Week 6 homework
Genetic circuits-Part I: Assembly technologies 🧩
DNA Assembly
Answer these questions about the protocol in this week’s lab:
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The components in the Phusion High-Fidelity PCR Master Mix, along with their purpose, are the following:
- Phusion PCR buffer: The buffer solution ensures optimal salt concentration and pH conditions for the amplification reaction to take place, while also providing the Mg+2 ions needed for the polymerase’s catalytic activity.
- Deoxyribonucleotides (dNTPs): This is a mix of A, T, G, and C nucleotides in equal concentrations (to prevent integration bias) which provides the building blocks necessary for the synthesis of the nascent DNA strands.
- Phusion polymerase: This is the enzyme that, upon given a free 3’ -OH group and a complementary DNA strand, will catalyze the actual amplification step, where new strands are synthesized.
- Dimethylsulfoxide (DMSO): DMSO can be added in especially challenging amplification reactions, in particular when the template sequence is particularly GC-rich. An increased GC content can lead to incomplete denaturation, as well as to the formation of secondary structures that lower PCR efficacy, therefore, a denaturing aid, such as DMSO, can reduce DNA melting temperature and non-specific binding for more effective amplification of the target sequence.
- Nuclease-free water: If needed, nuclease-free water can be added to the PCR solution to reach the final volume of the reaction. This secures that all ingredients have the appropriate concentration for the amplification to happen and no degrading enzymes can endanger the template DNA or the nascent strands’ integrity. The DNA template, as well as the primers, for the reaction are usually custom-designed and provided by the researcher.
2. What are some factors that determine primer annealing temperature during PCR?
The two primary factors determining primer annealing temperature are the primer’s length and its GC content. In general, the longer a primer is the higher its annealing temperature. The same holds true for a higher GC percentage, as in both cases the more hydrogen bonds formed between the template strand and the primer the stronger and more stable the annealing. Those two factors majorly influence the formation of secondary structures in primer molecules and primer dimers, which in turn affect PCR efficiency. However, the degree to which those two parameters influence primer annealing temperature is relative. For instance, a primer that is designed to introduce several point mutations at a short distance from one another (a “garland primer”) or a primer fabricated to insert an entirely novel small fragment to a sequence (a primer with a 5’ overhang), although seemingly longer, do not necessarily require a higher annealing temperature, at least not in the first cycles of the PCR, when the template DNA molecules outnumber the newly synthesized sequences. Similarly, apart from the GC content, the distribution of Gs and Cs in the primer can modulate its annealing point: primers with more than two or three sequential Gs and Cs accumulated at one point bind more firmly to their complementary strand and, thus, display higher Tms, than primers where Gs and Cs are more evenly spaced out and separated by brief arrays of As and Ts. The need for higher primer annealing temperatures can be alleviated by the addition of denaturing agents, such as DMSO as explained previously, which could also mildly impact the resulting primer Tm. Furthermore, the concentration of salts in the reaction, combined with their ionic strength, as well as the concentration of primers themselves, can influence annealing temperature too. An increased concentration of both can negate the electrostatic forces facilitating the formation of hydrogen bonds and favor non-specific binding respectively, resulting in the need for a higher annealing temperature.
3. There are two methods from this class that create linear fragments of DNA: PCR and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction enzyme digests show both similarities and differences. In terms of their respective protocols, conducting a PCR requires a thermocycler for the temperature to fluctuate during the different stages of the reaction, whereas restriction enzyme digests have to be incubated only at one specific temperature dictated by the restriction endonuclease (optionally, a second higher temperature, usually 80°C, can also be deployed for deactivating the enzyme). Additionally, the same three steps -denaturation, annealing, and extension- have to be repeated 25 - 30 times for a PCR, rendering it generally more time-consuming than a restriction digest, which does not involve any repeated steps. Apart from the DNA template, the buffer, and the respective enzyme in both techniques, PCR needs more reagents than a restriction endonuclease digestion, including the primers, the dNTPs, as well as the GC enhancers (like DMSO). Another difference lies in the lack of heat tolerance of restriction endonucleases used in digest reactions (as highlighted previously, they can be easily deactivated by increasing the reaction temperature), while DNA polymerases employed in PCRs are utilized exactly because they can withstand temperatures close to water’s boiling point and are even optimized for this specific property. On the other hand, there is a much wider variety of restriction enzymes that can be used for digests compared to the fewer variants of DNA polymerases commercially available. Lastly, while PCR requires the a priori designing of appropriate primers and restriction digests entail the selected endonuclease’s recognition site already existing in the DNA template, the products generated from both processes need column-based purification before being used in downstream applications.
Regarding their general use and purpose, as fundamental Molecular Biology techniques, they both generate linear DNA fragments. Nonetheless, in PCR, amplicons have blunt ends, whereas in restriction digests, digested DNA fragments can have 5’ or 3’ overhangs too. In many cases, both techniques are crucial for cloning designed DNA sequences, as they can isolate a particular DNA segment with great specificity and efficiency. Both techniques can be employed to confirm that the correct DNA fragment has indeed been inserted in an assembled construct after cloning is complete (and can be visualized with agarose gel electrophoresis), while they can also be combined on multiple occasions, for instance, to carry out a Gibson assembly or to degrade the remaining plasmid template through DpnI digestion after a PCR and before performing a bacterial transformation. On the contrary, PCR alone would be the preferred technique when the goal of the experiment is to insert a point mutation or a short new fragment to a known DNA sequence (with the suitable primers) or when the ultimate purpose of a reaction is simply to propagate and amplify DNA. PCR is a synthtic technique, leading to a much higher in vitro-synthesized nucleic acid yield and concentration than the initial one, which is, of course, not the case for restriction enzyme digestion, which can contain both in vitro- and in vivo-produced sequences. Concerning restriction enzyme digestion, it can be utilized when DNA needs to be broken down, for example, to decompose the plasmid template molecules after a PCR as mentioned above, or it can be employed for other cloning methods, such as Golden Gate assembly, which, traditionally, does not involve any PCR steps.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The first and most crucial step to ensure that PCR-amplified and digested DNA sequences are appropriate for Gibson cloning is to spend as much time as needed to design them correctly. During this phase, verifying that DNA segments are properly designed can be achieved by in silico simulating their assembly on a simulation software like Benchling. On a more practical note, apart from making sure to cautiously follow the steps of the designated protocols, PCR-amplified and digested DNA sequences should be purified through a column-based method and, ideally, have their concentration measured (for example, with a NanoDrop instrument) before any downstream processes. Lastly, prior to proceeding with the cloning, it is always a good idea to visualize the DNA amplicons and digested segments through agarose gel electrophoresis, to ensure that they were indeed generated and that they have the anticipated size as an indication that no mistakes had been made in previous experiments.
5. How does the plasmid DNA enter the E. coli cells during transformation?
For plasmid DNA to successfully enter E. coli cells during transformation, firstly, the correct positioning of the plasmid molecules on the periphery of the bacterial cells has to be ensured. Several chemical agents, such as MgCl2 and polyethylene glycol (PEG) 8000, are employed during the preparation of competent bacterial cells (bacteria that have been rendered available to receive exogenously procured DNA) to this end. In more detail, Mg+2 cations from MgCl2 help neutralize the negative charges of DNA’s phosphate backbone, allowing plasmids to approach and remain close to the bacterial cell walls, while extremely hydrophilic compounds, such as PEG, contribute to removing the aqueous coating of plasmid molecules, once again eliminating the barriers between the foreign DNA and the surface of the cells. This is further facilitated by keeping the cells about to be transformed on ice for approximately 10min. Once the plasmids are in position, a heat or electric shock during transformation effectively disrupts the integrity and continuity of the bacterial cell wall and cell membrane, which can result in circular DNA entering the cell. After the shock, bacteria are left to recover and repair their membranes, with a percentage of the cells having acquired an additional piece of DNA through the process.
6. Describe another assembly method in detail (such as Golden Gate Assembly).
- Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
- Model this assembly method with Benchling or Asimov Kernel!
For this section, I decided to describe Golden Gate assembly, as it is one of the methods I am most familiar with. It is a versatile, highly efficient, one-pot, scarless cloning method that allows joining up to 30 DNA segments in a single reaction. As visual aids, I will use two slides from a workshop presentation we organized with other members of iGEM Athens 2022 for the 18th Autumn Assembly of the European Pharmaceutical Students Association (EPSA) held in Athens in November 2022.
A specific category of restriction endonucleases, namely type IIS restriction enzymes (such as BsaI and BsmBI), constitutes the basis of Golden Gate assembly. What distinguishes type IIS endonucleases from traditional restriction enzymes is that they recognize a specific sequence of nucleotides but cleave DNA several nucleotides downstream (therefore outside) of their recognition site. This property enables the generation of custom four-nucleotide overhangs with the use of solely one restriction enzyme (Figure 6.1).
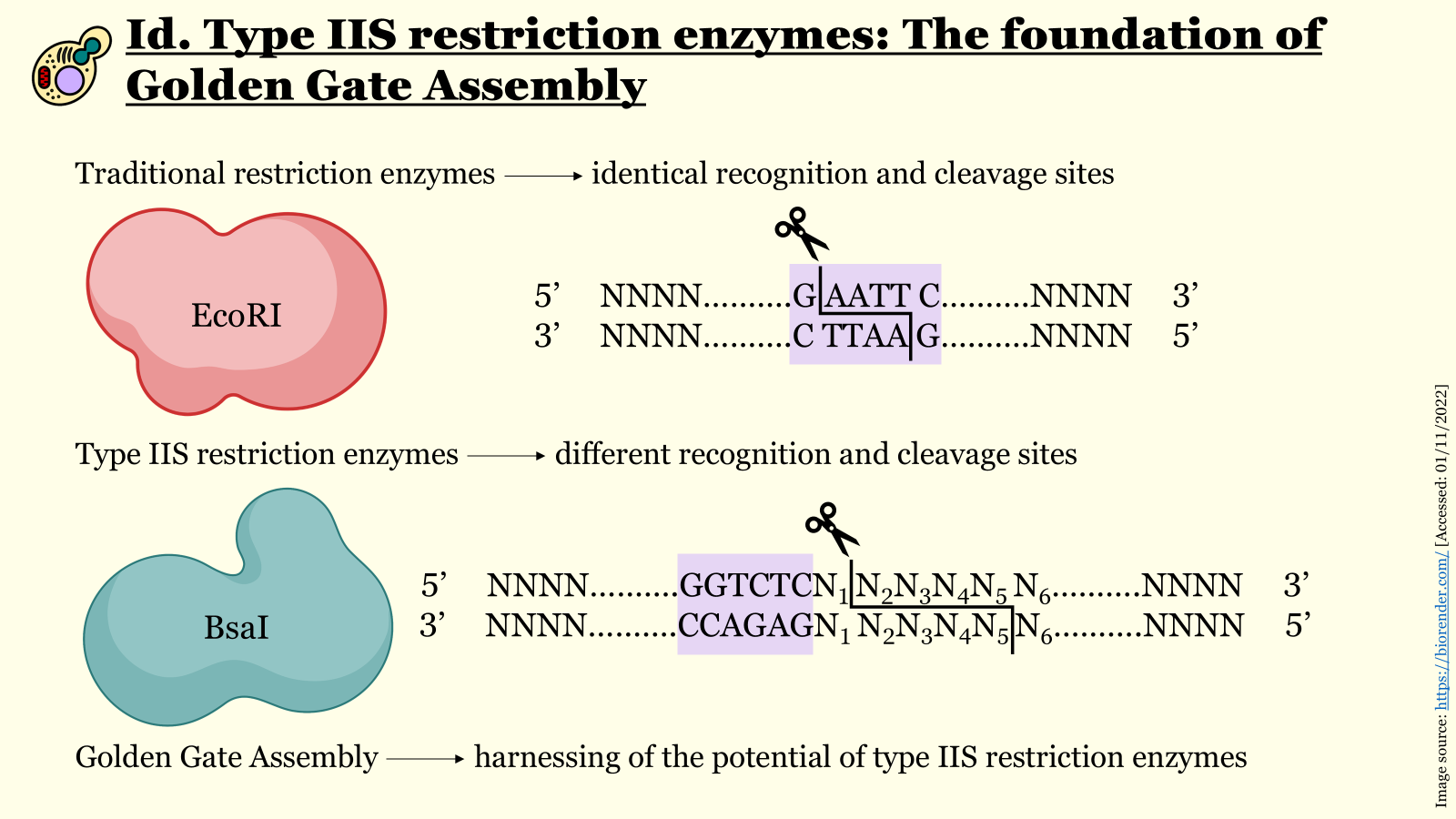 Figure 6.1 Slide from a workshop presentation of iGEM Athens 2022 for the 18th Autumn Assembly of EPSA explaining the mechanism of function for traditional restriction enzymes compared to type IIS endonucleases.
Figure 6.1 Slide from a workshop presentation of iGEM Athens 2022 for the 18th Autumn Assembly of EPSA explaining the mechanism of function for traditional restriction enzymes compared to type IIS endonucleases.
Due to the creation of custom overhangs by just one restriction endonuclease, complex DNA constructs consisting of up to 30 distinct parts can be arranged in a specific predesignated order and assembled with an extremely low error rate in a one-pot reaction. If the design of the individual DNA segments is executed correctly, meaning with recognition sites “outward” of cleavage sites for inserts and the inverse for the selected backbone (recognition sites “inward” of cleavage sites), all parts should be assembled seamlessly (without leaving any restriction sites behind) with the right configuration (Figure 6.2). Besides the orientation of the recognition and cleavage sites, another design prerequisite is that insert fragments must not contain internal recognition sites for the chosen type IIS endonuclease, which can be removed via site-directed mutagenesis prior to assembly.
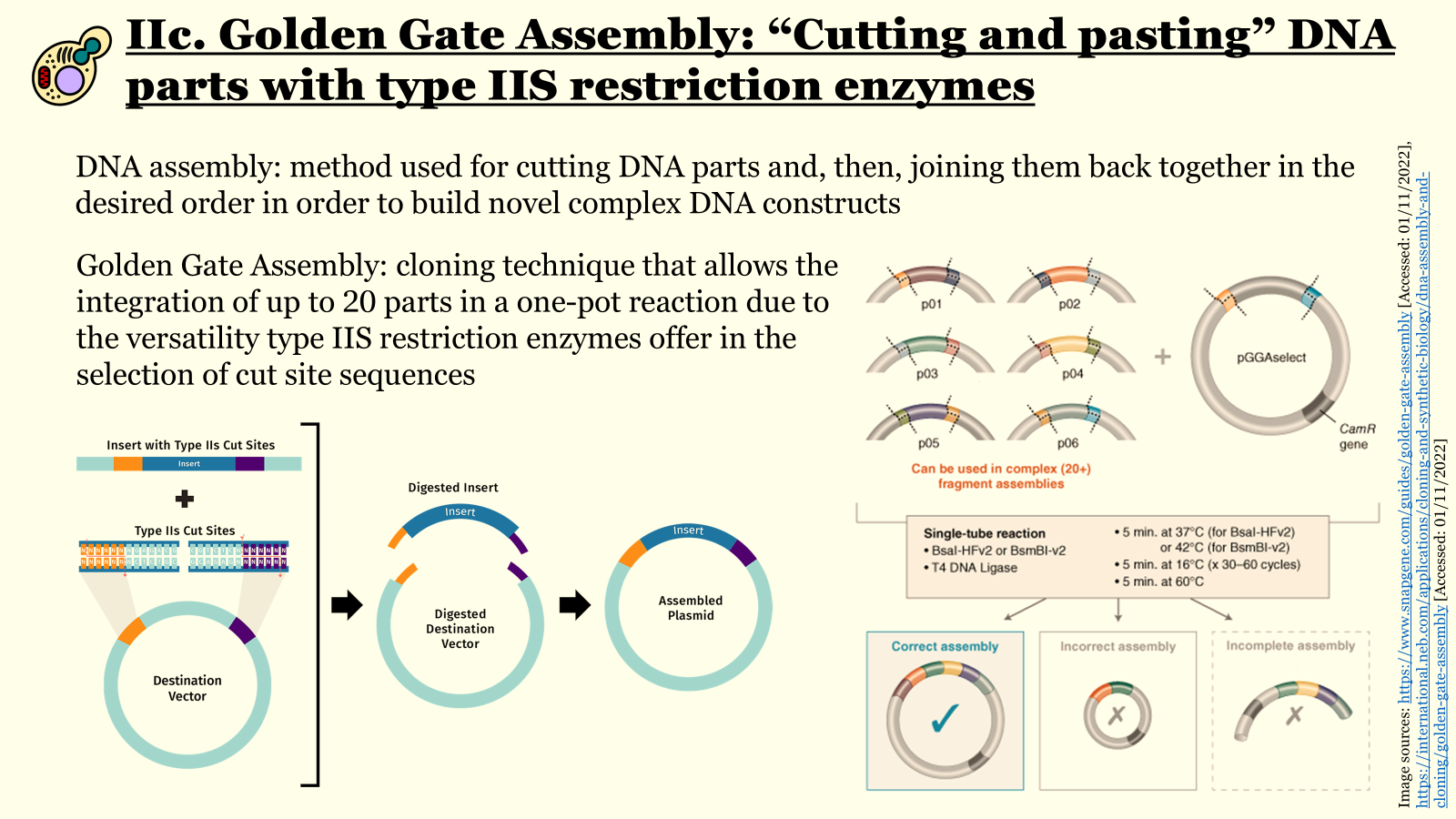 Figure 6.2 Slide from a workshop presentation of iGEM Athens 2022 for the 18th Autumn Assembly of EPSA illustrating how Golden Gate assembly enables building complex genetic circuits in a one-pot reaction.
Figure 6.2 Slide from a workshop presentation of iGEM Athens 2022 for the 18th Autumn Assembly of EPSA illustrating how Golden Gate assembly enables building complex genetic circuits in a one-pot reaction.
Apart from the selected type IIS restriction enzyme(s), a T4 DNA ligase is included in the master mix too, so that both digestion and ligation occur in a single tube, typically through 30 - 60 consecutive cycles at the appropriate temperatures in a standard thermocycler. This protocol ensures that molecules of the desired final construct accumulate over time, as they lack the type IIS restriction sites, whereas incorrect products are re-digested. This recycling property secures high efficiency which, along with all the aforementioned advantages of Golden Gate assembly, render this cloning method ideal for various Synthetic Biology applications.
To model Golden Gate Assembly, I will assemble a simplified version of one of the transcriptional units I have designed for the idea I will probably pursue for my individual final project. This simplified version of the genetic circuit enables the expression of a reflectin from the common cuttlefish Sepia officinalis, namely the protein REF8 1 2, induced by the constitutive J23102 promoter from the Anderson Collection, mediated by the Elowitz RBS, and terminated by the double terminator B0015. Instead of writing a long passage explaining every step of the process and capturing screenshots throughout the document it, I thought it would be better to record my Benchling session of assembling the construct in a short video. As I had already uploaded all the genetic parts I needed and optimized their sequences, the video consists of roughly two parts: the first demonstrates the addition of recognition and cut sites for BsaI (the type IIS enzyme I used for the assembly) to both 5’- and 3’-sides of all parts and the second shows the actual assembly with Benchling’s respective tool. Lastly, in the interest of sharing some general information, I utilized the level 1 vector pTU1-A-lacZ as a plasmid backbone and standard Golden Gate Assembly fusion sites for all internal junctions.
Figure 6.3 Video recording the building process of the pTU1-A-REF8 vector for REF8 expression through Golden Gate Assembly. Figure created in Benchling.
Sepia officinalis mRNA for reflectin 8 (gene REF8) - Nucleotide - NCBI. https://www.ncbi.nlm.nih.gov/nuccore/HE687206 ↩︎
Bassaglia Y, Bekel T, Da Silva C, et al. ESTs library from embryonic stages reveals tubulin and reflectin diversity in Sepia officinalis (Mollusca — Cephalopoda). Gene. 2012;498(2):203-211. doi:10.1016/j.gene.2012.01.100 ↩︎