Week 5 homework
Protein design-Part II 💻
Part 1: SOD1 binder peptide design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
This week, the assignment entails designing short peptides that bind mutant SOD1 and then deciding which ones are worth advancing toward therapy by using three models developed in the Chatterjee Lab.
A. Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card, generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
- To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Upon retrieving the Homo sapiens SOD1 (HsSOD1) peptide sequence from its UniProt page, I noticed that the alanine residue that is mutated in the A4V protein variant is in position 5 and not in position 4 of the peptide chain. This means that the methionine in position 1 of the nascent protein molecule is post-translationally cleaved during the protein’s maturation process 1. Based on this and after incorporating the A4V mutation into the peptide chain, the SOD1 sequence I decided to use for this week’s assignment is the following:
>sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2, A4V variant ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
As the length and the number of the binding peptides had already been defined by the assignment, I thought it would be interesting to experiment with the k value, which, in the script we were given, was set to 3 2. As we can design only four peptides, I decided to “sacrifice” model confidence to a certain degree (and report higher perplexity values) in favor of diversity by increasing the k value to 4. The results of this analysis, including the known SOD1-binding peptide FLYRWLPSRRGG, are presented below, in Table 5.1.
Table 5.1 Peptide binders generated by PepMLM for the A4V mutant of SOD1, along with their perplexity scores and the known SOD1-binding peptide FLYRWLPSRRGG.
| Peptide sequence | Control or test | Peptide perplexity | |
|---|---|---|---|
| 0 | FLYRWLPSRRGG | Control | n/a |
| 1 | WLSPATVAARKX | Test | 7.249112 |
| 2 | WRYGAVGAKLWX | Test | 9.529020 |
| 3 | HRYVWTAARHKX | Test | 13.445100 |
| 4 | WRYGVAGVAHKX | Test | 9.256418 |
B. Evaluate binders with AlphaFold3
- After navigating to the AlphaFold Server, for each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
AlphaFold3 requires a defined peptide sequence to visualize protein-peptide interactions, which, I did not obtain for my test peptides, as can be seen by the “X” at position 12 in all four of them (possibly due to the increased k value I used). This provided a “loophole” I could capitalize on to experiment with several amino acid residues with different biochemical properties. So, I chose eight different amino acids out of the 20 proteinogenic ones, each representing a different chemical profile:
- arginine (R), as an amino acid harboring a positively charged side chain
- aspartic acid (D), as an amino acid harboring a negatively charged side chain
- serine (S), as an amino acid harboring a polar but uncharged side chain
- cysteine (C), as an amino acid harboring a S-containing side chain
- glycine (G), as the only achiral amino acid
- proline (P), because it has a straight-up weird structure and belongs to its own category
- leucine (L), as an amino acid harboring a non-aromatic hydrophobic side chain and
- tryptophan (W), as an amino acid harboring an aromatic and spatially-challenging-to-accomodate hydrophobic side chain.
Based on this rationale, instead of four, I tried 32 different peptides on AlphaFold3’s server, eight for each peptide template. After screening each peptide template with the eight different amino acids I had selected above, I decided to proceed with the amino acid combination that produced the highest ipTM score in each case. The results of this process are displayed in Table 5.2. I also experimented with simulating multiple copies of the same selected peptide interacting with SOD1 A4V. Nevertheless, this introduced a new level of complexity, as it had to integrate peptide-peptide interactions in the model too, so I opted not to continue with this approach.
Table 5.2 Peptide binders selected after a binding-based screening on AlphaFold3 for the A4V mutant of SOD1, along with their ipTM scores and including the known SOD1-binding peptide FLYRWLPSRRGG (iteration 1).
| Peptide sequence | Control or test | ipTM | |
|---|---|---|---|
| 0 | FLYRWLPSRRGG | Control | 0.89 |
| 1 | WLSPATVAARKC | Test | 0.90 |
| 2 | WRYGAVGAKLWC | Test | 0.90 |
| 3 | HRYVWTAARHKW | Test | 0.90 |
| 4 | WRYGVAGVAHKW | Test | 0.91 |
Upon obtaining the four peptide binders presented in Table 5.2, I wondered if I could further improve them by replacing the final amino acid with a different one exhibiting similar biochemical properties, which, however, I arbitrarily chose not to include in the first iteration of my binding screening. For this second iteration, I swapped cysteine with methionine (M) for the first two peptides and tryptophan with phenylalanine (F) and tyrosine (Y) for the third and fourth PepMLM-generated peptides. As a result, the first peptide was further optimized with a methionine as its final amino acid residue (Table 5.3).
Table 5.3 Peptide binders selected after a binding-based screening on AlphaFold3 for the A4V mutant of SOD1, along with their ipTM scores and including the known SOD1-binding peptide FLYRWLPSRRGG (iteration 2).
| Peptide sequence | Control or test | ipTM | |
|---|---|---|---|
| 0 | FLYRWLPSRRGG | Control | 0.89 |
| 1 | WLSPATVAARKM | Test | 0.91 |
| 2 | WRYGAVGAKLWC | Test | 0.90 |
| 3 | HRYVWTAARHKW | Test | 0.90 |
| 4 | WRYGVAGVAHKW | Test | 0.91 |
All peptides (0 - 4, as numbered in Table 5.3) appear to localize near the N-terminus of both monomers in the homodimer (Figure 5.1C-G). They also seem to approach the dimer interface, rather than engage the β-barrels, and they are all surface-bound (Figure 5.1C-G).
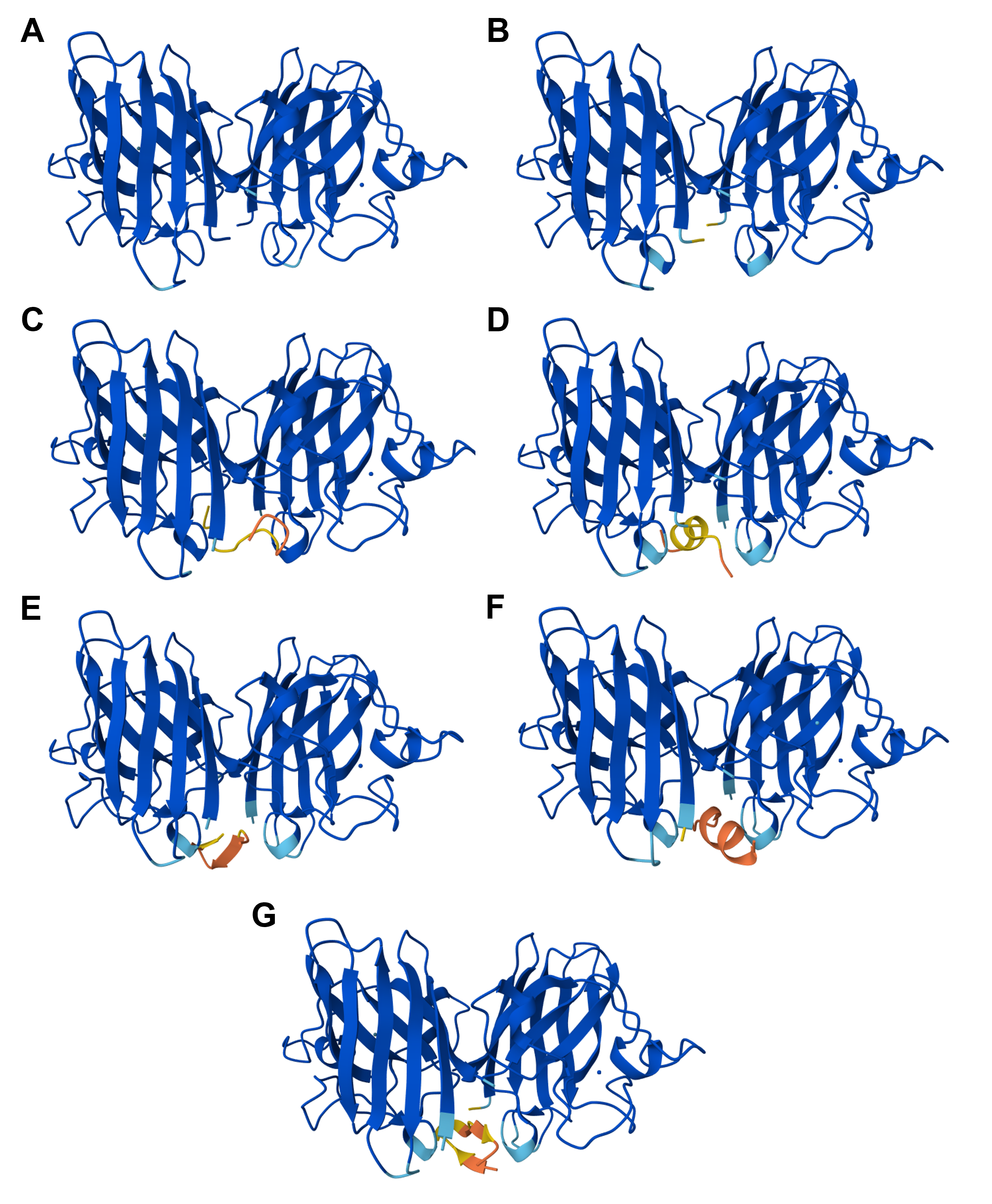 Figure 5.1 3D visualizations of SOD1 and SOD1 A4V with and without interacting with already known and PepMPL-generated binding peptides as computed by the AlphaFold3 server: (A) SOD1, (B) SOD1 A4V, (C) SOD1 A4V with the known binding peptide FLYRWLPSRRGG (peptide 0), (D) SOD1 A4V with the binding peptide WLSPATVAARKM (peptide 1), (E) SOD1 A4V with the binding peptide WRYGAVGAKLWC (peptide 2), (F) SOD1 A4V with the binding peptide HRYVWTAARHKW (peptide 3), and (G) SOD1 A4V with the binding peptide WRYGVAGVAHKW (peptide 4). Figure generated with AlphaFold3.
Figure 5.1 3D visualizations of SOD1 and SOD1 A4V with and without interacting with already known and PepMPL-generated binding peptides as computed by the AlphaFold3 server: (A) SOD1, (B) SOD1 A4V, (C) SOD1 A4V with the known binding peptide FLYRWLPSRRGG (peptide 0), (D) SOD1 A4V with the binding peptide WLSPATVAARKM (peptide 1), (E) SOD1 A4V with the binding peptide WRYGAVGAKLWC (peptide 2), (F) SOD1 A4V with the binding peptide HRYVWTAARHKW (peptide 3), and (G) SOD1 A4V with the binding peptide WRYGVAGVAHKW (peptide 4). Figure generated with AlphaFold3.
Among the PepMLM-generated peptides shown in Table 5.3, all four of them were assigned an ipTM score >0.8, indicating confident, high-quality, and successful predictions of the complex’s structure by the model. More importantly, peptides 1 - 4 were given higher ipTM scores than the known SOD1-binding peptide FLYRWLPSRRGG, which can be translated as a more accurate prediction of the relative positions among the components of the complex. Peptides 1 and 4 in particular scored 0.91 compared to the 0.89 ipTM score of peptide 0, rendering them promising alternatives at this stage of the analysis. As an additional measure of the peptides’ performance, I calculated their combined score as well, defined with the formula 0.8 x ipTM + 0.2 x pTM, to include their pTM metrics too 3. Once again, peptides 1 - 4 scored consistently higher than FLYRWLPSRRGG, with peptides 2 and 3 receiving a combined score of 0.904 over peptide 0’s 0.896, along with peptides 1 and 4 being assigned 0.914 for the same measure.
C. Evaluate properties of generated peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes:
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties? Choose one peptide you would advance and justify your decision briefly.
To better simulate the binding target, which in this case, is an ion-recruiting protein homodimer, I inserted two copies of SOD1 A4V’s amino acid sequence in the designated field of the PeptiVerse interface. The predicted properties of the PepMLM-generated peptides are summarized below, in Table 5.4.
Table 5.4 SOD1 A4V-binding peptides’ properties as predicted by PeptiVerse, with the peptides either being known binders or having been generated with PepMLM.
| Peptide sequence | Solubility | Haemolysis | Binding affinity | Molecular weight | Net charge (pH 7) | Isoelectric point | Hydrophobicity | |
|---|---|---|---|---|---|---|---|---|
| 0 | FLYRWLPSRRGG | Soluble (100%) | Non-haemolytic (95.3%) | Weak binding (6.098pKd/pKi) | 1,507.7Da | 2.76 | 11.71 | -0.71 GRAVY |
| 1 | WLSPATVAARKM | Soluble (100%) | Non-haemolytic (98.4%) | Weak binding (5.368pKd/pKi) | 1,330.6Da | 1.76 | 11.00 | 0.24 GRAVY |
| 2 | WRYGAVGAKLWC | Soluble (100%) | Non-haemolytic (92.4%) | Medium binding (7.831pKd/pKi) | 1,409.7Da | 1.75 | 9.31 | 0.15 GRAVY |
| 3 | HRYVWTAARHKW | Soluble (100%) | Non-haemolytic (98.3%) | Weak binding (5.572pKd/pKi) | 1,610.8Da | 2.93 | 11.00 | -1.28 GRAVY |
| 4 | WRYGVAGVAHKW | Soluble (100%) | Non-haemolytic (96.8%) | Medium binding (7.799pKd/pKi) | 1,429.6Da | 1.85 | 9.99 | -0.29 GRAVY |
According to PeptiVerse’s predictions as presented in Table 5.4, all screened peptides are soluble (100% probability) and non-haemolytic (> 92% probability). It would also appear that higher ipTM scores do not necessarily correlate with a strong binding affinity to the protein variant. This is especially the case for peptide 1, which, despite receiving a 0.91 ipTM score, is anticipated to have weak binding affinity to the target. After obtaining the data above from PeptiVerse, I would choose to proceed with one the two peptides that displayed medium binding affinity, namely either peptide 2 or peptide 4. Since the discrepancy between their binding affinities is relatively small, I would advance peptide 4, which was assigned a 0.91 score with AlphaFold3 and demonstrates a higher probability of not causing haemolysis (96.8% > 92.4%).
D. Generate optimized peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
- After opening the moPPit Colab linked from the HuggingFace moPPIt model card, make a copy and switch to a GPU runtime.
- In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
For the generation of binding peptides through moPPit, I chose the “Haemolysis” and “Solubility” criteria with a weight of 2, as I considered them less significant than “Specificity”, to which I assigned a weight of 3, as well as “Affinity” and “Motif”, whose weights I increased to 7, since those two are the principal factors determining the structure of the peptide binders for this assignment. More specifically, for the “Motif” criterion, I designated amino acids 1 - 10, 106 - 112, and 140 - 153 as positions that should be primarily taken into account. The first region is where the A4V mutation resides, while the second seems to be related to the formation of two short β-sheets beneath the β-barrel regions; those structures are present in the mutant variant but not in the wildtype one (Figure 5.1B compared to A). Additionally, the C-terminus of the protein, here represented by residues 140 - 153, appears to be the main interface for the homodimer formation and stabilization, hence its inclusion as a region that should influence the generation of binding peptides. The newly-designed peptides can be found below (Table 5.5, Figure 5.2).
Table 5.5 Peptide binders for the A4V mutant of SOD1 generated by moPPit, along with their ipTM scores.
| Peptide sequence | ipTM | |
|---|---|---|
| 1 | KKKCGVLVVVHD | 0.89 |
| 2 | AVTMKRKPLFCQ | 0.92 |
| 3 | PKSQKVKTCVAQ | 0.89 |
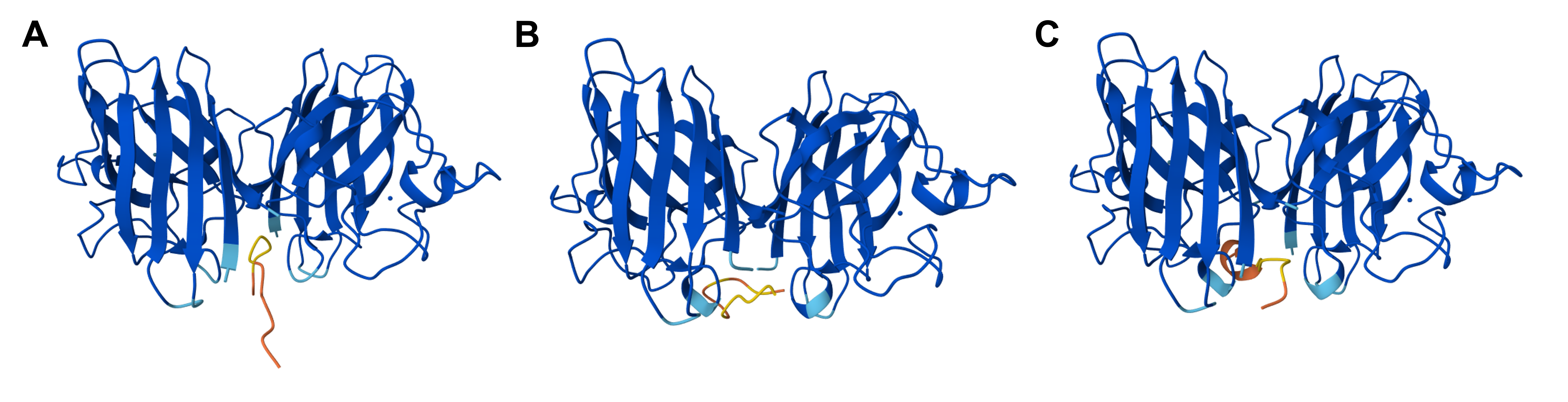 Figure 5.2 3D visualizations of SOD1 A4V interacting with moPPit-generated binding peptides as computed by the AlphaFold3 server: (A) SOD1 A4V with the binding peptide KKKCGVLVVVHD (peptide 1_moPPit), (B) SOD1 A4V with the binding peptide AVTMKRKPLFCQ (peptide 2_moPPit), and (C) SOD1 A4V with the binding peptide PKSQKVKTCVAQ (peptide 3_moPPit). Figure generated with AlphaFold3.
Figure 5.2 3D visualizations of SOD1 A4V interacting with moPPit-generated binding peptides as computed by the AlphaFold3 server: (A) SOD1 A4V with the binding peptide KKKCGVLVVVHD (peptide 1_moPPit), (B) SOD1 A4V with the binding peptide AVTMKRKPLFCQ (peptide 2_moPPit), and (C) SOD1 A4V with the binding peptide PKSQKVKTCVAQ (peptide 3_moPPit). Figure generated with AlphaFold3.
Before advancing any of the moPPit-generated peptides to clinical studies, I would first evaluate them through the PeptiVerse to assess their biochemical properties and screen for possible unintended effects. The results of this analysis are presented in Table 5.6.
Table 5.6 SOD1 A4V-binding peptides’ properties as predicted by PeptiVerse, with the peptides having been generated with moPPit.
| Peptide sequence | Solubility | Haemolysis | Binding affinity | Molecular weight | Net charge (pH 7) | Isoelectric point | Hydrophobicity | |
|---|---|---|---|---|---|---|---|---|
| 1 | KKKCGVLVVVHD | Soluble (100%) | Non-haemolytic (95.7%) | Tight binding (9.909pKd/pKi) | 1,324.6Da | 1.84 | 9.20 | 0.36 GRAVY |
| 2 | AVTMKRKPLFCQ | Soluble (100%) | Non-haemolytic (98.0%) | Weak binding (6.635pKd/pKi) | 1,421.8Da | 2.78 | 10.06 | -0.09 GRAVY |
| 3 | PKSQKVKTCVAQ | Soluble (100%) | Non-haemolytic (98.4%) | Medium binding (8.371pKd/pKi) | 1,316.6Da | 2.95 | 9.81 | -0.76 GRAVY |
Stevens JC, Chia R, Hendriks WT, et al. Modification of superoxide dismutase 1 (SOD1) properties by a GFP tag–implications for research into amyotrophic lateral sclerosis (ALS). PLoS ONE. 2010;5(3):e9541. doi:10.1371/journal.pone.0009541 ↩︎
Chen T, Dumas M, Watson R, et al. PepMLM: Target Sequence-Conditioned Generation of Therapeutic Peptide Binders via Span Masked Language Modeling. arXiv. August 11, 2024. doi:10.48550/arxiv.2310.03842 ↩︎
Omidi A, Møller MH, Malhis N, Bui JM, Gsponer J. AlphaFold-Multimer accurately captures interactions and dynamics of intrinsically disordered protein regions. Proc Natl Acad Sci USA. 2024;121(44):e2406407121. doi:10.1073/pnas.2406407121 ↩︎