Week 4 homework
Protein design-Part I 💻
Part 1: Conceptual questions
Answer any nine of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (On average, an amino acid is ~100 Daltons.)
Depending on the type of meat, as well as the manner it is processed prior to consumption, 500g of meat contain approximately 100 - 130g of protein. Assuming that this protein consists entirely of amino acids (meaning, excluding metal ions, such as iron or zinc, which can be found bound to protein molecules, or glycans and other moieties added to proteins through post-translational modifications), then 100-130g of amino acids = 6.02 - 7.83x1025Da approximately. Therefore, if the molecular weight of one amino acid is on average ~100Da, then 500g of meat contain (6.02 - 7.83x1025Da)/100Da = 6.02 - 7.83x1023 amino acid molecules.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When humans consume proteins from beef or fish, the proteins do not remain as they are, but are digested and disassembled into oligopeptides and, eventually, into their building blocks, namely amino acids. Amino acids can be further broken down, however, the majority of them are diverted to the synthesis of nascent protein molecules in the organism/cells that have absorbed those amino acids from food consumption. The kind of proteins that will be synthesized with these amino acids is dictated by the organism’s (in this case, human) genes, in combination with intercellular interactions and other environmental cues in general, since an organism’s genome is what determines their developmental program, in other words, what makes a cow a cow, a fish a fish, and a human a human.
- Why are there only 20 natural amino acids?
There are 20 standard/canonical or proteinogenic amino acids encoded by the genetic code. It is very likely that, in the last four billion years of Earth’s existence, since the first organic molecules started to emerge, many different amino acid molecules have occurred. Nevertheless, this combination of 20 amino acids seems to have been favored by what can be considered a deeply fundamental form of natural selection. The main factors that led to this set of 20 amino acids comprising the proteinogenic code of all life on the planet can be traced to the diversity of sizes and chemical properties they display, the prevalence of the atoms that constitute their components, the stability they provide to protein folding and structure, all balanced by their biosynthetic cost. 1
- Can you make other non-natural amino acids? Design some new amino acids.
Designing new amino acids primarily entails identifying what modifications can be introduced to the 20 natural ones. Among the most obvious ones is replacing one of the usual elements found in organic compounds (C, H, O, N, S, P) with another element from the periodic table. Based on this concept, I decided to swap the C in alanine’s side chain methyl group with Si (Figure 4.1A), which has similar properties but is associated with digital computing. For the second amino acid, I focused on amino acids with aromatic rings, which have always fascinated me as chemical moieties due to their conjugated π electrons and extraordinary properties. Sticking with the aromatic theme, I replaced the benzene ring in phenylalanine’s side chain with azulene (Figure 4.1B). Azulene is an aromatic group with a seven- and five-membered ring and has a distinctive blue hue, which could be useful for detection assays. Finally, to design the third amino acid (which will also ensure my being locked away in Chemistry prison for all eternity), I joined two copies of the basic amino acid skeleton (the core C with one amino and one carboxyl group) together to generate what could be described as a “double” amino acid (Figure 4.1C). Although very unorthodox and probably chemically unstable, especially in its trans form (Figure 4.1C, bottom), the “double” amino acid is envisioned to function as an adapter enabling connecting amino acids in unusual ways, such as generating peptides with two N- or two C-termini.
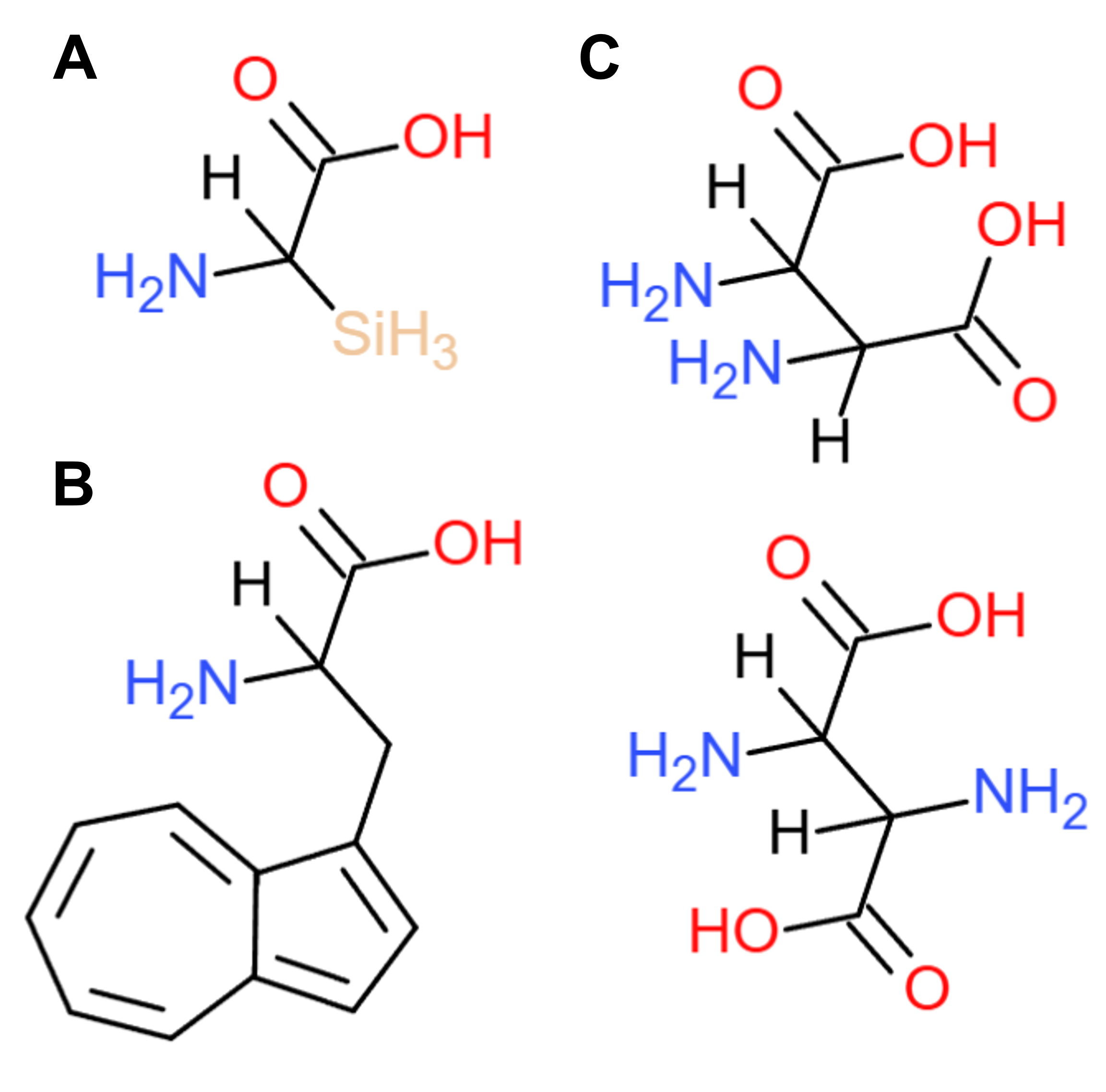 Figure 4.1 Schematics of new non-natural amino acids: (A) alanine with the C in its methyl group replaced with Si, (B) phenylalanine with the benzene in its side chain swapped with azulene, and (C) the “double” amino acid, both as a cis (top) and trans (bottom) isomer. Figure drawn on ChemDoodle.
Figure 4.1 Schematics of new non-natural amino acids: (A) alanine with the C in its methyl group replaced with Si, (B) phenylalanine with the benzene in its side chain swapped with azulene, and (C) the “double” amino acid, both as a cis (top) and trans (bottom) isomer. Figure drawn on ChemDoodle.
- Where did amino acids come from before enzymes that make them, and before life started?
In 1953, Stanley Miller and Harold Urey conducted what is now known as the “Miller-Urey experiment”, which supports the prebiotic (prior to the emergence of life) synthesis of amino acids. In more detail, the researchers replicated the chemical conditions of Earth’s early atmosphere by combining methane, ammonia, hydrogen, and water vapor in a flask. By stimulating those compounds with electric sparks (recreating lightning), Miller and Urey were able to detect simple amino acids, such as glycine, alanine, and valine, under the simulated early Earth conditions, demonstrating that complex organic molecules could form spontaneously from inorganic precursors in the prebiotic “primordial soup”. 2
Another hypothesis claims the introduction of amino acids to Earth from space through meteorites. Among them, the Murchison meteorite 3, which fell near Murchison, Victoria, in Australia in 1969, has been found to contain over 90 amino acids, including 19 also found on Earth, suggesting an extraterrestrial origin for organic compounds.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Natural proteins consist of L-amino acids (rather than D-amino acids), meaning that the chirality (or the molecular orientation) of amino acids in naturally occurring proteins tends to be left-handed rather than right-handed 4. Due to this property, structural stability favors the formation of right-handed or clockwise-rotating α-helices with L-amino acids 5 6, with the exception of regions rich in glycine, which is the only achiral of the 20 proteinogenic amino acids. Based on this rationale, a peptide chain composed of D-amino acids would display a structural propensity towards left-handedness and would, therefore, most likely form left-handed or counterclockwise-rotating α-helices.
- Why are most molecular helices right-handed?
Similarly to proteins’ “preference” towards L-amino acids, the dominant forms of DNA and RNA consist of nucleotides containing D-sugars, D-deoxyribose and D-ribose respectively. In both cases of proteins and nucleic acids, the chirality of their building blocks thermodynamically favors right-handed helical structures, which minimize steric hindrances and optimize hydrogen bonding among monomers. In other words, the inherent geometry of the amino acids and sugars found in naturally occurring proteins, as well as DNA and RNA molecules, renders right-handed α-helices (in the case of proteins) and the right-handed β-form double helix (in the case of DNA) more structurally stable and energetically efficient, as they require less energy for maintenance, than their left-handed counterparts 5 6.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
In physiologically folded proteins, β-sheets are arranged in a parallel or antiparallel orientation to form zigzag polypeptide chains. They tend to be positioned side-by-side, as this conformation is mediated by hydrogen bonds between the amino- and carboxyl-groups of neighbouring sheets. However, in pathogenic states (which often involve the misfolding of the protein too), this very property favors the “edge-to-edge” aggregation of multiple β-sheets, potentially leading to the formation of insoluble amyloid fibrils. Another mechanism that drives β-sheet aggregation occurs from the shape of this type of protein secondary structure, which forces the hydrogen bonds between neighbouring domains to exist on the same plane, while the side chains of hydrophobic amino acid residues (which are common in β-sheets) are inclined to protrude out of either side of that plane. These hydrophobic moieties can interact with the hydrophobic side chains of amino acids from another sheet, creating hydrophobic spaces between them. It is, therefore, thermodynamically favorable for β-sheets to gradually be stacked on top of each other, which, once again, can result in their aggregation 7 8 9.
- Design a β-sheet motif that forms a well-ordered structure.
For this task, at first I thought to draw inspiration from nature and, specifically, from naturally occurring proteins characterized by a well-ordered structure despite being rich in β-sheets. One such category of proteins are fluorescent proteins, for instance GFP, whose wild-type structural configuration forms a fluorophore-protecting can-shaped cavity termed a “β-barrel”. I thought I could experiment with increasing the stability of the β-barrel, while retaining most of its intrinsic structural properties. One way to approach this would be to swap several of the hydrophobic amino acids located in its barrel-forming region (approximately amino acids 13 - 227) with prolines. Amino acid residues with hydrophobic R-groups contribute both with their side chains but also with their amino- and carboxyl-moieties to β-sheet aggregation propensity as explained in the previous question. On the contrary, proline does not have a hydrophobic R-group, as the amino acid’s cyclic side chain curls back into its own nitrogen backbone, thus preventing the amino-group’s hydrogen from interacting with other amino acids through hydrogen bonds 10 11 12. However, I then proceeded to reject this idea, as this well-ordered β-sheet pattern already exists and has been optimized by nature through evolution. I similarly rejected other ideas involving symmetric naturally generated protein motifs, such as the ones found in viral capsids.
At this point, I wondered what other intricate β-sheet motifs had already been discovered in nature. I was intrigued to find one called “the greek key”, which comprises a protein supersecondary structure consisting of four adjacent antiparallel β-strands connected non-sequentially by hairpin loops in a shape reminiscent of the meander pattern (also known as “the greek key”) 13. Wanting to honor greek heritage as well, I designed a secondary structure motif abstractly inspired by the swirls commonly observed at the bottom part of traditional greek antefixes (“ακροκέραμα”), which are ornamental ceramic tiles placed at the edge of the roof (Figure 4.2B, top). This motif contains eight β-sheets, divided into two columns and positioned antiparallel to each other in pairs, while connected by hairpin loops in the sequence 1-3-2-4-5-7-6-8 (Figure 4.2A). Upon a second glance, I realized that this pattern also resembles the capital, meaning the upper part (“κιονόκρανο”), of columns sculpted in ionic style (Figure 4.2B, bottom). This motif is, of course, imaginary, therefore it would have to be artificially constructed within a protein molecule to test for its structural integrity and conclude whether it qualifies or not as a well-ordered protein supersecondary structure.
 Figure 4.2 Visual representation of the antefix-derived β-sheet secondary structure motif and its inspirational source. (A) The pattern contains eight β-sheets, divided into two columns and positioned antiparallel to each other in pairs non-sequentially. (B) This β-sheet motif was inspired by the spirals displayed at the bottom part of traditional greek antefixes (top) or bilaterally in ionic column capitals (bottom). Figure from the ceramic art workshop “Akrokeramo”.
Figure 4.2 Visual representation of the antefix-derived β-sheet secondary structure motif and its inspirational source. (A) The pattern contains eight β-sheets, divided into two columns and positioned antiparallel to each other in pairs non-sequentially. (B) This β-sheet motif was inspired by the spirals displayed at the bottom part of traditional greek antefixes (top) or bilaterally in ionic column capitals (bottom). Figure from the ceramic art workshop “Akrokeramo”.
Part 2: Protein analysis and visualization
Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
- Briefly describe the protein you selected and why you selected it.
For this assignment, I selected reflectin from Sepia officinalis, the common cuttlefish, as it contributes, among other proteins and structures, to the formation of intricate iridescent patterns that distinguish several cephalopods, such as cuttlefish, squids, and octopuses. In general, reflectins enable rapid, dynamic camouflage by controlling light reflection, as they possess high refractive indices and form nanostructured photonic films called iridophores, which can shift color in response to neurotransmitters 14. I am deeply fascinated by those patterns and the way they are created, so I drew inspiration from this natural phenomenon for one of my final project ideas, hence my interest in delving more into this particular protein.
- Identify the amino acid sequence of your protein.
- How long is it? What is the most frequent amino acid?
- How many protein sequence homologs are there for your protein?
- Does your protein belong to any protein family?
I retrieved reflectin’s amino acid sequence from Sepia officinalis (SoREF8) from its corresponding page on NCBI:
>CCI88216.1 reflectin [Sepia officinalis] MNRFMNRYRPMFNNMHNNMYNNMYRGRYRGMMEPMSRMTMDFQGRYMDSQGRMVDPRYYDYYGRWNDYDR YYGKSMFNYGWMMDGDRYNNYYRWMDFPERYMDMSGYQMDMYGRWMDMQGRHCNPFNQWGHNRYGQSFNY NYGRNMFYPERWMDMSNYSMDMQGRYMDRWGRHCNPFSQNMNWYGRYWNYPGYNNYYYNRHMYYPERYFD MSNWQMDMQGRWMDMQGRHNNPYWYNWYGRQMYYPYQNNWYGRWDYPGMDYSNWQMDMQGRWMDMQGRYM DPWMSDYSYNN
The peptide chain is 291aa long, with the most frequent amino acid being tyrosine (Y) as it appears in the sequence 47 times.
UniProt’s BLAST tool identified 20 results with a sequence identity of 50 - 88% that appear to be homologs of SpREF8 from other appearance-shifting cephalopods, such as the Pharaoh cuttlefish (Sepia pharaonis), the East Asian common octopus (Octopus sinensis), and the common octopus (Octopus vulgaris).
Reflectins belong to a unique family of intrinsically disordered proteins (IDPs) found in cephalopods. They are highly unusual, rich in aromatic and sulfur-containing amino acids, for example, tryptophan (W) and methionine (M), and can self-assemble into diverse nanostructures 14.
- Identify the structure page of your protein in RCSB.
- When was the structure solved? Is it a good quality structure? (A good quality structure is one with good resolution, the smaller the better (Resolution: 2.70 Å)).
- Are there any other molecules in the solved structure apart from protein?
- Does your protein belong to any structure classification family?
The query about reflectin on RCSB did not yield any results, even when computed structure models (CSMs) were included. However, there was a 3D model of the protein’s structure on its UniProt page (Figure 4.3). No other molecules seem to participate in this structure, which is consistent with the protein’s property to interact mainly with photons.
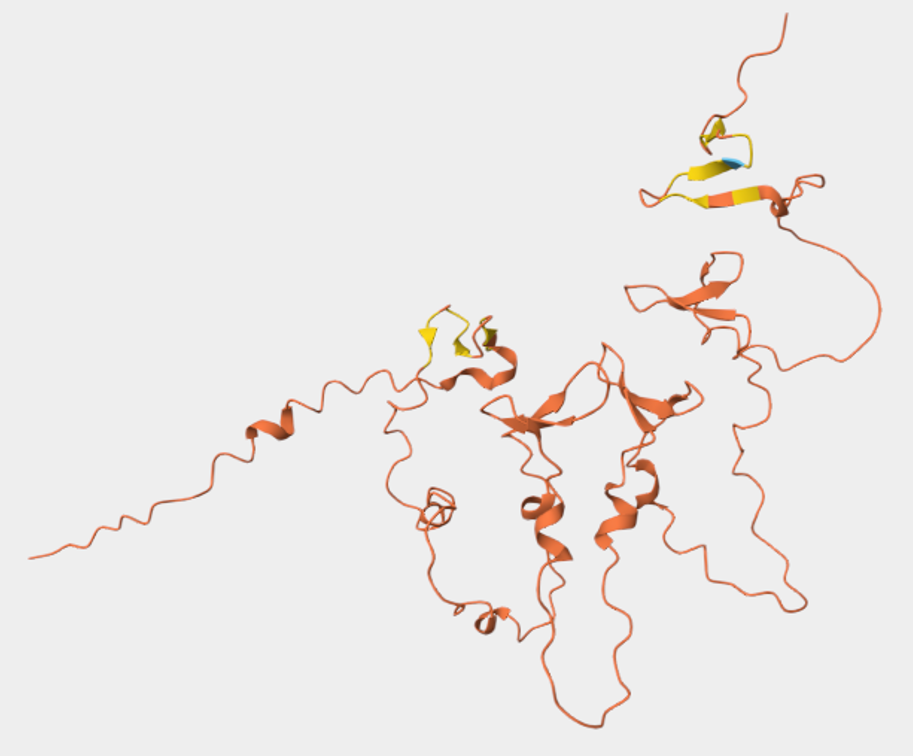
Figure 4.3 Snapshot of reflectin’s (SoREF8) 3D structure visualization. Figure from its UniProt page.
- Open the structure of your protein in any 3D molecule visualization software.
- Visualize the protein as “cartoon”, “ribbon”, and “ball and stick”.
- Color the protein by secondary structure. Does it have more helices or sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Below you can find the 3D structure visualizations of SoREF8 in the “cartoon”, “ribbon” (Figure 4.4A), and “ball and stick” (or “liquorish”) (Figure 4.4B) models as rendered in PyMOL.
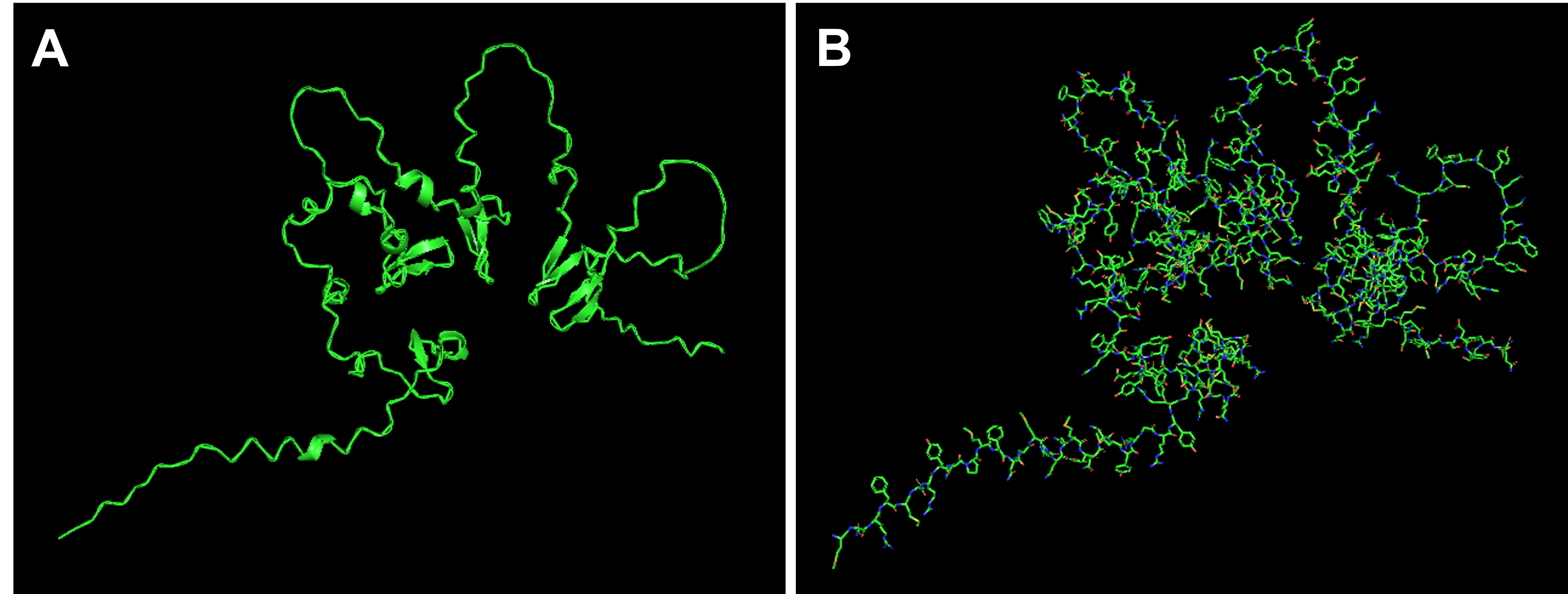 Figure 4.4 Visualizations of reflectin’s (SoREF8) 3D structure in the “cartoon” and “ribbon” model (A), as well as in the “ball and stick” (or “liquorish”) model (B). Figure generated with the PyMOL software.
Figure 4.4 Visualizations of reflectin’s (SoREF8) 3D structure in the “cartoon” and “ribbon” model (A), as well as in the “ball and stick” (or “liquorish”) model (B). Figure generated with the PyMOL software.
When coloring the protein based on its secondary structures, it appears that it has an almost equal number of very short α-helices (Figure 4.5, red) and β-sheets (Figure 4.5, yellow), while the largest part of the protein retains a less structurally defined loop form (Figure 4.5, green).
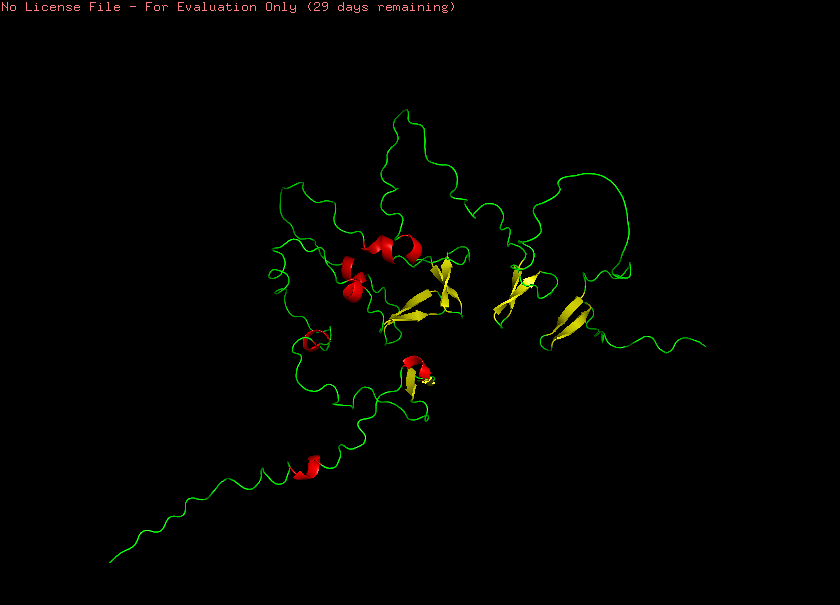 Figure 4.5 Visualization of reflectin’s (SoREF8) secondary structures, including α-helices (depicted in red), β-sheets (highlighted in yellow), and loops (marked with green). Figure generated with the PyMOL software.
Figure 4.5 Visualization of reflectin’s (SoREF8) secondary structures, including α-helices (depicted in red), β-sheets (highlighted in yellow), and loops (marked with green). Figure generated with the PyMOL software.
Furthermore, reflectin seems to have more hydrophilic and charged amino acids than hydrophobic residues in its peptide chain, which is consistent with the respective reflectin protein family’s high content of aromatic and sulfur-containing amino acids, such as tryptophan (W) and methionine (M) (Figure 4.6).
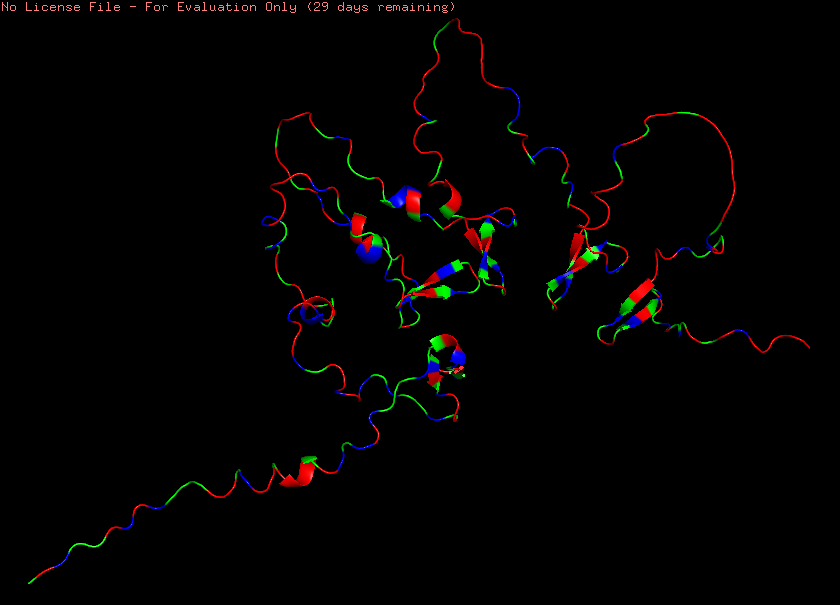 Figure 4.6 Visualization of reflectin’s (SoREF8) structure based on its amino acid hydrophobicity: hydrophobic residues are illustrated in green, whereas hydrophilic/polar and charged ones are displayed in red and blue respectively. Figure generated with the PyMOL software.
Figure 4.6 Visualization of reflectin’s (SoREF8) structure based on its amino acid hydrophobicity: hydrophobic residues are illustrated in green, whereas hydrophilic/polar and charged ones are displayed in red and blue respectively. Figure generated with the PyMOL software.
Lastly, upon visualizing reflectin’s (SoREF8) surface, the protein does form large gaping holes where its loops are located, however, their size renders them highly unsuitable for binding ligands (Figure 4.7). After a quick 3D inspection of the entire molecule’s surface, I could not find any binding pockets, which also aligns with the absence of complex secondary structures in the protein. Complex secondary structures, often facilitated by long stretches of hydrophobic amino acid residues, which are absent in reflectin’s peptide sequence, enable the folding of the amino acid chain into pores and cavities that can serve as binding sites for specific ligands. The absence of binding pockets in the case of reflectin does not come as a surprise though, as the principal element these proteins have to interact with are rays of light.
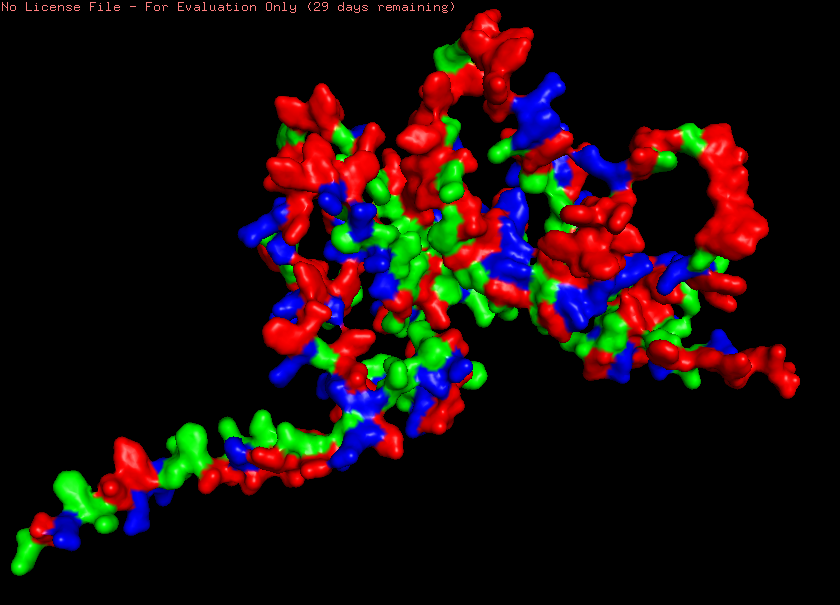 Figure 4.7 Visualization of reflectin’s (SoREF8) surface, where no binding pockets can be seen. Figure generated with the PyMOL software.
Figure 4.7 Visualization of reflectin’s (SoREF8) surface, where no binding pockets can be seen. Figure generated with the PyMOL software.
Part 3: Using ML-based protein design tools
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU. Then, choose your favorite protein from the PDB.
3.1 Protein language modeling
Deep mutational scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern? (Choose a residue and a mutation that stands out.)
Model esm2_t30_150M_UR50D was deployed to generate the deep mutational scan of reflectin (SoREF8), which revealed several regions that are highly sensitive to mutations in the protein. More specifically, positions 17 - 22, 40 - 48, 97 - 122, 145 - 153, 158 - 165, 207 - 230, and 264 - 278, which also appear darker in the mutation scan heatmap, indicate protein regions with an integral role in reflectin’s structure and function, being, therefore, less tolerant to amino acid substitutions (Figure 4.8). Those regions may also correspond to short patterns repeated across the protein’s amino acid sequence. Additionally, some points that stand out in the heatmap as favorable mutations are glutamine (Q) amino acid residue at positions 110 and 157, as well as a M amino acid residue at position 207 (denoted with yellow color in Figure 4.8).
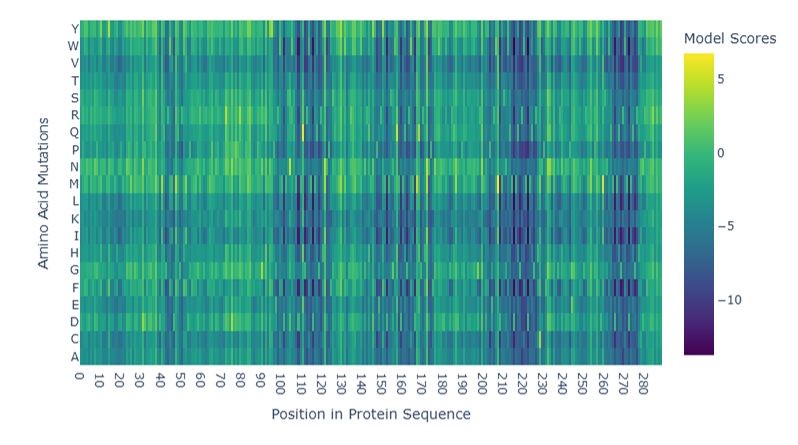 Figure 4.8 Mutation scan heatmap of reflectin from Sepia officinalis. Figure generated by utilizing Evolutionary Scale Modeling version 2 (esm2_t30_150M_UR50D).
Figure 4.8 Mutation scan heatmap of reflectin from Sepia officinalis. Figure generated by utilizing Evolutionary Scale Modeling version 2 (esm2_t30_150M_UR50D).
b. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to the experiment.
Unfortunately, I could not find any experimental scans for SoREF8 in the literature. Nevertheless, I managed to discover a paper investigating the structural properties of another member of the reflectin family, namely a truncated version of reflectin A1 from the longfin inshore squid (Doryteuthis pealei), which displays N-terminal and internal motifs distinctive of many other reflectin proteins 15. The truncated variant of this specific reflectin protein (RfA1TV) contains an ERYMD amino acid sequence at positions 37 - 41, which appears to play a significant role in the formation of a helix crucial for granting the biomolecule its reflective properties 15. In SoREF8, a similar sequence of the amino acids GRYMD can be seen between positions 40 and 48, which were identified as a region critical for the protein’s function by the mutational scan in the previous question.
Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins?
In various cases, protein clusters created by the t-SNE analysis shown below (Figure 4.9) consist of proteins with a common property, such as proteins that associate with DNA, including topoisomerases, DNA repair proteins, DNA polymerases, and helicases, or, in another example, receptors of various molecules, like cytokines, developmental regulators (e.g. Hedgehog), or even motor proteins (e.g. myosin). Proteins in Figure 4.9 seem to be clustered based on function and biochemical properties rather than species, as homologs of proteins that serve similar purposes but originate from evolutionarily distant organisms (for instance, from Escherichia coli, Drosophila melanogaster, and Homo sapiens) tend to be grouped together in the plot. Thus, it would appear that embedding a dataset of proteins in reduced dimensionality retains their biochemical profile and spatially distributes them according to functional (and, therefore, probably also structural) similarity.
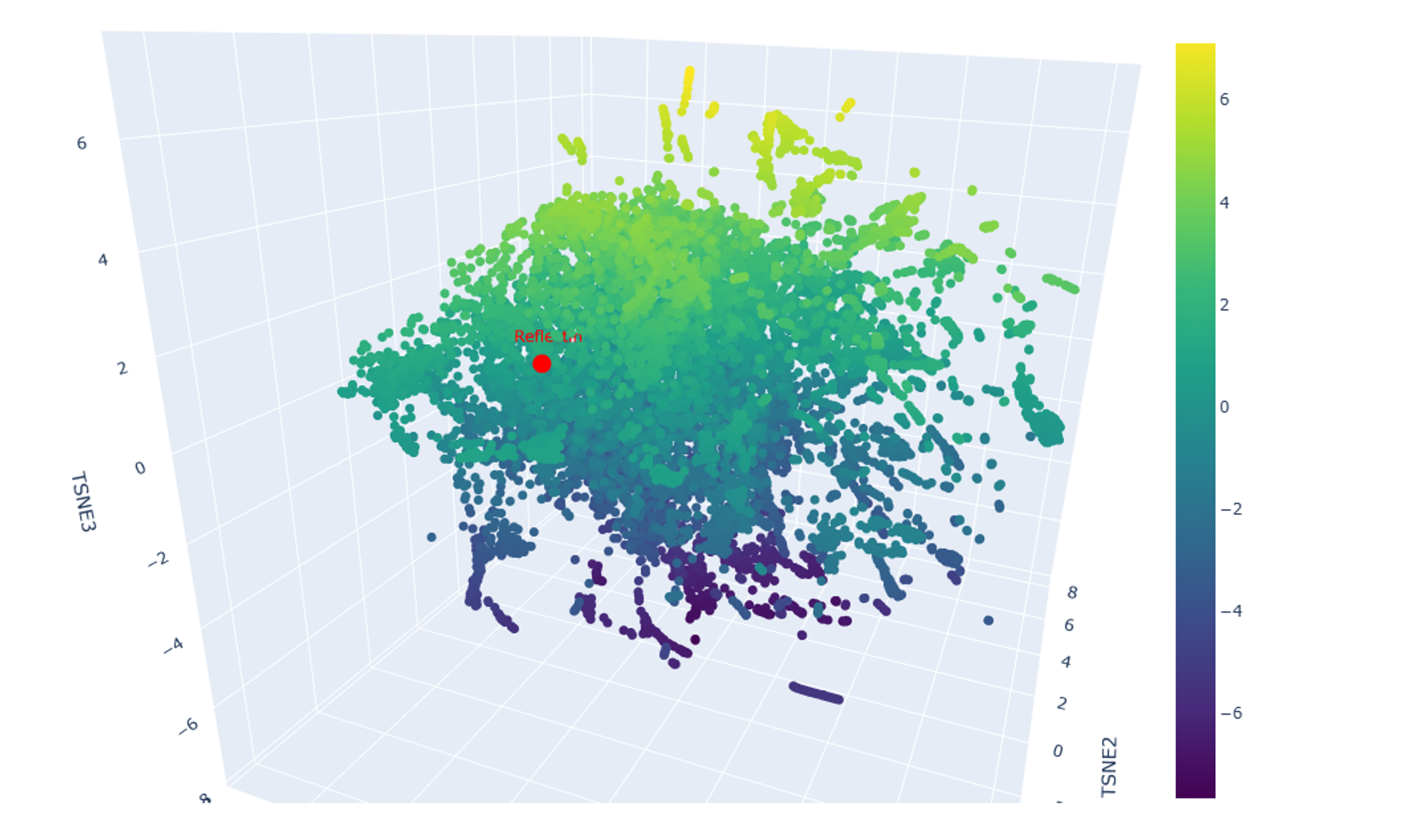 Figure 4.9 3D t-SNE visualization of 15,178 protein sequence embeddings, including reflectin (SoREF8) highlighted in red.
Figure 4.9 3D t-SNE visualization of 15,178 protein sequence embeddings, including reflectin (SoREF8) highlighted in red.
b. Place your protein in the resulting map and explain its position and similarity to its neighbors.
As I mentioned earlier, reflectins belong to a very unique and distinct family of proteins, as they reflect and refract light through self-assembling into intricate nano-layers within cells. Based on that, I was expecting SoREF8 to be positioned close to another cluster of proteins related to the emergence of structural color, which are mainly of bacterial origin. However, the latent space analysis visualized in Figures 4.9 and 4.10 was created with a dataset of 15,178 protein sequences, which, of course, did not include all the protein molecules ever discovered, only a portion of them, which is probably the reason no proteins associated with structural color were present in the plot or, at least, close to where reflectin was placed. On the contrary, the closest neighbour to reflectin appears to be a cluster of proteins mostly from multicellular organisms with bilateral symmetry, such as Hyalophora cecropia, Gallus gallus, Mus musculus, and Homo sapiens (Figure 4.10, black box). This cluster is particularly enriched in proteins acting as mediators or receptors facilitating immune responses (e.g. hemolin, IgA Fc receptor, interleukin-1 receptor, immunoglobulin heavy chain). Such a correlation seems bizarre, especially when taking into consideration reflectin’s loose secondary structure (Figures 4.3 - 4.7), allowing it to interact with photons, and the very intricate, characteristic, and β-sheet-abundant secondary structure of proteins of the immune system, which contributes to the formation of nooks and cavities for epitope recognition 16.
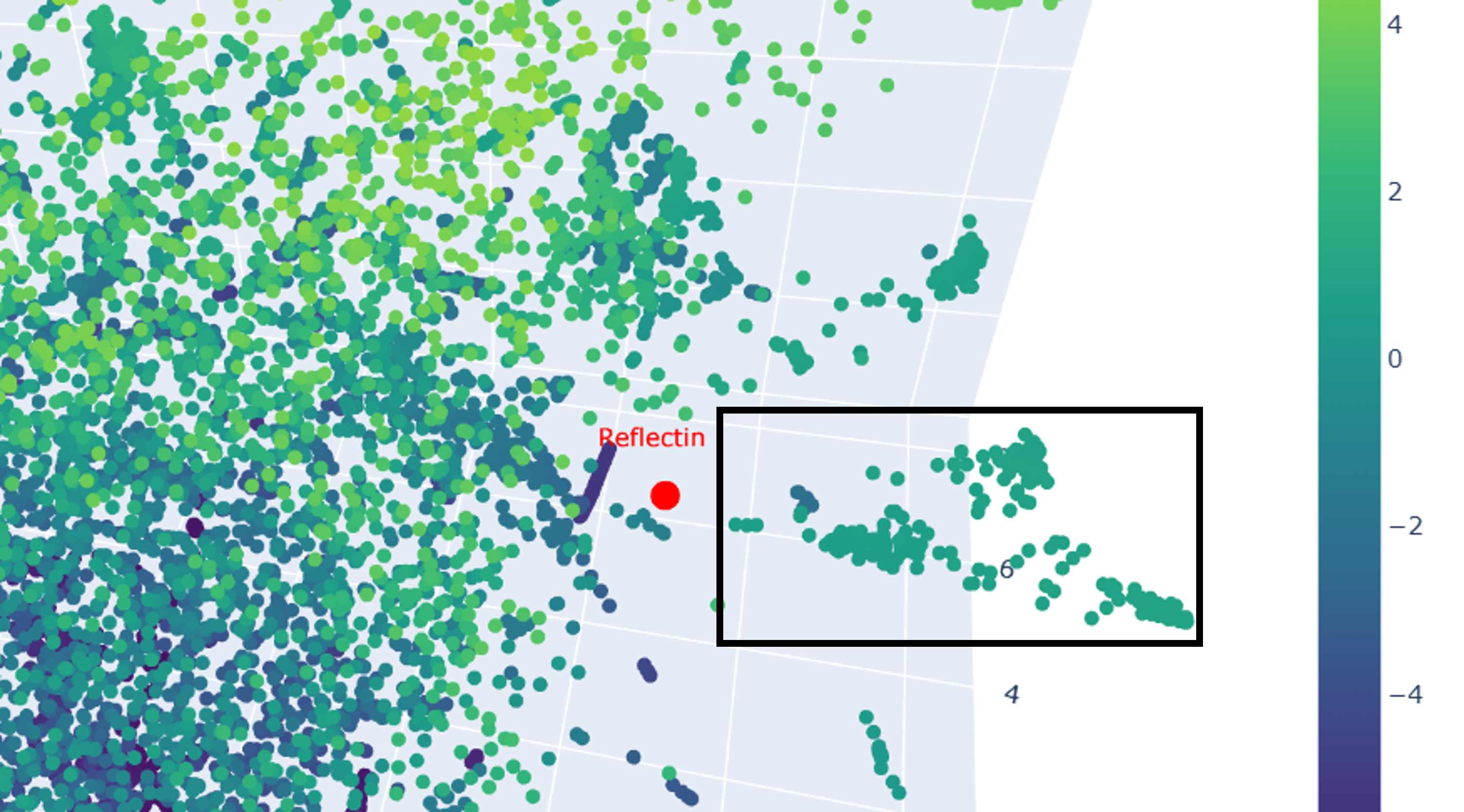 Figure 4.10 Close-up of the region surrounding reflectin in the 3D t-SNE visualization. Once again, reflectin (SoREF8) is shown in red, while its neighbouring protein cluster, enriched in proteins partaking in immune responses, is encapsulated in a black frame.
Figure 4.10 Close-up of the region surrounding reflectin in the 3D t-SNE visualization. Once again, reflectin (SoREF8) is shown in red, while its neighbouring protein cluster, enriched in proteins partaking in immune responses, is encapsulated in a black frame.
3.2 Protein folding
Folding a protein
a. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
By comparing Figures 4.3 and 4.11, I would say that wild-type reflectin’s coordinates as predicted by ESMFold’s algorithm do not match the structure provided by UniProt. However, this could possibly be explained if we take into account that SoREF8 belongs to a special family of intrinsically disordered proteins, hence the challenge of reliably and repeatedly generating its tertiary structure through separate computational tools.
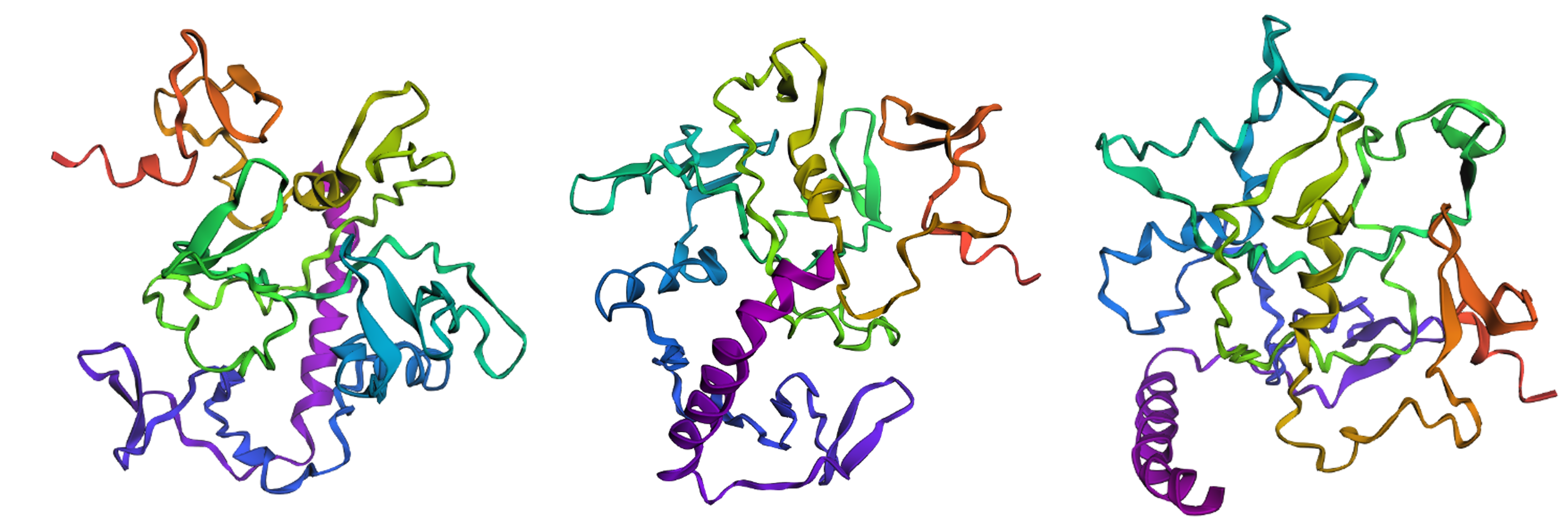 Figure 4.11 Illustration of wild-type SoREF8’s structure from three different points of view. Figure generated by ESMFold.
Figure 4.11 Illustration of wild-type SoREF8’s structure from three different points of view. Figure generated by ESMFold.
b. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
For this exercise, I will be comparing the ESMFold-predicted structures of different mutated variants of reflectin with the ESMFold-reconstructed wild-type protein.
Firstly, I experimented with three separate point mutations in SoREF8’s amino acid sequence either in regions “outside” of segments critical for the protein’s function (according to the deep mutational scan performed above) or “inside” those segments. For most of the point mutations I introduced, I decided to target amino acids significant to the structure of reflectins in general, therefore I mainly converted aromatic and sulfur-containing residues to alanine, which is small and chiral, while featuring a chemically inert, non-bulky methyl side group. In both cases of swapping amino acids outside and inside the regions identified by the mutational scan, the tertiary structure of reflectin (Figure 4.12A and B) did not seem to diverge dramatically from the wild-type (Figure 4.11), showing that the protein’s structure is quite resilient to point mutations in general.
Secondly, I tried converting entire segments of wild-type reflectin’s amino acid sequence into alanine arrays, once again both in regions outside and inside the domains that were identified as crucial for the protein’s folding and function. For this part, I converted whole arrays of 10 to 15 sequential amino acid residues to a series of alanines and, in contrast to the point mutations, these modifications resulted in noticeable changes in reflectin’s 3D structure. In both cases, but more intensely when the substitutions were positioned within the areas that are crucial for folding, reflectin’s tertiary structure became more elongated and enriched in α-helices (Figure 4.12C and D) compared to the wild-type (Figure 4.11), indicating that the protein cannot tolerate modifications of large amino acid segments.
Finally, I decided to swap three specific amino acids in the wild-type sequence with different ones, based on the mutations that were suggested by the deep mutational scan as favorable for optimized folding of the protein, namely replacing the residue at positions 110 and 157 with Q and the one at position 207 with M (Figure 4.8). This intervention did not appear to particularly affect the protein’s structure (Figure 4.12E compared to the wild-type in Figure 4.11) and this version of reflectin would most likely have to be experimentally tested in the wet lab to verify whether it actually contributes to optimized folding and, therefore, to optimized function.
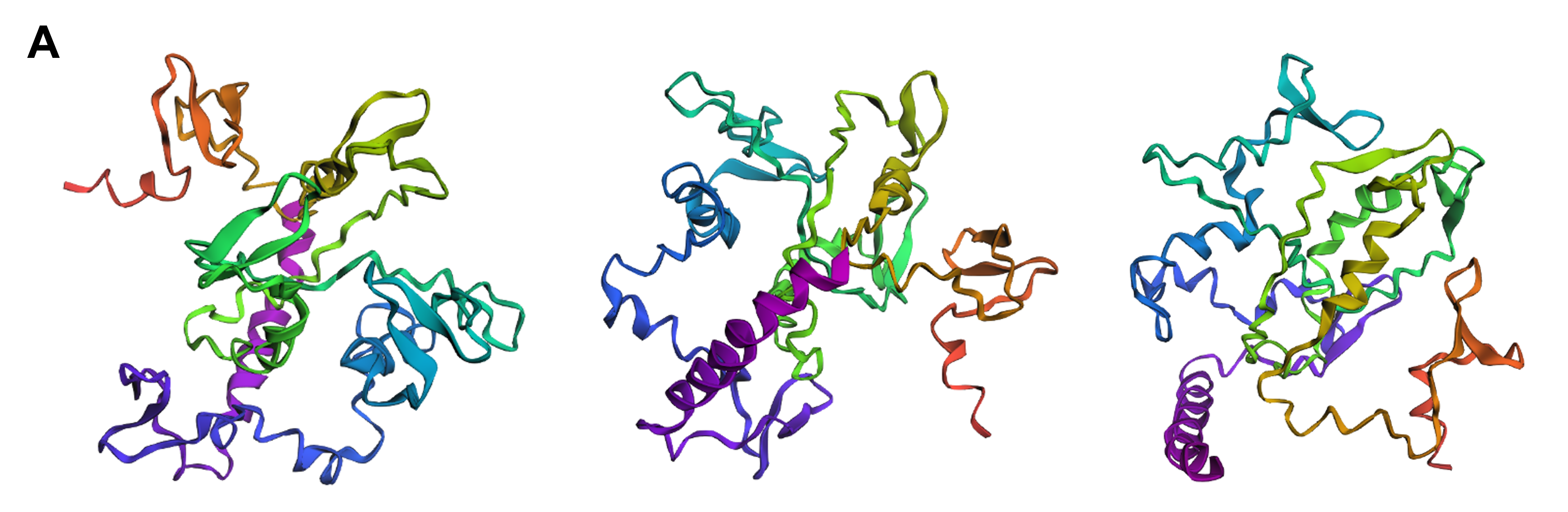
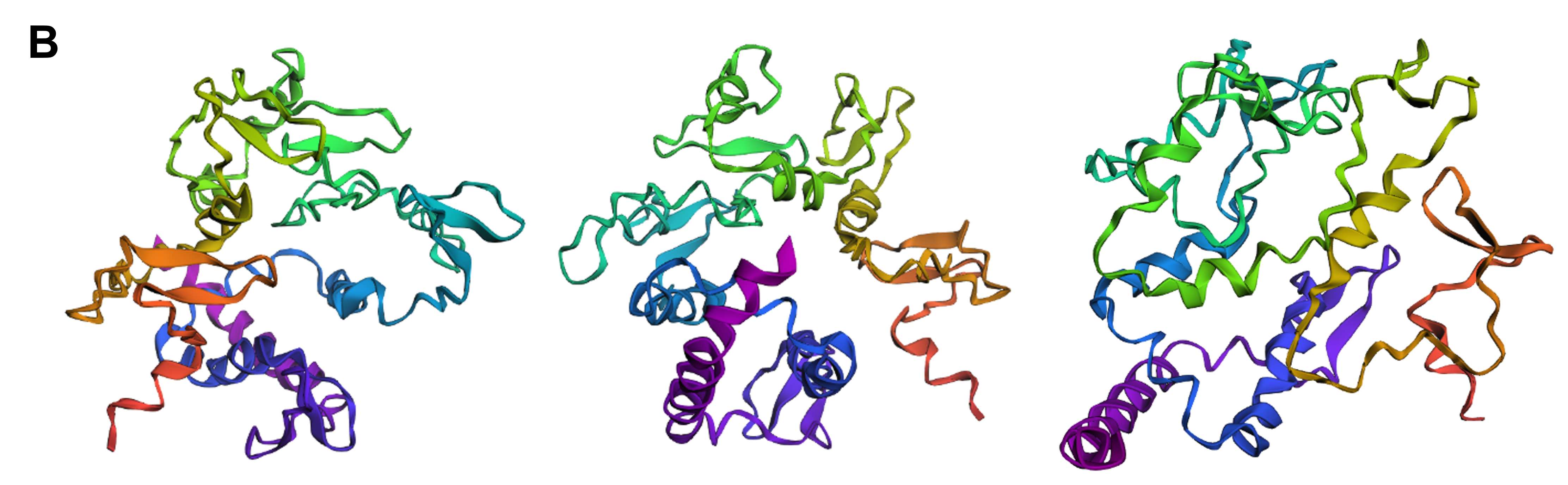
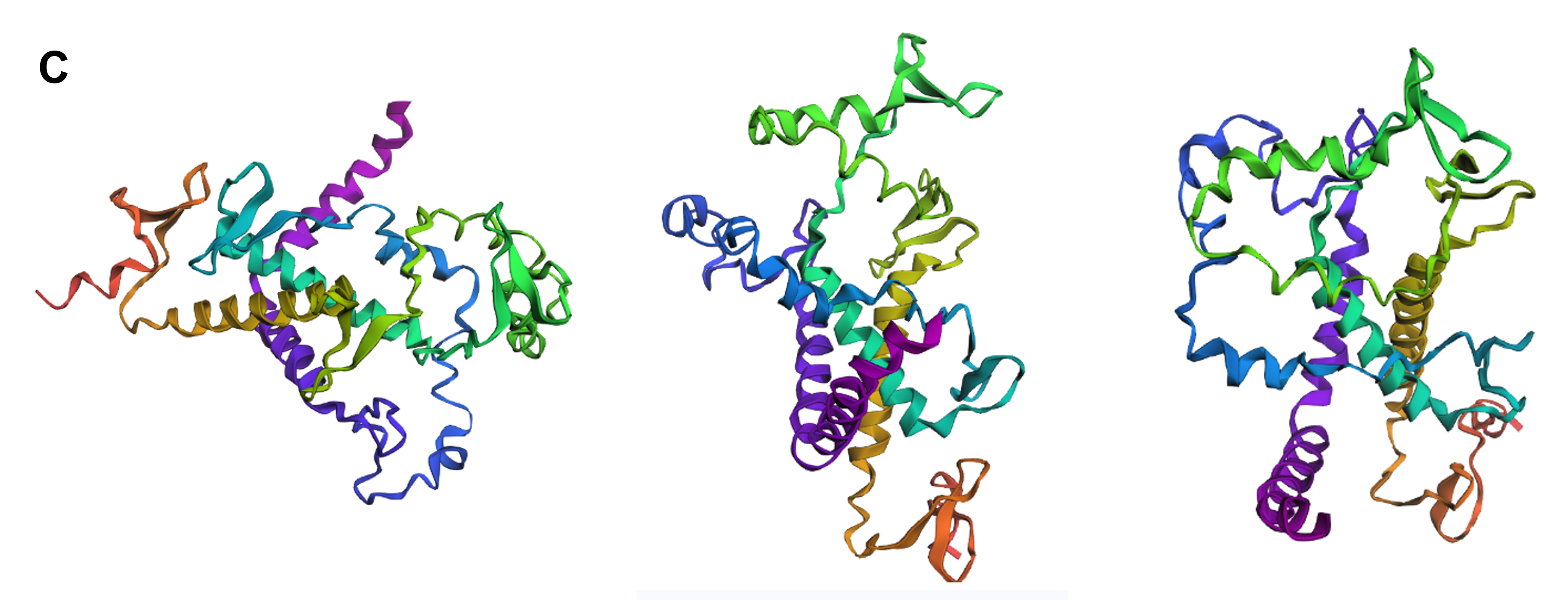
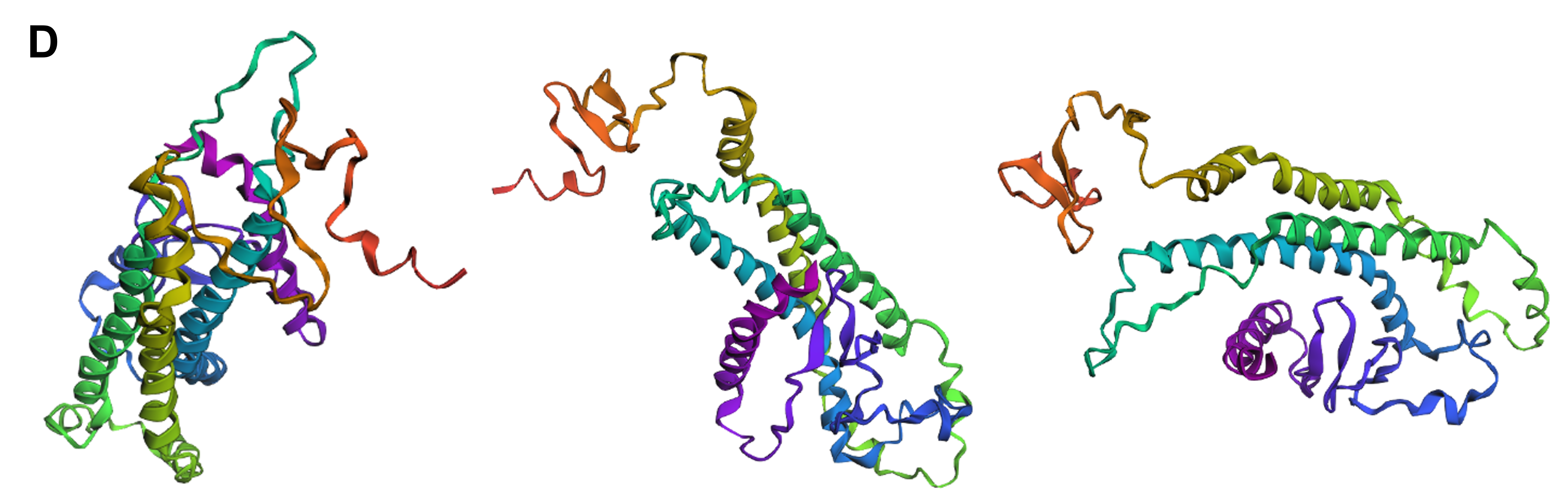
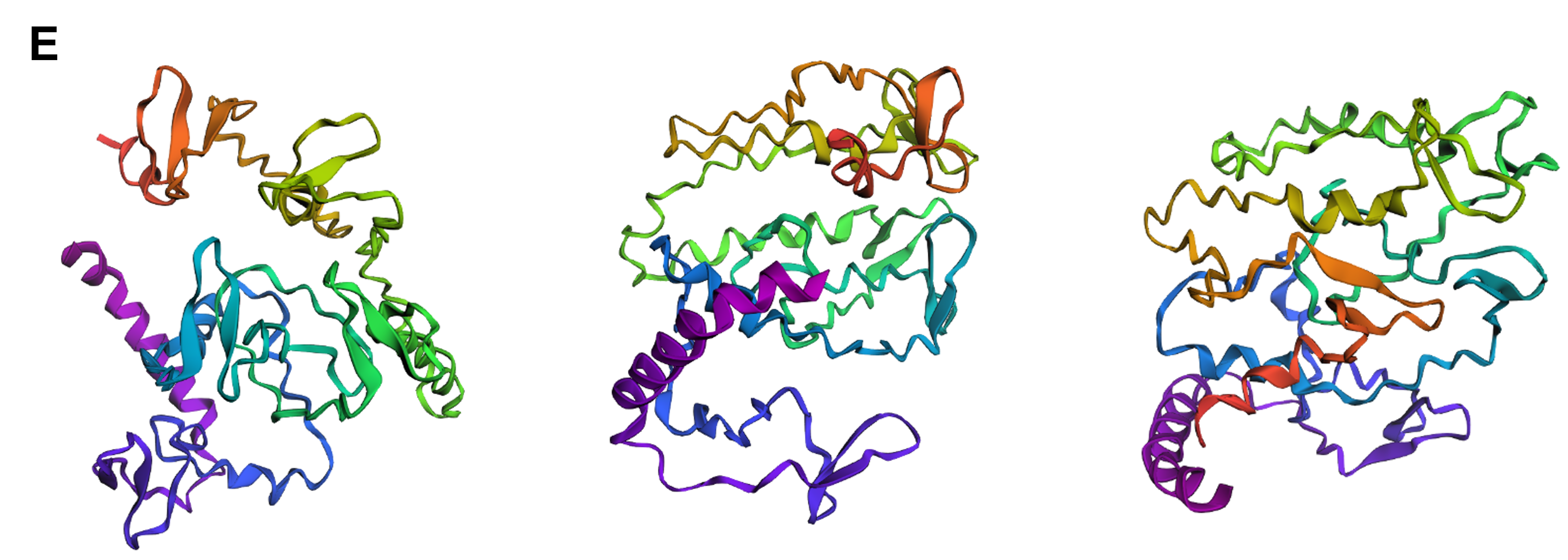 Figure 4.12 Tertiary structure visualization of different mutated variants of SoREF8 from three different points of view as predicted by ESMFold: (A) SoREF8 Y58A_N182A_Q247A, (B) SoREF8 M104A_D161A_W222A, (C) SoREF8 30 - 40A_125 - 140A_235 - 250A, (D) SoREF8 100 - 110A_150 - 165A_215 - 230A, and (E) SoREF8 D110Q_N157Q_R207M. Figure generated by ESMFold.
Figure 4.12 Tertiary structure visualization of different mutated variants of SoREF8 from three different points of view as predicted by ESMFold: (A) SoREF8 Y58A_N182A_Q247A, (B) SoREF8 M104A_D161A_W222A, (C) SoREF8 30 - 40A_125 - 140A_235 - 250A, (D) SoREF8 100 - 110A_150 - 165A_215 - 230A, and (E) SoREF8 D110Q_N157Q_R207M. Figure generated by ESMFold.
3.3 Protein generation
Inverse-folding a protein
a. Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
As there is no tertiary structure submitted to PDB for SoREF8, I used the ESMFold prediction generated above as a prompt for ProteinMPNN. By deploying model v_48_020, ProteinMPNN produced the following amino acid sequence for SoREF8 based on the probability heatmap demonstrated in Figure 4.13:
Reflectin [Sepia officinalis] amino acid sequence created by ProteinMPNN LAALLAALLPLLLLAAALAALLALLLGLPLLLLPLSLLTKNENGEWYDALGRLVDPRLLDPLGRLLLAELLLPGLPLLLLLLLPPELLALLLLLLLGPLALLDLSNFSLNSEGNWLDSEGNEVDPLSLLKYLPNGLLLPLLLLPLLLLPPLLLDLSKYTKNEEGEWLNEEGEVVDPLSLPLDEEEELLELPGLPLELLLLPLLLPLPLLDLSNFTLNEEGEWLNENGEPVDPEELLRLLLLPLDLLDPEALAPLKIEGFDFSKLYKNEEGEWYDENGNKINLEELEKLLNK
The BLASTp algorithm detected an 18.54% identity between the original SoREF8 amino acid sequence and the one predicted by ProteinMPNN. This discrepancy can mainly be attributed to the computational tool’s interpretation of reflectin’s probability heatmap (Figure 4.13). In particular, it appears that the ProteinMPNN software tends to place L and A residues in positions where there is probabilistically no prevalent amino acid (such as reflectin’s N-terminus in this case), while it has also replaced all M, C, H, and Q residues contained in the wild-type variant with one of the remaining 16 amino acids. Both types of intervention have drastically skewed the frequency with which each amino acid residue appears in the original and computationally generated sequences, leading to two fundamentally different peptide chains for the same protein molecule.
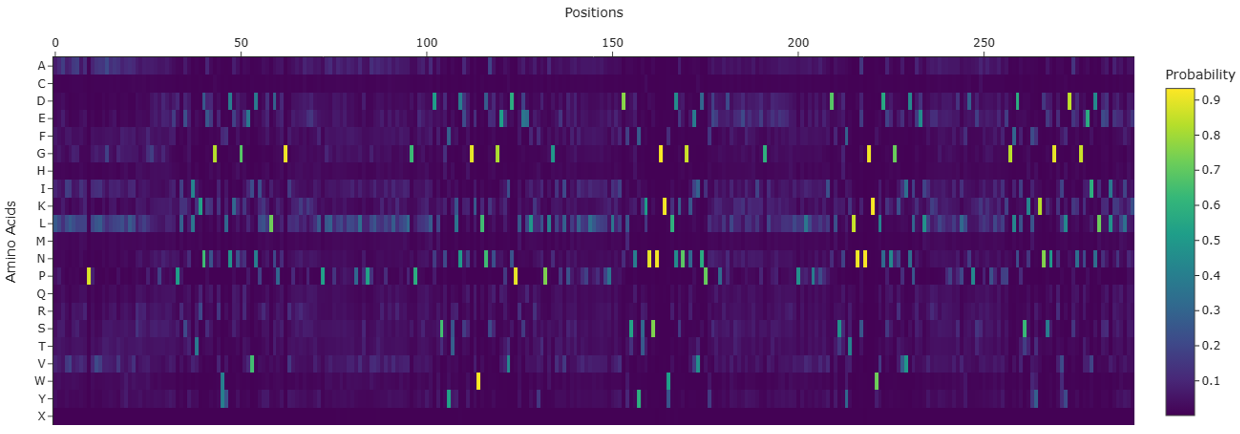 Figure 4.13 Heatmap illustrating the probability of each amino acid residue appearing at one of the 291 positions of reflectin’s peptide chain based on the wild-type’s tertiary structure as recreated by ESMFold. Figure generated with ProteinMPNN.
Figure 4.13 Heatmap illustrating the probability of each amino acid residue appearing at one of the 291 positions of reflectin’s peptide chain based on the wild-type’s tertiary structure as recreated by ESMFold. Figure generated with ProteinMPNN.
b. Input this sequence into ESMFold and compare the predicted structure to your original.
Compared to the tertiary structure of the original sequence as predicted by ESMFold (Figure 4.11), the folding of the ProteinMPNN-suggested peptide chain for reflectin seems to engage more α-helices and fewer β-sheets (Figure 4.14). The larger number of α-helices in the computationally recreated protein molecule renders it more compact (Figure 4.14) than the wild-type one, resulting in a 3D structure that is readily distinguishable from the original’s folding motif.
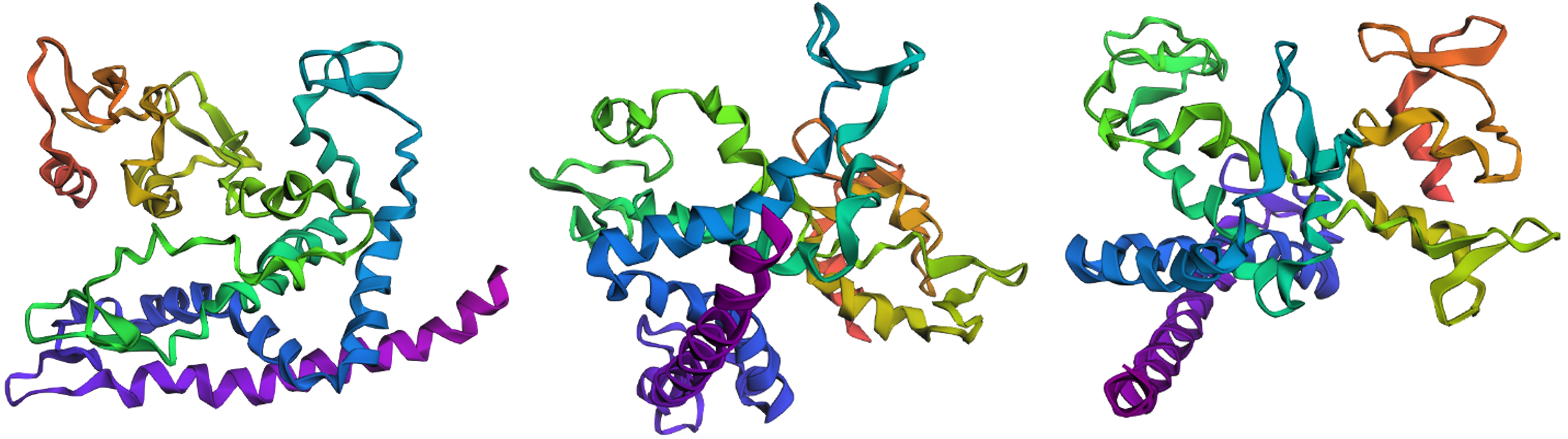 Figure 4.14 Visualization of the 3D structure for the amino acid sequence generated by ProteinMPNN with the ESMFold prediction of SoREF8’s structure as an input from three different points of view. Figure created with ESMFold.
Figure 4.14 Visualization of the 3D structure for the amino acid sequence generated by ProteinMPNN with the ESMFold prediction of SoREF8’s structure as an input from three different points of view. Figure created with ESMFold.
Doig AJ. Frozen, but no accident - why the 20 standard amino acids were selected. FEBS J. 2017;284(9):1296-1305. doi:10.1111/febs.13982 ↩︎
Miller SL. A Production of Amino Acids Under Possible Primitive Earth Conditions. Science. 1953;117(3046):528-529. doi:10.1126/science.117.3046.528 ↩︎
Kvenvolden KA, Lawless JG, Ponnamperuma C. Nonprotein amino acids in the Murchison meteorite. Proc Natl Acad Sci USA. 1971;68(2):486-490. doi:10.1073/pnas.68.2.486 ↩︎
Egli M, Zhang S. Making sense of helices: right and wrong models in science and art. Mol Front J. 2023;07(01n02):71-81. doi:10.1142/S2529732523500086 ↩︎
Rzepa H. Why are α-helices in proteins mostly right-handed? Henry Rzepa’s Blog. Published November 29, 2019. https://www.ch.ic.ac.uk/rzepa/blog/?p=3802 ↩︎ ↩︎
Cole BJ, Bystroff C. Alpha helical crossovers favor right-handed supersecondary structures by kinetic trapping: the phone cord effect in protein folding. Protein Sci. 2009;18(8):1602-1608. doi:10.1002/pro.182 ↩︎ ↩︎
Aggregation Prone Regions (APRs). VIB Switch Laboratory. https://switchlab.org/aprs ↩︎
Liu L, Klausen LH, Dong M. Two-dimensional peptide-based functional nanomaterials. Nano Today. 2018;23:40-58. doi:10.1016/j.nantod.2018.10.008 ↩︎
Eskandari S, Guerin T, Toth I, Stephenson RJ. Recent advances in self-assembled peptides: Implications for targeted drug delivery and vaccine engineering. Advanced Drug Delivery Reviews. 2016;110-111:169-187. doi:10.1016/j.addr.2016.06.013 ↩︎
Samuel D, Kumar TK, Ganesh G, et al. Proline inhibits aggregation during protein refolding. Protein Sci. 2000;9(2):344-352. doi:10.1110/ps.9.2.344 ↩︎
Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci USA. 2002;99(5):2754-2759. doi:10.1073/pnas.052706099 ↩︎
Shamsir MS, Dalby AR. Beta-sheet containment by flanking prolines: molecular dynamic simulations of the inhibition of beta-sheet elongation by proline residues in human prion protein. Biophys J. 2007;92(6):2080-2089. doi:10.1529/biophysj.106.092320 ↩︎
Zhang C, Kim SH. A comprehensive analysis of the Greek key motifs in protein β-barrels and β-sandwiches. Wiley Online Library. Published online August 15, 2000. doi:10.1002/1097-0134(20000815)40:3 ↩︎
Kramer RM, Crookes-Goodson WJ, Naik RR. The self-organizing properties of squid reflectin protein. Nat Mater. 2007;6(7):533-538. doi:10.1038/nmat1930 ↩︎ ↩︎
Umerani MJ, Pratakshya P, Chatterjee A, et al. Structure, self-assembly, and properties of a truncated reflectin variant. Proc Natl Acad Sci USA. 2020;117(52):32891-32901. doi:10.1073/pnas.2009044117 ↩︎ ↩︎
Janeway CA Jr, Travers P, Walport M, Shlomchik MJ. The structure of a typical antibody molecule. Immunobiology - NCBI Bookshelf. Published 2001. https://www.ncbi.nlm.nih.gov/books/NBK27144/ ↩︎