAGAAGGTGCTACTTTACGGTTCTCCTGTGATAGTCCCTACTACTTGATTGGTCAAGAAACATTAACCTGCCAAGGTAATGGTCAGTGGAATGGACAGATACCACAATGTAAGAACTTGGTCTTTTGCCCTGACCTGGATCCTGTAAACCATGCTGAACACAAGGTTAAAATTGGTGTTGAACAAAAATATGGTCAGTTTCCTCAAGGAACTGAAGTTACCTATACGTGTTCGGGTAACTACTTCTTGATGGGTTTTGATACCTTAAAATGCAACCCTGATGGGTCTTGGTCAGGATCACAGCCATCCTGTGTTAAAGTGGCAGACAGAGAGGTCGACTGTGACAGTAAAGCTGTAGACTTCTTGGATGATGTTGGTGAACCGGTCAGGATCCACTGTCCTGCTGGCTGTTCTTTGACAGCTGGTACTGTTTGGGGTACAGCCATATACCATGAGCTTTCCTCCGTGTGCCGCGCAGCCATCCATGCTGGCAAGCTTCCAAACTCTGGAGGAGCTGTCCATGTTGTGAACAATGGCCCGTACTCCGACTTTCTGGGTTCCGACCTGAATGGTATAAAATCGGAAGAGTTGAAGTCTCTTGCCAGAAGTTTTAGATTCGATTATGTCCGTTCCTCCACAGCAGGTAAGTCAGGATGCCCTGATGGATGGTTTGAGGTAGACGAGAACTGTGTGTATGTTACATCAAAGCAGAGAGCATGGGAAAGAGCTCAAGGTGTGTGCACCAATATGGCTGCTAGACTTGCTGTGCTGGACAAAGATGTAATTCCAAACTCGTTGACTGAGACTCTAAGAGGGAAAGGTTTAACCACCACGTGGATAGGATTGCATAGACTAGATGCTGAGAAGCCCTTTATTTGGGAGTTAATGGATCGTAGTAATGTGGTTCTGAATGATAACCTAACCTTCTGGGCCTCTGGTGAACCTGGAAATGAAACTAACTGCGTATATATGGACATCCAAGATCAGTTGCAGTCTGTGTGGAAAACCAAGTCATGTTTTCAGCCATCTAGTTTTGCTTGCATGATGGATCTGTCAGATAGAAATAAAGCCAAGTGCGATGATCCTGGATCATTGGAAAATGGACACGCCACACTTCATGGACAATCCATTGATGGTTTCTATGCTGGTTCTTCTATAAGGTACAGCTGCGAGGTTCTCCACTACCTCAGTGGAACTGAAACCGTAACTTGTACCACAAATGGCACTTGGAGTGCTCCGAAACCGCGATGTATCAAAGTCATCACCTGCCAAAACCCCCCTGTACCATCATATGGTTCTGTGGAAATCAAACCCCCAAGTAGAACTAACTCGATAAGTCGTGTTGGGTCACCTTTCTTGAGGTTGCCAAGATTACCCCTCCCATTAGCTAGAGCAGCCAAGCCTCCTCCAAAGCCTAGATCCTCACAACCCTCTACTGTGGACTTGGCCTCTAAGGTTAAATTGCCTGAAGGTCATTACCGTGTCGGGTCTAGGGCCATCTACACGTGCGAGTCGAGATACTACGAACTATTGGGATCTCAAGGCAGAAGATGTGACTCTAACGGAAACTGGTCCGGTCGGCCAGCGAGCTGTATTCCAGTTTGCGGACGGTCAGATTCTCCTCGTTCTCCTTTTATCTGGAATGGTAATTCTACAGAAATTGGTCAGTGGCCGTGGCAGGCAGGAATCTCTAGATGGCTTGCAGACCACAATATGTGGTTTCTCCAATGTGGAGGATCTCTATTGAATGAGAAGTGGATCGTCACTGCTGCCCATTGTGTCACCTACTCTGCTACTGCTGAGATTATTGACCCCAATCAATTTAAAATGTATCTGGGCAAGTACTACCGTGATGACTCCAGAGATGATGACTATGTACAAGTAAGAGAGGCTCTTGAGATCCACGTCAATCCTAACTACGACCCCGGCAATTTGAACTTTGACATAGCCTTGATTCAACTGAAAACTCCTGTTACTTTGACTACACGAGTCCAACCAATTTGTCTGCCTACTGACATCACGACAAGAGAACATTTGAAGGAGGGAACATTAGCAGTTGTTACGGGTTGGGGTTTGAATGAAAACAACACCTATTCAGAGACTATTCAACAAGCTGTGTTGCCTGTTGTTGCAGCCAGCACCTGCGAAGAGGGGTACAAGGAGGCAGACTTACCACTGACTGTTACAGAGAACATGTTCTGTGCAGGTTACAAGAAGGGACGTTATGATGCCTGCTCCGGTGACAGCGGAGGACCTTTAGTGTTTGCTGATGATTCCCGTACCGAAAGGAGATGGGTCTTGGAAGGGATTGTCAGCTGGGGCAGTCCCTCCGGATGTGGAAAGGCGAACCAGTATGGTGGCTTCACTAAAGTTAACGTTTTCCTGTCATGGATTAGACAATTCATTTAA
3.4. You have a sequence! Now what? What technologies could be used to produce this protein from your DNA? Describe in your words how the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
For the expression of the codon-optimized version of CrFactor C in P. pastoris, the first step would be to replace the regulatory elements integrated in the cassette generated above, as they have been selected for bacterial expression, with parts that would be recognized in a yeast cell, such as the methanol-activated AOX1 promoter, an appropriate Kozak sequence, and the AOX1 terminator. After assembling the new genetic cassette for P. pastoris expression, it would have to be inserted into an integrative vector (probably from the pPICZα series), which would also carry a selection marker, for instance, zeocin, in conjunction with the expression cassette for the protein of interest. Subsequently, this integrative vector would be employed for the transformation of the yeast cells. In a portion of the successfully transformed yeast cells, the expression cassette-selection marker sequence would then be incorporated into the organism’s genome and, through the antibiotic resistance conferred by the selection marker, those positive transformants could be identified. By utilizing zeocin in particular, the most highly expressing strains can be readily isolated through increasing the dose of the antibiotic, as the resistance provided by zeocin is directly proportional to the number of selection marker genes integrated, which is a direct indication for the number of genes encoding the protein of interest integrated as well. The highly-expressing positive transformants would, afterwards, be cultured in the presence of methanol, which can strongly induce the transcription of the codon-optimized CrFactor C gene, whose mRNA would then be translated (ensured by the Kozak consensus) into CrFactor C protein molecules. Lastly, the nascent CrFactor C protein would be transferred to the endoplasmic reticulum (ER) and the Golgi apparatus for post-translational modifications.
For proteins with extended post-translational modification requirements, such as factor C, a cell-free expression system would not be recommended. However, a T7-based in vitro transcription method, coupled with a highly active and reliable in vitro eukaryotic translation system, such as rabbit reticulocyte lysate (RRL), would be an appealing alternative. For the expression of recombinant CrFactor C, though, the translation system should also be supplemented with microsomal membranes to secure the capacity for glycosylation 10.
3.5. How does it work in nature/biological systems?
- Describe how a single gene codes for multiple proteins at the transcriptional level.
At the transcriptional/post-transcriptional level, the same gene, or rather, the same transcript, can code for multiple proteins through the mechanism of alternative splicing. More specifically, in the majority of eukaryotic organisms, genes consist of both exons, genetic segments that can be translated, and introns, genetic segments that are not translated but have regulatory roles. A crucial step in the transcript maturation process is the excision of introns, which is performed by the spliceosome, a large ribonucleoprotein complex found within the nucleus of the cell. Apart from removing the non-coding introns, the spliceosome also contributes to joining the remaining regions, namely the exons, from the precursor mRNA to generate the mature transcript. However, during this process, one or more different exons can be omitted, if, for instance, they are excised along with their flanking introns, leading to multiple variations of the mature mRNA (depending on the exons it contains) and, thus, to multiple proteins. Another mechanism through which a transcript can encode multiple proteins are multicistronic genes in prokaryotes. Multicistronic genes are frequently found clustered in prokaryotic operons, where a single transcription process, initiated by the gene’s sole promoter, leads to the synthesis of a long transcript with more than one start codons. Each start codon results in the expression of a different protein, albeit with all translation products from one multicistronic gene usually serving metabolic functions of the same metabolic pathway.
More clearly at the translational level, a single gene can code for multiple proteins if it contains more than one reading frames in eukaryotes too. The reading frames can also overlap, however, each one has its own start and stop codon. Lastly, at the post-translational level, a protein in a eukaryotic cell can exist in several distinct versions. Even though those versions are not considered different molecules, as they occurred from the same gene, a protein can be altered after it has been biosynthesized through post-translational modifications (PTMs), which include, but are not limited to, the cleavage of the initial methionine amino acid residue and the addition of other chemical moieties, such as glycans or phosphate groups.
- Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated protein!
The alignment of the gene for the codon-optimized CrFactor C, the transcribed RNA, and the resulting protein is illustrated in Figure 2.3. As the gene is fairly large, I opted to show only the part of the gene that codes for the protein’s N-terminus.
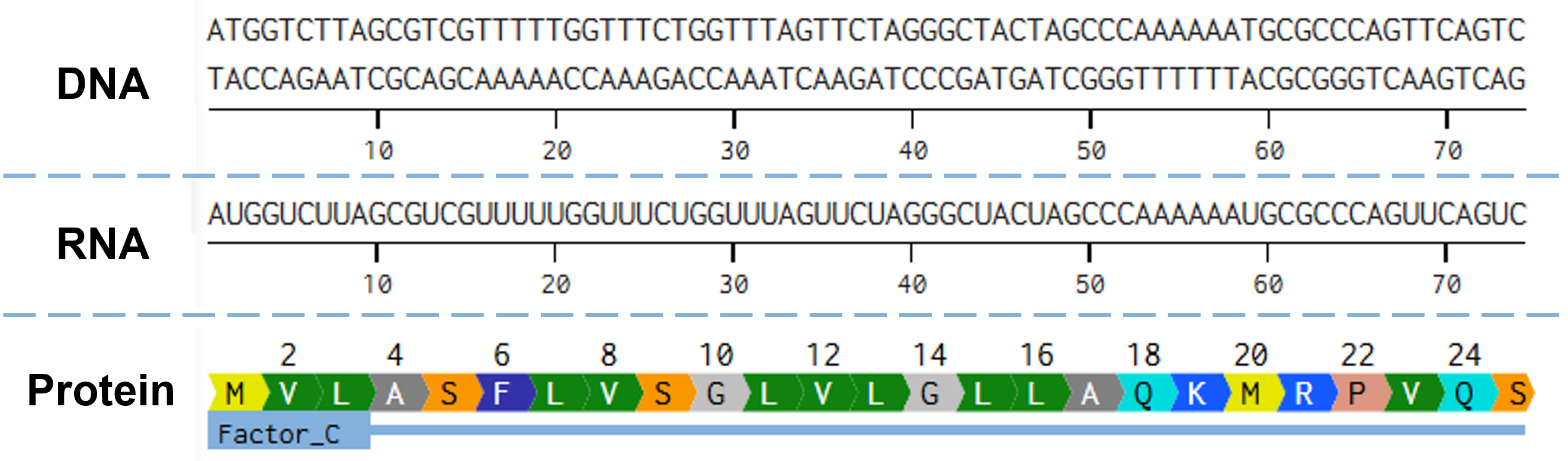 Figure 2.3 Alignment of the gene encoding CrFactor C, as it has been codon optimized for expression in P. pastoris, with its mature mRNA transcript, and the resulting CrFactor C protein. Figure generated with Benchling.com.
Figure 2.3 Alignment of the gene encoding CrFactor C, as it has been codon optimized for expression in P. pastoris, with its mature mRNA transcript, and the resulting CrFactor C protein. Figure generated with Benchling.com.
Part 4: Prepare a Twist DNA Synthesis Order
By following the instructions on preparing a DNA synthesis order for Twist, a plasmid containing the CrFactor C sequence codon-optimized for P. pastoris was generated (Figure 2.4).
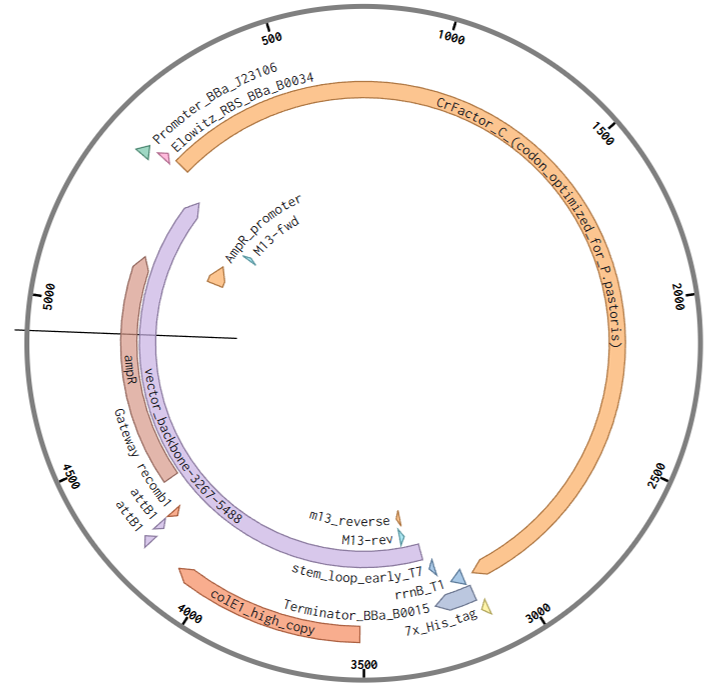
Figure 2.4 Snapshot of the plasmid map for pTwist(amp, high copy)-CrFactor_C generated for the Twist DNA synthesis order. Plasmid map created on Benchling.com.
Part 5: DNA read/write/edit
5.1 DNA Read (i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
In continuation of the CrFactor C project, once again, expressing the codon-optimized sequence in P. pastoris would require assembling the genetic cassette, including the gene and all its flanking regulatory elements. Sequencing the assembled construct constitutes an important step before proceeding with the transformation in order to verify that the cloning was indeed successful and that the newly assembled expression cassette is identical to the designed one.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also, answer the following questions:
- Is your method first-, second- or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
For whole-plasmid sequencing of the integrative vector carrying the CrFactor C codon-optimized sequence, I would choose Oxford Nanopore sequencing. It is a third-generation sequencing technology that combines high speed, reliability, and read accuracy (98-99%), as well as low cost.
Another advantage of this technology is that it requires minimal library preparation, essentially omitting time-consuming steps, such as PCR or reverse transcription of RNA into DNA. Sample preparation involves a single, rapid “tagmentation” step facilitated by a hyperactive transposase complex. The complex, also called a transposome, is pre-loaded with synthetic, double-stranded adapters to simultaneously fragment DNA or RNA strands and ligate sequencing adapters, which, for Nanopore sequencing specifically, carry a motor protein as well that will help thread the nucleotide chains through the nanopores in the flow cell. The entire process of transposome-based library preparation lasts no more than 5min.
Once preparation of the sequencing library is completed, the sample can be loaded onto the flow cell, which contains thousands of nanopores (nano-scale protein channels) embedded in an electrically resistant membrane. The membrane separates two chambers, each filled with an electrolyte solution, and allows the flow of ions between the two chambers exclusively through its integrated nanopores. The ionic current induced by the flow of charged particles through each nanopore is monitored and measured with an individual electronic sensor. The sensor detects and reports the disruptions in the ionic current caused by the passing through of individual nucleotide bases, each of which displays a unique electrical fingerprint, hence electrical disruptive pattern, depending on its biochemical properties. Specialized base-calling algorithms interpret those disruptions by generating data in the form of nucleotide sequences in real time. Therefore, the output of the technology is an electrical current graph depicting “squiggles” of specific amplitude, which can then be translated into sequencing data consisting of readable nucleotide chains. Those sequencing data can be further processed afterwards by employing alignment, assembly, and analysis bioinformatics tools 11.
5.2 DNA Write (i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize!
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
To actually create the genetic circuit for the expression of CrFactor C in P. pastoris described above, each genetic element would first have to be individually synthesized by a DNA synthesis company, such as Twist. Apart from the codon-optimized sequence of the CrFactor C gene previously shown, those genetic elements include:
- the AOX1 promoter from P. pastoris, combined with its native Kozak sequence (last six nucleotides)
CTGTTCTAACCCCTACTTGACAGCAATATATAAACAGAAGGAAGCTGCCCTGTCTTAAACCTTTTTTTTTATCATCATTATTAGCTTACTTTCATAATTGCGACTGGTTCCAATTGACAAGCTTTTGATTTTAACGACTTTTAACGACAACTTGAGAAGATCAAAAAACAACTAATTATTGAAAGAATTCCGAAACGand - the AOX1 terminator from P. pastoris
TCAAGAGGATGTCAGAATGCCATTTGCCTGAGAGATGCAGGCTTCATTTTTGATACTTTTTTATTTGTAACCTATATAGTATAGGATTTTTTTTGTCATTTTGTTTCTTCTCGTACGAGCTTGCTCCTGATCAGCCTATCTCGCAGCTGATGAATATCTTGTGGTAGGGGTTTGGGAAAATCATTCGAGTTTGATGTTTTTCTTGGTATTTCCCACTCCTCTTCAGAGTACAGAAGATTAAGTGAGA
Both aforementioned parts have been sourced from the iGEM Registry of Standard Biological Parts. After receiving the individually synthesized parts, the promoter, Kozak consensus, coding sequence, and terminator would have to be assembled into the genetic cassette for the expression of the CrFactor C. For this, I would use a reliable cloning technique, such as Golden Gate assembly, which capitalizes on type IIS restriction enzymes’ property to cleave a double-stranded DNA molecule downstream of their recognition site. This property allows the generation of custom four-nucleotide overhangs with the use of solely one restriction enzyme, enabling the assembly of complex DNA constructs (consisting of up to 30 distinct parts) seamlessly, with an extremely low error rate in a one-pot reaction. This method requires a lot of caution in its design phase, so that the appropriate flanking regions that will generate the suitable overhangs for the assembly step are added to each segment with the right orientation (alternatively, they can also be added after the initial synthesis of the parts through PCR). Additionally, it is important to remove any internal recognition sites for the selected type IIS endonuclease through site-directed mutagenesis before construct assembly. With proper design and execution of the previously mentioned laboratory techniques, the genetic cassette for the expression of the recombinant CrFactor C in P. pastoris can, therefore, be synthesized.
5.3 DNA Edit (i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Although it would probably be more rational to simply redo the cassette assembly with the desired DNA elements, I could use a gene editing method, such as CRISPR, to swap the bacteria-specific regulatory parts in the pTwist(amp, high copy)-CrFactor_C plasmid generated above for the Twist order with regulatory parts with a similar function but specialized for gene expression in P. pastoris. Those genetic segments would include the promoter, RBS, and terminator already integrated into the vector, which would be replaced by the AOX1 promoter, its native Kozak sequence, and the AOX1 terminator respectively. This way, an already assembled genetic construct can be repurposed for heterologous expression in a different organism (in this case, one that is more appropriate for the expression of the selected gene). At a later stage, the swapped regulatory sequences can be altered again with new ones, for instance, a stronger promoter and a more reliable terminator, to optimize transcription and obtain more robust protein synthesis.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions:
- How does your technology of choice edit DNA? What are the essential steps?
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The CRISPR-Cas9 system is a tremendously versatile and powerful genome-editing technology that can introduce precise modifications in DNA. The principle, as well as the main components of the system, have been adapted from bacterial cells, where transcripts from the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) array are combined with Cas (CRISPR-associated) proteins to recognize and neutralize foreign genetic material, such as bacteriophage DNA, thus functioning as a natural defense mechanism.
At its core, CRISPR editing relies on guiding a DNA-cutting enzyme to a specific sequence in the genome, where it introduces a targeted break that the cell then repairs, often resulting in a desired genetic modification. More specifically, a guide molecule called a single-guide RNA (sgRNA or gRNA), which is designed to be complementary to the target DNA sequence, forms a complex with the Cas9 endonuclease (Figure 2.5A and B). Once the complex is delivered into the target cells, the gRNA directs Cas9 to the genetic target through base pairing. Importantly, the target site must be adjacent to a short motif known as the PAM (protospacer adjacent motif), which is required for Cas9 binding. Upon recognition, Cas9 generates a double-strand break in the DNA (Figure 2.5C), which in turn activates the cell’s natural repair pathways. The repair mechanisms include non-homologous end joining (NHEJ) or homology-directed repair (HDR), with the former often introducing brief insertions or deletions to the target site, which can be disruptive to the gene’s function. The latter, on the other hand, enables incorporating a DNA template to obtain more precise editing (Figure 2.5D).
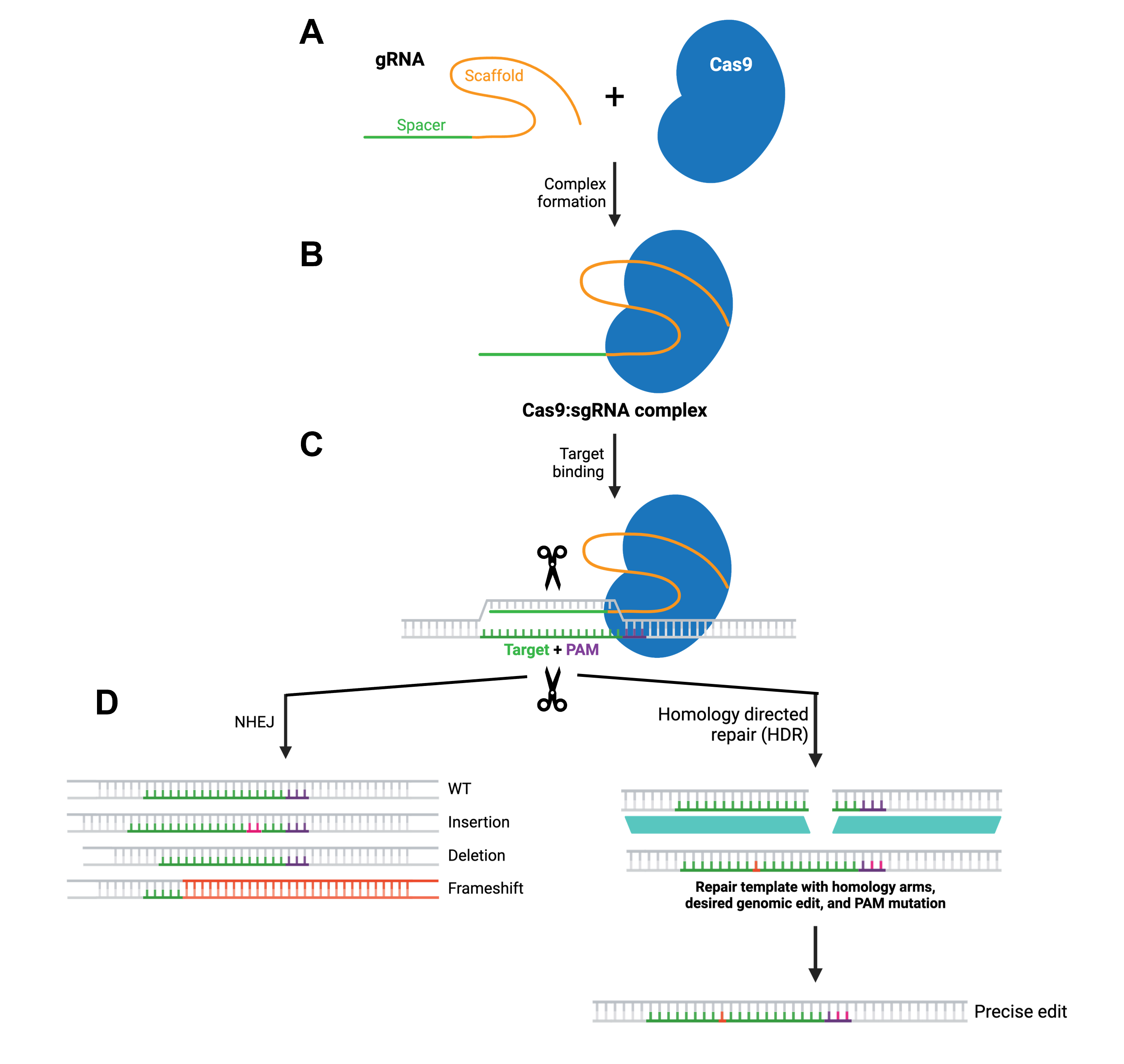 Figure 2.5 Overview of the CRISPR-Cas9 gene editing mechanism. Figure modified from Addgene’s CRISPR guide.
Figure 2.5 Overview of the CRISPR-Cas9 gene editing mechanism. Figure modified from Addgene’s CRISPR guide.
Preparation for a CRISPR experiment involves several design and input considerations. First, the target DNA sequence must be computationally analyzed to identify sites that can be specifically targeted by gRNA molecules and are located near a PAM sequence. The selected sgRNA sequence is then synthesized and often inserted into a plasmid vector that also encodes Cas9. Alternatively, Cas9 protein and sgRNA can be delivered directly as a ribonucleoprotein complex. If the experiment aims to integrate a precisely altered sequence via HDR, a donor DNA template containing the intended modification should be designed as well, typically with homologous arms flanking the edit site. Additional inputs may include primers for verification, plasmids for delivery (as already mentioned), appropriate host cells depending on the application, as well as availability of delivery methods depending, in turn, on host cell type.
Despite its potential, CRISPR has several limitations, the main one concerning off-target effects, where the Cas9-sgRNA complex binds and cuts at unintended genomic sites, potentially causing unwanted mutations. Improved gRNA design and optimized Cas variants have reduced this risk, without, however, entirely eliminating it. Efficiency can be another shortcoming of the method, particularly for HDR-based edits, which are often less efficient than NHEJ and are usually influenced by the cell cycle stage. Additionally, delivery of CRISPR components into certain cell types or tissues remains challenging. There are also biological constraints, such as immune responses to Cas proteins and variability in editing outcomes between cells. Lastly, ethical considerations and regulatory frameworks can confine CRISPR implementation, especially in human germline editing.
Real Science. Why Horseshoe Crab Blood Is So Valuable. 2020. Accessed February 15, 2026. https://www.youtube.com/watch?v=oXVnuG3zO_0 ↩︎
BerkiGEM2007Present1 - 2007.igem.org. Accessed February 16, 2026. https://2007.igem.org/BerkiGEM2007Present1 ↩︎
Maloney T, Phelan R, Simmons N. Saving the horseshoe crab: A synthetic alternative to horseshoe crab blood for endotoxin detection. PLoS Biol. 2018;16(10):e2006607. doi:10.1371/journal.pbio.2006607 ↩︎
Ding JL, Navas MA, Ho B. Molecular cloning and sequence analysis of factor C cDNA from the Singapore horseshoe crab, Carcinoscorpius rotundicauda. Mol Marine Biol Biotechnol. 1995;4(1):90-103. ↩︎ ↩︎
Ding JL, Ho B. Endotoxin detection–from limulus amebocyte lysate to recombinant factor C. Subcell Biochem. 2010;53:187-208. doi:10.1007/978-90-481-9078-2_9 ↩︎
UniProt. UniProt. Accessed February 15, 2026. https://www.uniprot.org/uniprotkb/Q26422/entry ↩︎
factor C=endotoxin-sensitive intracellular serine protease zymogen {cl - Nucleotide - NCBI. Accessed February 15, 2026. https://www.ncbi.nlm.nih.gov/nuccore/S77063 ↩︎
Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28(1):292. doi:10.1093/nar/28.1.292 ↩︎
Codon Usage Database. Accessed February 8, 2026. https://www.kazusa.or.jp/codon/ ↩︎
Beckler GS, Thompson D, Van Oosbree T. In vitro translation using rabbit reticulocyte lysate. Methods Mol Biol. 1995;37:215-232. doi:10.1385/0-89603-288-4:215 ↩︎
BestDx Academy. Nanopore Sequencing. 2023. Accessed February 15, 2026. https://www.youtube.com/watch?v=FYEWrUVJ2as ↩︎