HTGAA - Week 2: DNA read, write and edit

My Homework
WEEK 2 - CUT, COPY AND STITCH
This week explores the read–write–edit toolkit: sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Recitation (Wed, Feb 11)
DNA Gel, restriction enzymes, Benchling intro, Twist intro
(▶️Recording |
💻Slides)
Ice Kiattisewee
Documentation
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
Instructions
Part 0: Basics of Gel Electrophoresis
Attend or watch all lecture and recitation videos. Optionally watch bootcamp.


Part 1: Benchling & In-silico Gel Art
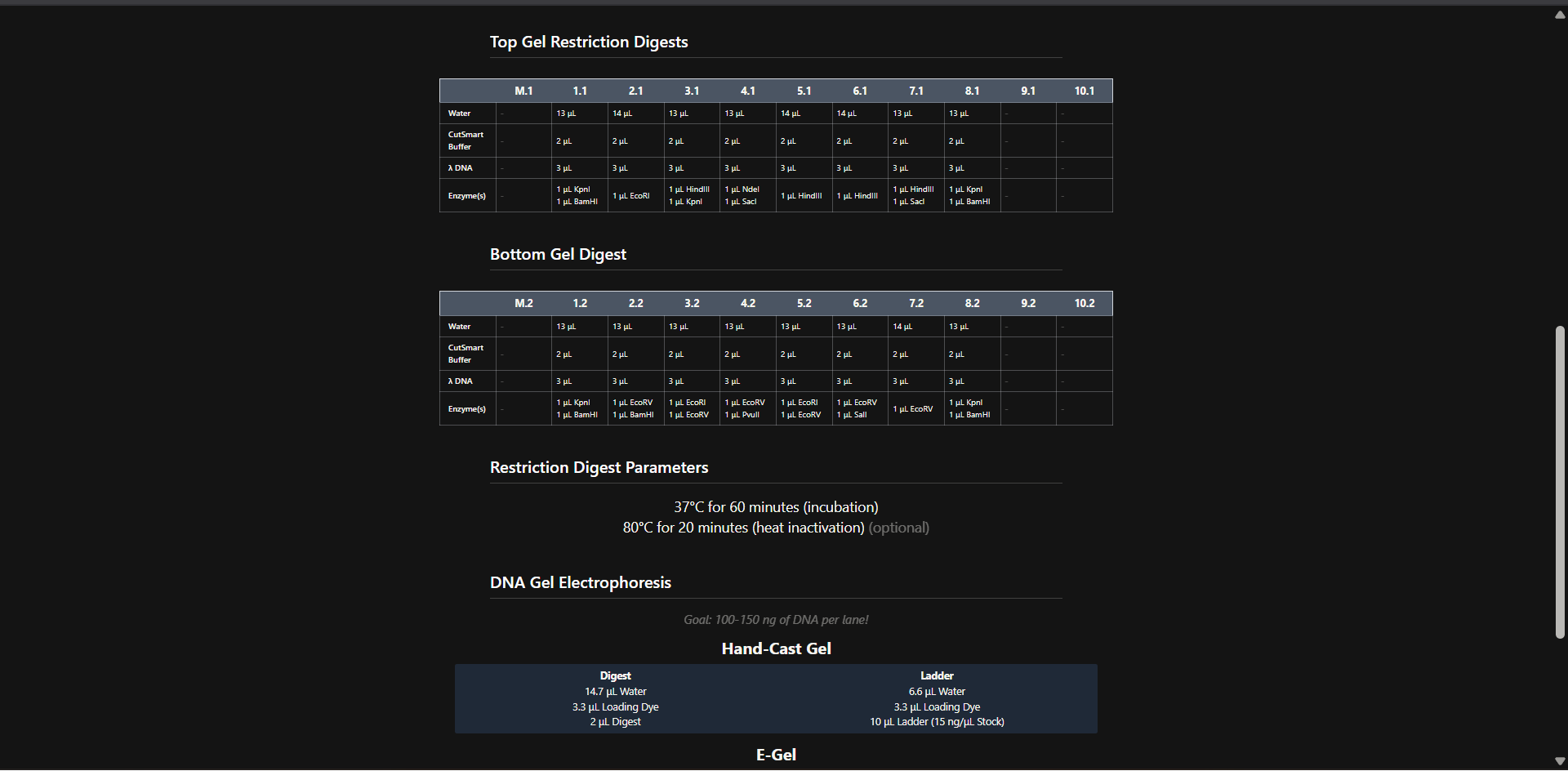
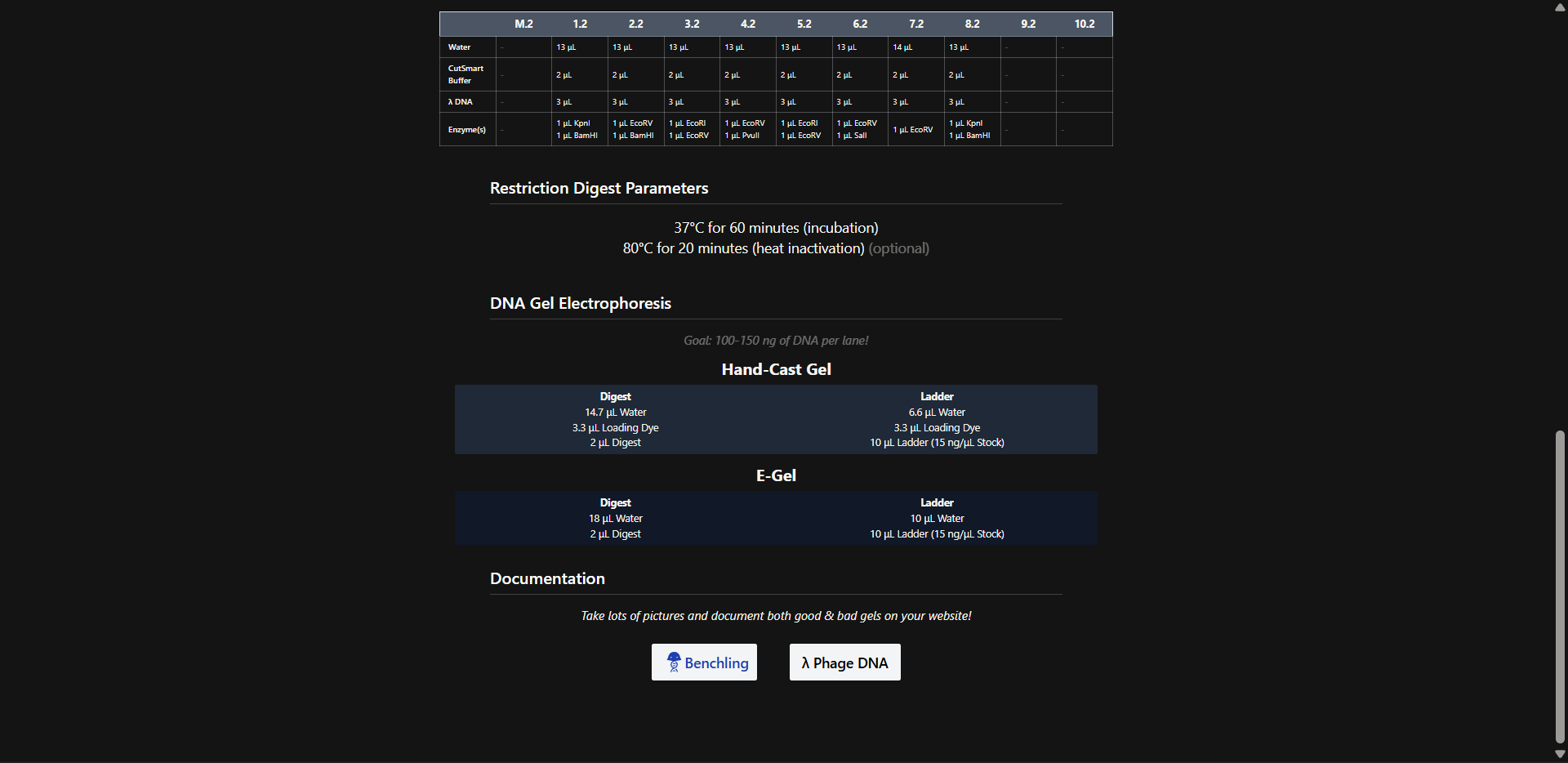
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview:
- Make a free account at benchling.com
- Import the Lambda DNA.
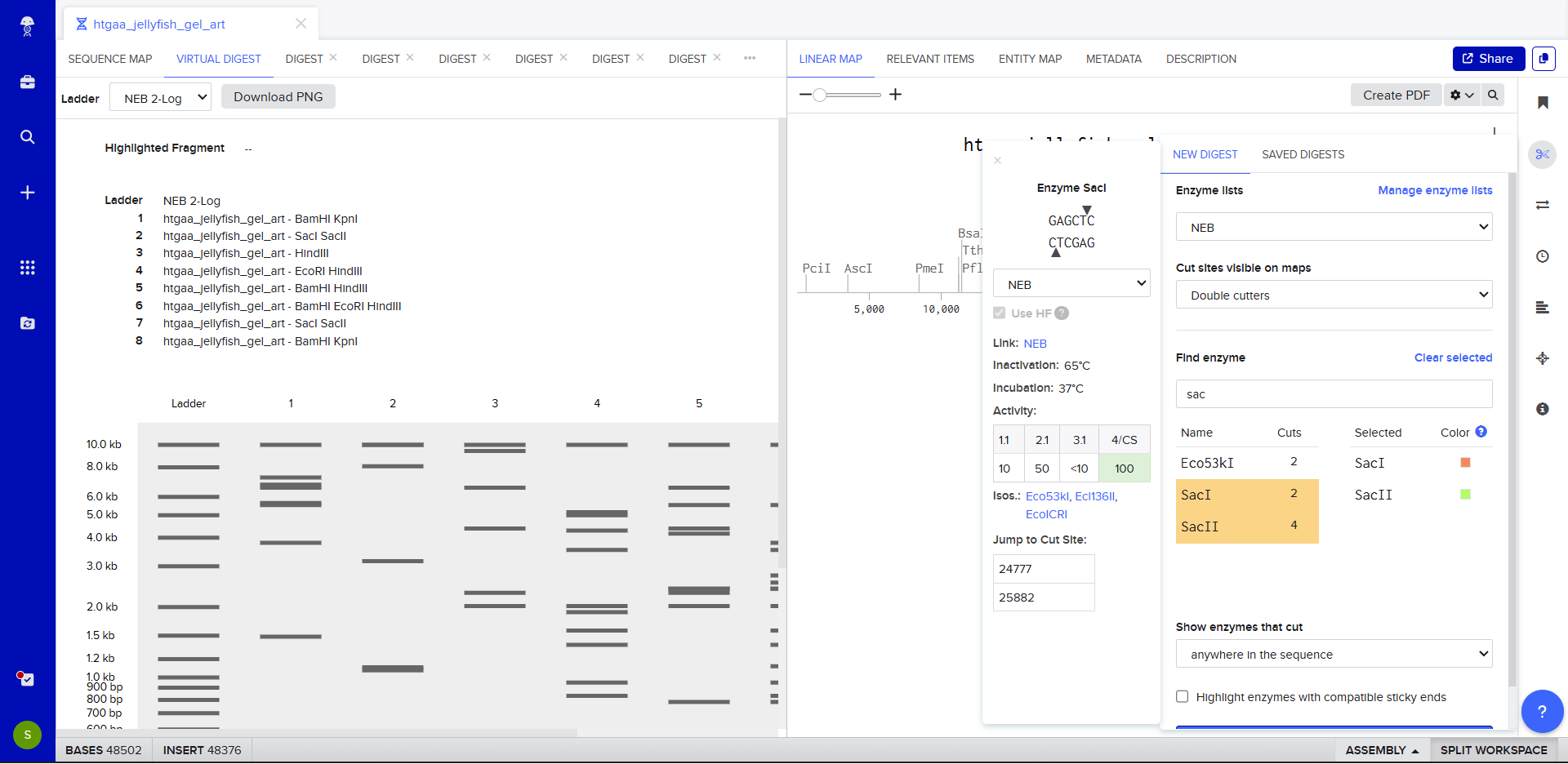
- Simulate Restriction Enzyme Digestion with the following Enzymes:
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- SalI
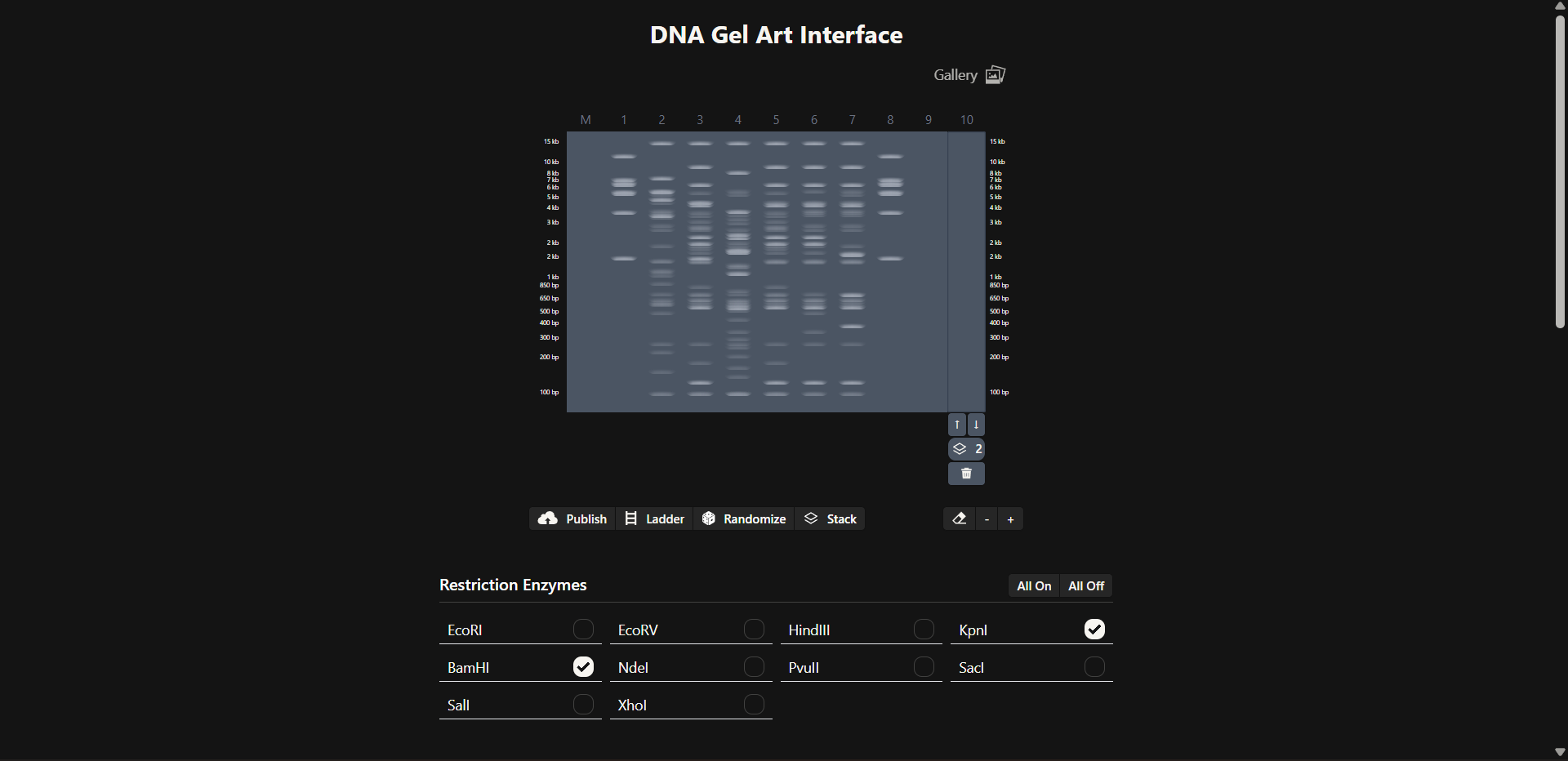
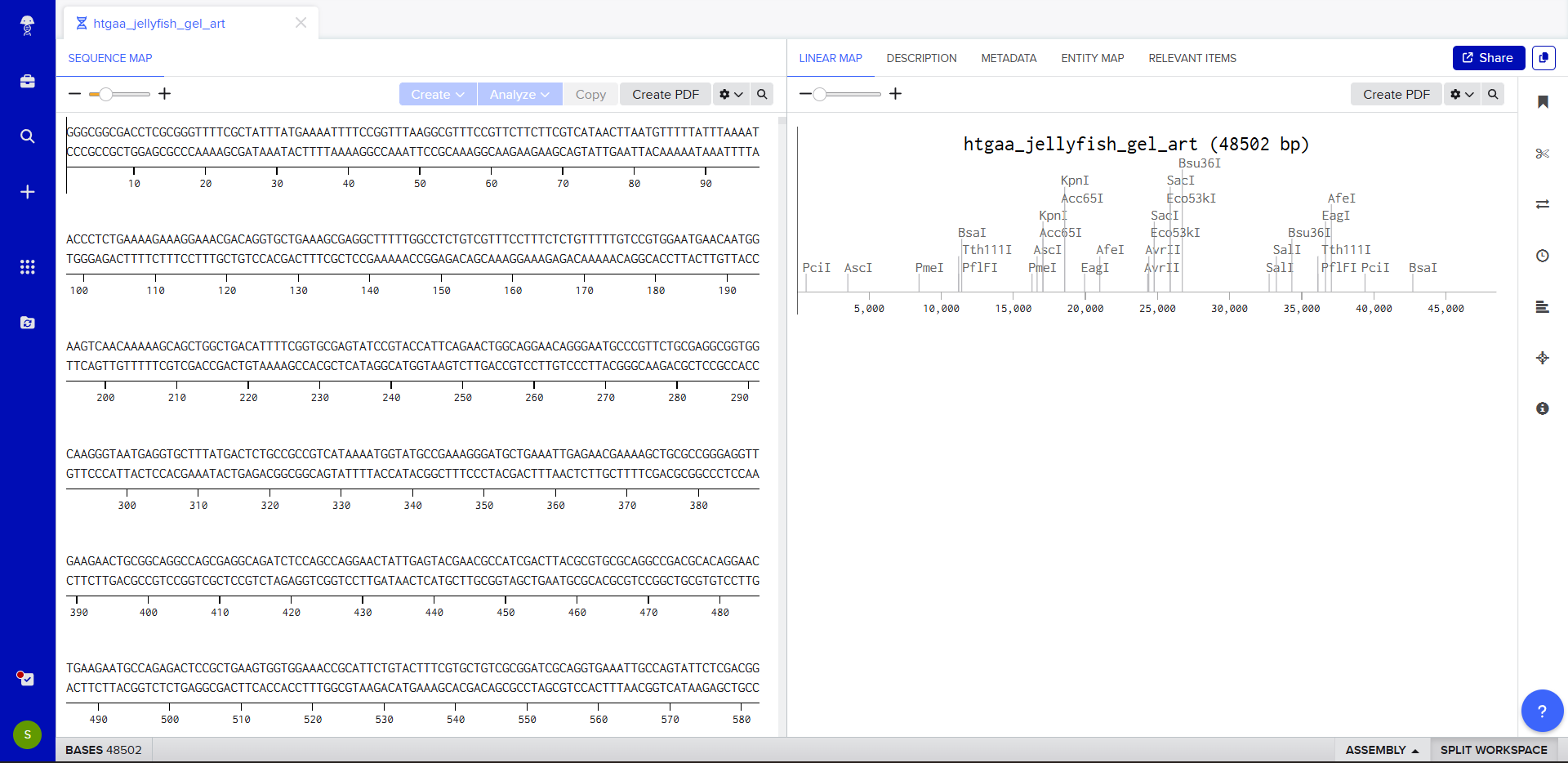
- Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
- You might find Ronan’s website a helpful tool for quickly iterating on designs!
Ronan Donovan’s electrophoresis simulation
Attempt to generate a Jellyfish design!
| 
| 
|
|---|
Benchling simulation
Generating a gel design with Benchling enzymes on site
| 
| 
|
|---|
| 
|
|---|
Final download:
Yes well, we tried that jellyfish (hope you can see it too!), mad respect to Paul Vanouse.
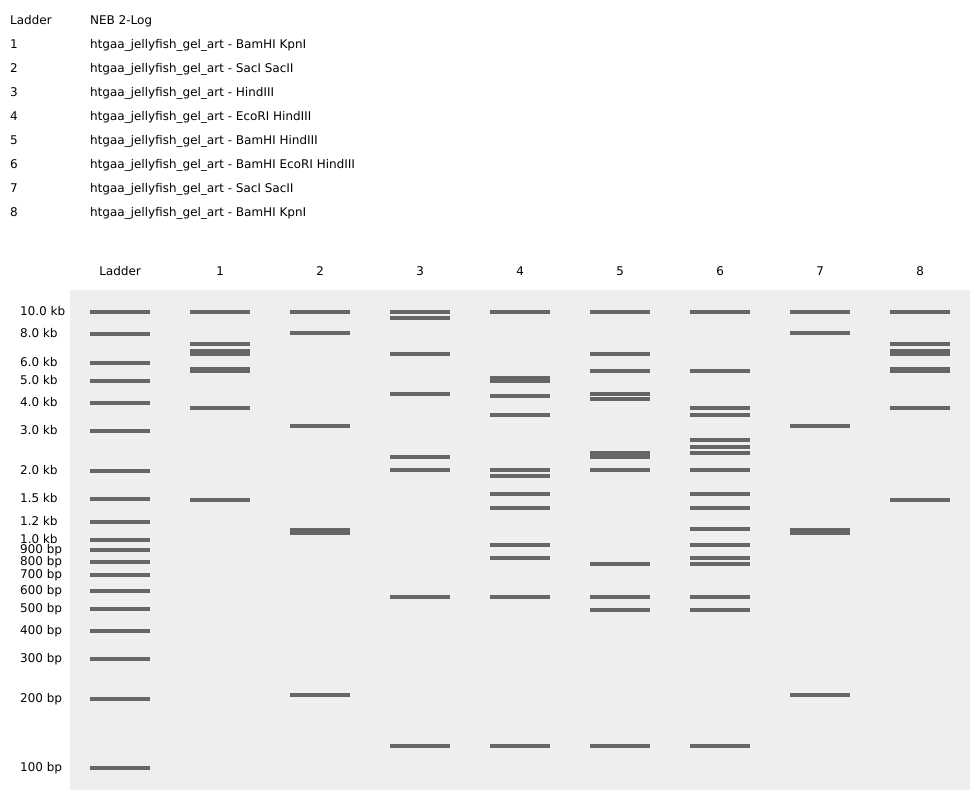
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Optional (for those with Lab access) |
Perform the lab experiment you designed in Part 1 and outlined in the Gel Art: Restriction Digests and Gel Electrophoresis protocol.
Part 3: DNA Design Challenge
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
3.1. Choose your protein.
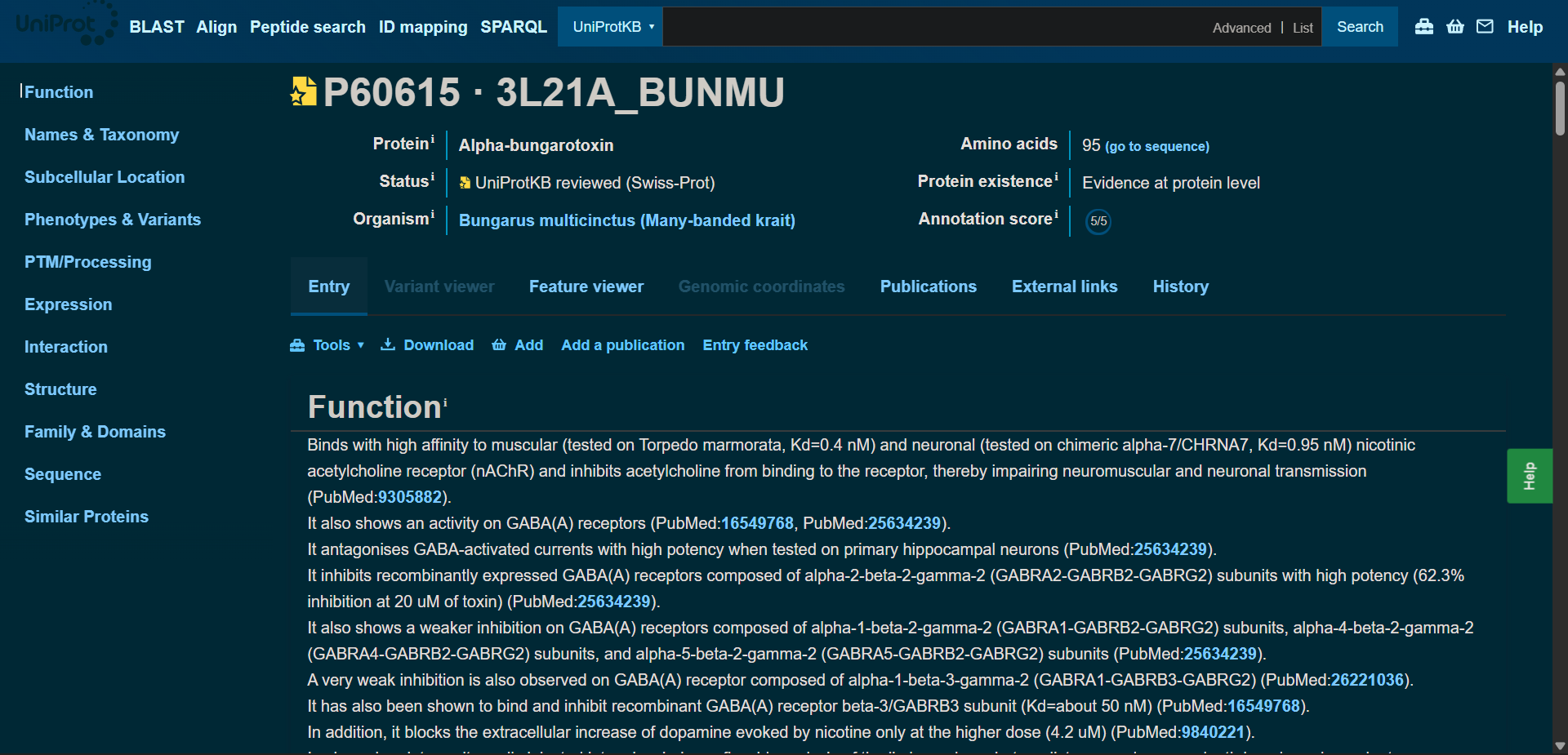
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
[Example from our group homework, you may notice the particular format — The example below came from UniProt]
>sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
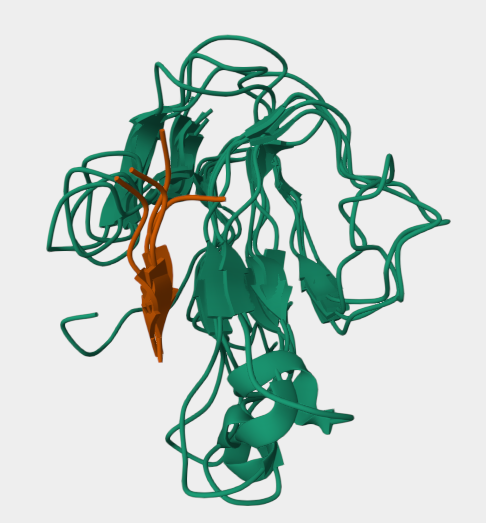
Chosen protein: α-Bungarotoxin
I chose α-bungarotoxin, a neurotoxin found in the venom of the snake Bungarus multicinctus.

Why?
- It is an extremely potent toxin.
- It binds almost irreversibly to nicotinic acetylcholine receptors.
- It causes paralysis by blocking neuromuscular transmission.
- It is widely used in neurobiological research.
- It is relatively small (95 amino acids), which makes it manageable for sequence analysis.
| 
|
|---|---|
| 
|
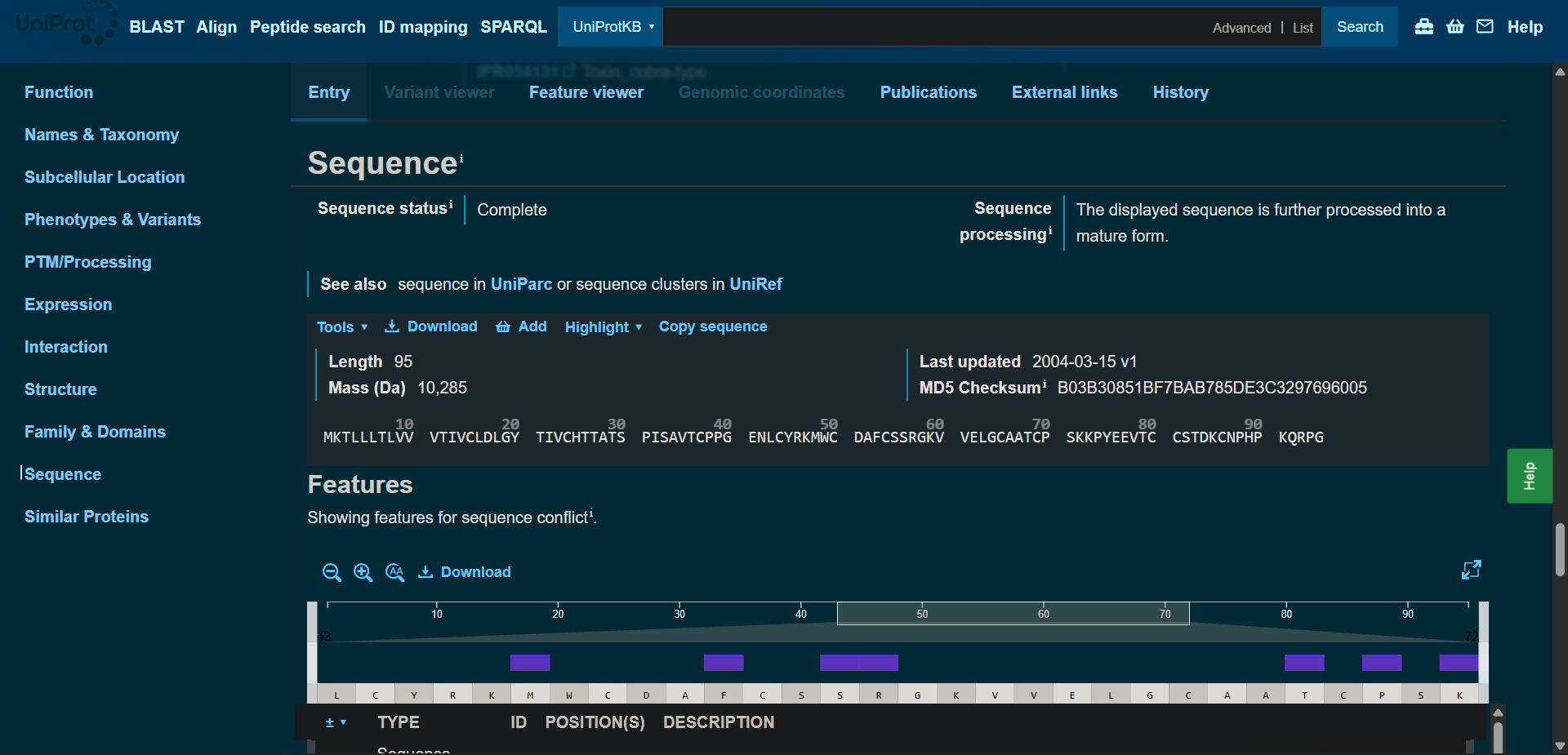
Protein sequence (UniProt-style format)
P60615|3L21A_BUNMU Alpha-bungarotoxin
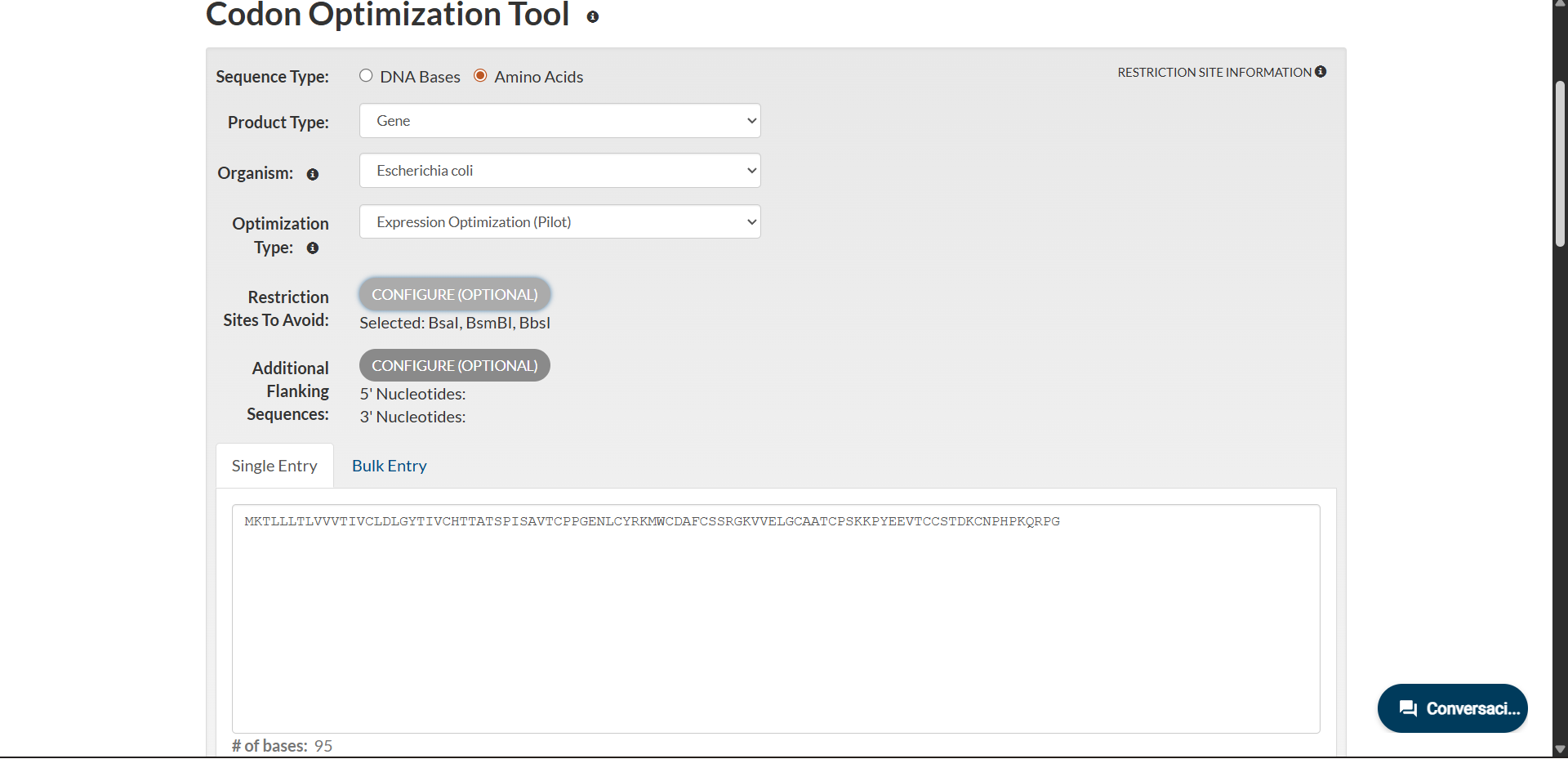
>sp|P60615|3L21A_BUNMU Alpha-bungarotoxin OS=Bungarus multicinctus OX=8616 PE=1 SV=1 MKTLLLTLVVVTIVCLDLGYTIVCHTTATSPISAVTCPPGENLCYRKMWCDAFCSSRGKVVELGCAATCPSKKPYEEVTCCSTDKCNPHPKQRPG
(95 amino acids; cysteine-rich protein with multiple disulfide bonds)
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
[Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI]
Lysis protein DNA sequence
atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
As discussed in class, due to the degeneracy of the genetic code, multiple codons can encode the same amino acid. Therefore, reverse translation from a protein sequence to a DNA sequence is not unique.
Reverse translation was performed using the Bioinformatics reverse translation tool. Due to codon degeneracy, the resulting DNA sequence represents one possible coding sequence corresponding to the selected protein.
| 
|
|---|
Complete pdf of the reverse translation: [pdf]
A possible nucleotide sequence corresponding to the selected protein is shown below:
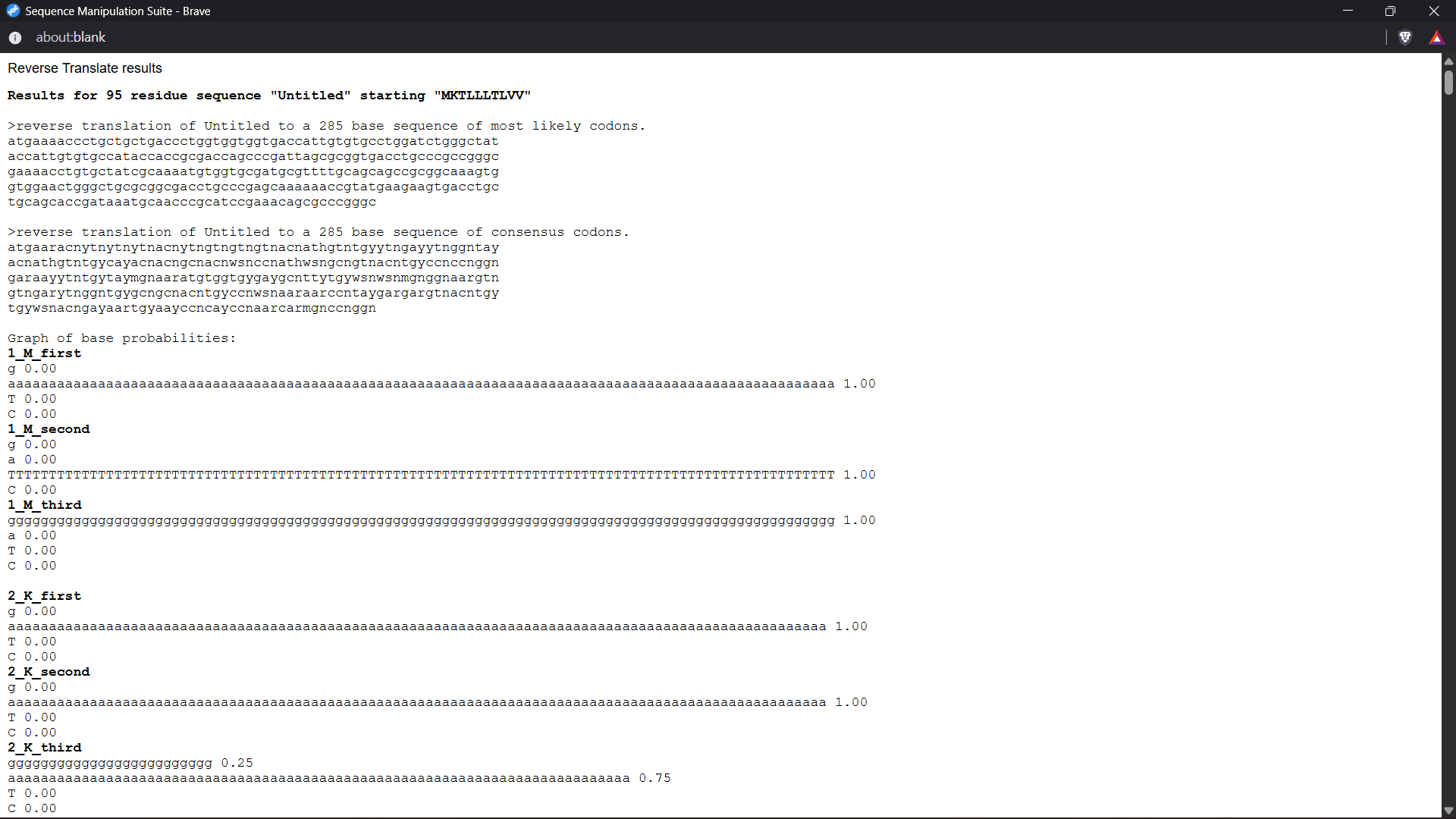
Reverse translation of Untitled to a 285 base sequence of most likely codons
atgaaaaccctgctgctgaccctggtggtggtgaccattgtgtgcctggatctgggctataccattgtgtgccataccaccgcgaccagcccgattagcgcggtgacctgcccgccgggcgaaaacctgtgctatcgcaaaatgtggtgcgatgcgttttgcagcagccgcggcaaagtggtggaactgggctgcgcggcgacctgcccgagcaaaaaaccgtatgaagaagtgacctgctgcagcaccgataaatgcaacccgcatccgaaacagcgcccgggc
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
[Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI]
Lysis protein DNA sequence with Codon-Optimization
ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
Why is codon optimization necessary?
Although the genetic code is degenerate, meaning that multiple codons can encode the same amino acid, organisms do not use synonymous codons with equal frequency. Each organism has a preferred codon usage bias that reflects the abundance of its tRNAs.
If a gene containing rare codons is introduced into a heterologous host organism, several issues may arise:
- Reduced translation efficiency
- Lower protein yield
- Increased risk of ribosome stalling
- Potential misfolding due to slowed or irregular translation kinetics
Therefore, codon optimization is performed to adapt the coding sequence to the codon usage preferences of the chosen host organism, improving translation efficiency and overall protein production.
Selected organism for optimization
The coding sequence was optimized for expression in: Escherichia coli
Why E. coli?
- It is the most widely used bacterial expression system.
- It is cost-effective, fast-growing, and easy to genetically manipulate.
- It is ideal for recombinant protein production.
Although α-bungarotoxin is a cysteine-rich protein containing multiple disulfide bonds, specialized strains (e.g., oxidative cytoplasm strains) or periplasmic targeting strategies can facilitate proper folding.
Codon-optimized sequence for E. coli
Codon optimization was performed using the Expression Optimization (Pilot) algorithm provided by Integrated DNA Technologies. The amino acid sequence was optimized for expression in Escherichia coli while avoiding BsaI, BsmBI and BbsI restriction sites.
| 
| 
|
|---|
The resulting optimized coding sequence (285 bp) is:
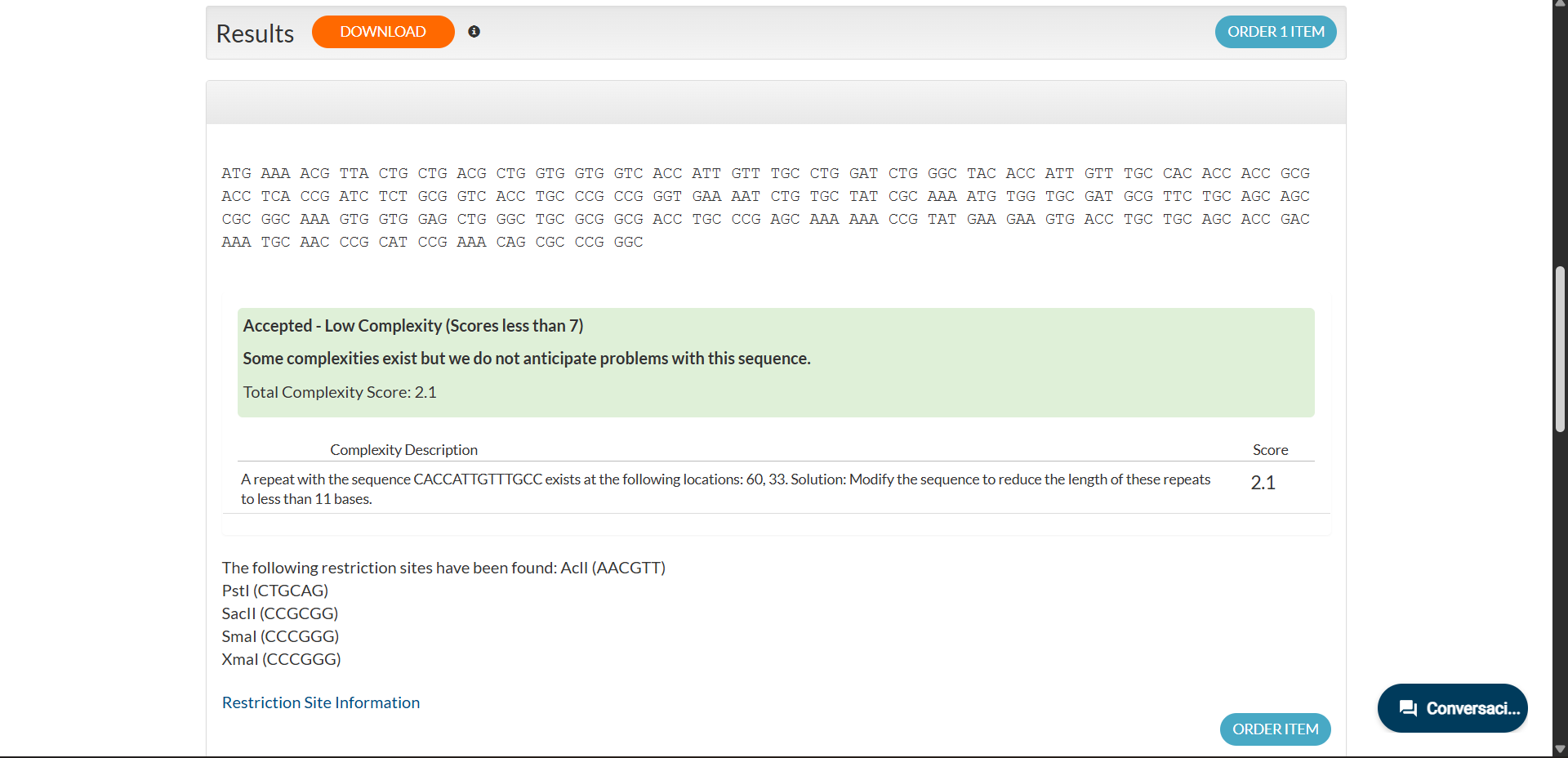
ATG AAA ACG TTA CTG CTG ACG CTG GTG GTG GTC ACC ATT GTT TGC CTG GAT CTG GGC TAC ACC ATT GTT TGC CAC ACC ACC GCG ACC TCA CCG ATC TCT GCG GTC ACC TGC CCG CCG GGT GAA AAT CTG TGC TAT CGC AAA ATG TGG TGC GAT GCG TTC TGC AGC AGC CGC GGC AAA GTG GTG GAG CTG GGC TGC GCG GCG ACC TGC CCG AGC AAA AAA CCG TAT GAA GAA GTG ACC TGC TGC AGC ACC GAC AAA TGC AAC CCG CAT CCG AAA CAG CGC CCG GGC
Sequence analysis indicated low complexity (score 2.1), suggesting no anticipated synthesis issues. Internal restriction sites unrelated to the cloning strategy were detected but do not interfere with the intended design.
3.4. We have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Once the codon-optimized DNA sequence has been obtained, several biotechnological strategies can be used to produce the corresponding protein. These methods rely on the fundamental biological processes of transcription and translation. Protein production can be achieved using either cell-dependent systems or cell-free expression systems.


3.5. [Optional] How does it work in nature/biological systems?
- Describe how a single gene codes for multiple proteins at the transcriptional level.
- Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
In natural biological systems, a single gene can give rise to multiple protein products. Although the classical view suggests that one gene encodes one protein, several regulatory mechanisms allow diversification at the transcriptional and post-transcriptional levels.
Mechanisms that allow one gene to produce multiple proteins
1. Alternative splicing
In eukaryotic organisms, genes contain exons and introns. During RNA processing, introns are removed and exons are joined together. However, different combinations of exons can be assembled, producing distinct mRNA variants from the same gene.
This process, known as alternative splicing, results in different protein isoforms with potentially different functions.
2. Alternative promoters
A single gene may contain multiple promoter regions. Depending on which promoter is activated, transcription may begin at different start sites, generating mRNAs with different 5′ ends. This can influence translation efficiency or alter the protein sequence.
3. Alternative translation initiation sites
Some mRNAs contain more than one possible start codon (AUG). Ribosomes may initiate translation at different positions, leading to proteins of different lengths.
4. RNA editing
In certain organisms, specific nucleotides in the RNA sequence are chemically modified after transcription. This can change codons and therefore alter the amino acid sequence of the final protein.
Alignment example: DNA → RNA → Protein
Using our optimized coding sequence as an example:
DNA (coding strand)
5′- ATG AAA ACG TTA CTG CTG -3′
Transcribed mRNA
(Thymine is replaced by uracil)
5′- AUG AAA ACG UUA CUG CUG -3′
Translated protein
Met – Lys – Thr – Leu – Leu – Leu
This alignment illustrates the central dogma of molecular biology:
DNA → RNA → Protein
In prokaryotes such as Escherichia coli, transcription and translation are coupled and occur simultaneously in the cytoplasm. In contrast, in eukaryotic cells, transcription occurs in the nucleus and translation occurs in the cytoplasm after RNA processing.
Part 4: Preparing a Twist DNA Synthesis Order
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
SECTION A. BENCHLING
This is a practice exercise, not necessarily the real Twist order!
4.1. Creating a Twist account, and Benchling account
4.2. Building a DNA Insert Sequence
We’ll make a sequence that will allow E. coli to glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein):
- In Benchling, we select New DNA/RNA sequence
- Now name the insert sequence and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
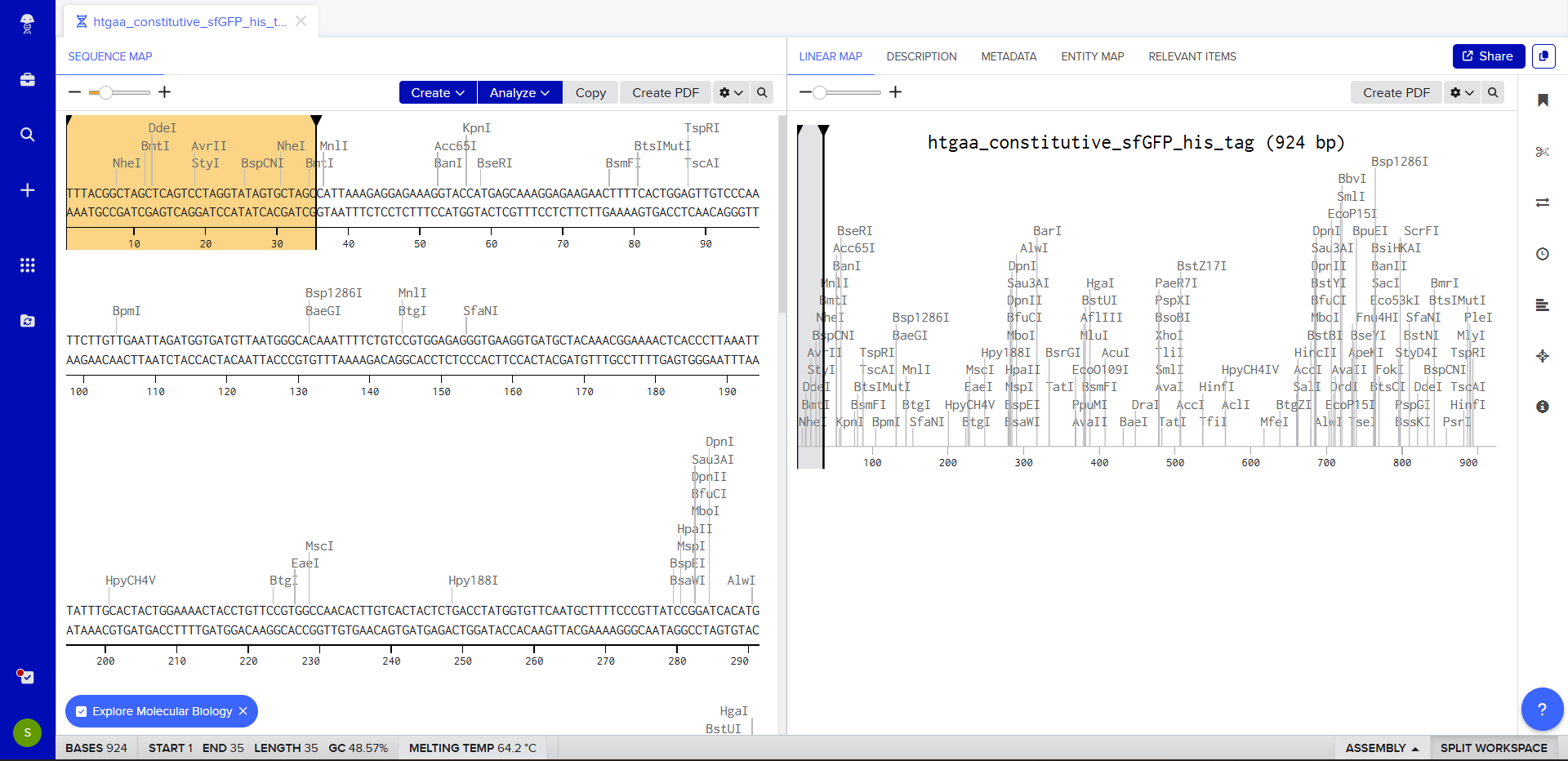
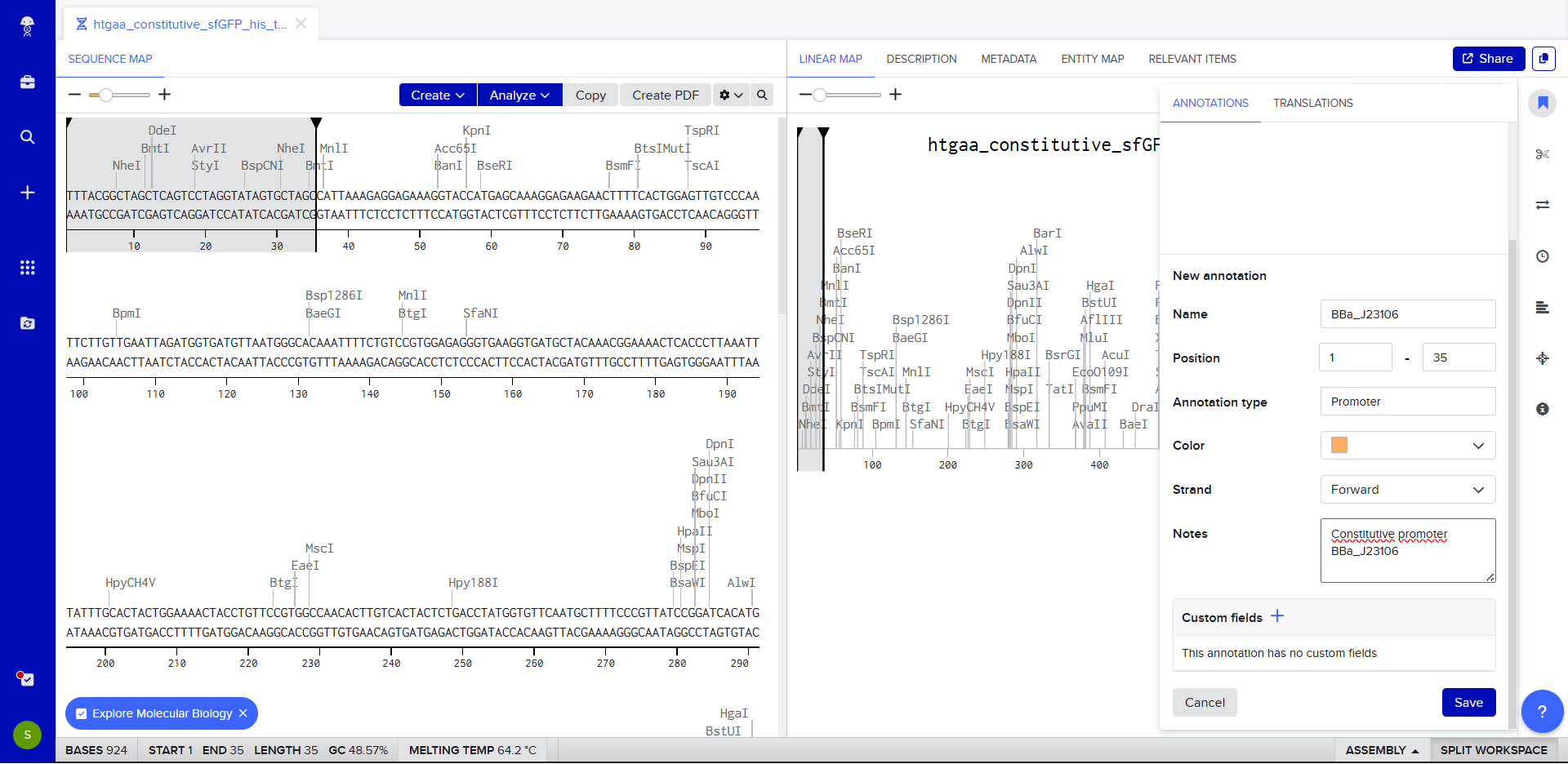
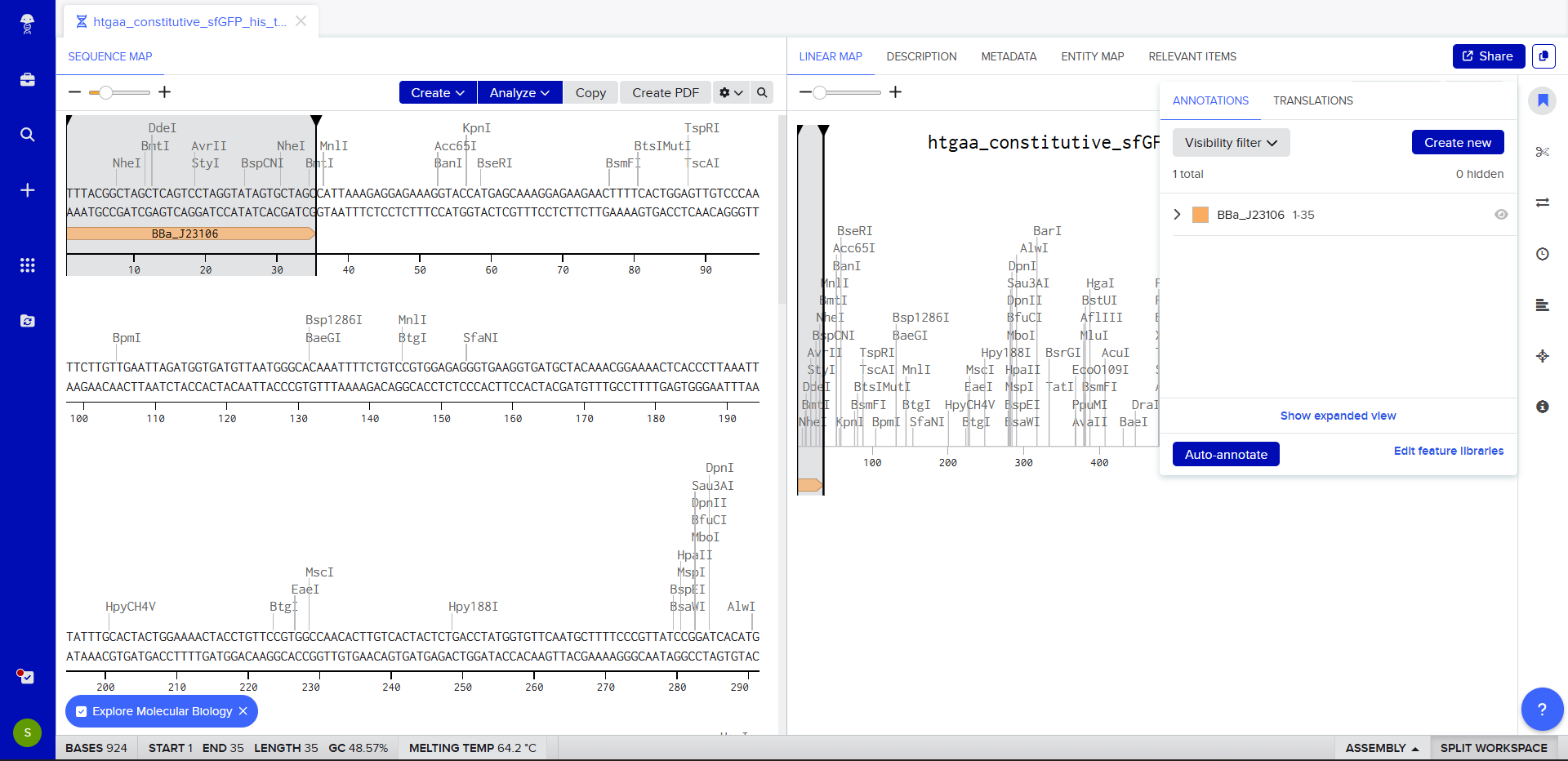
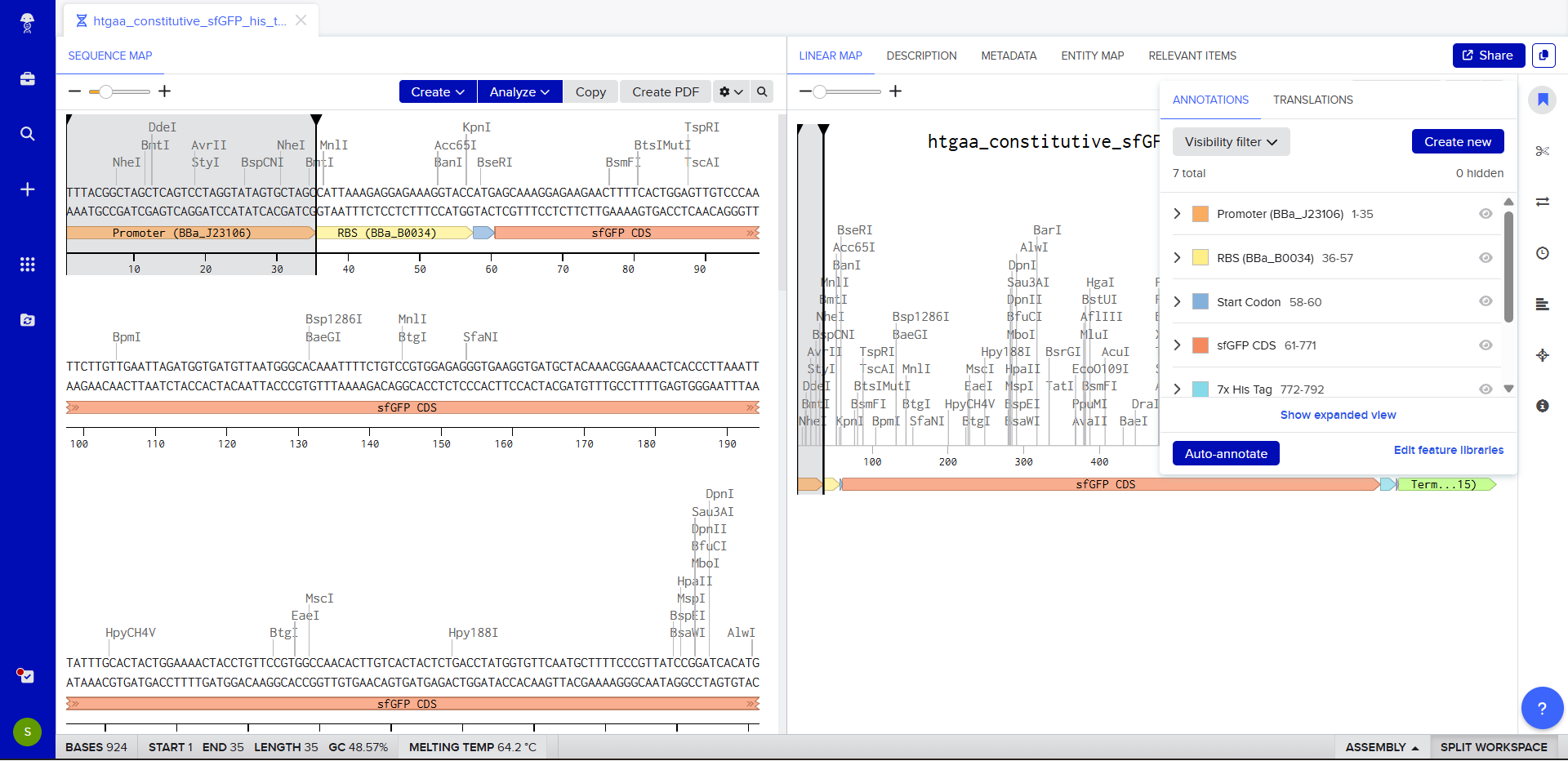
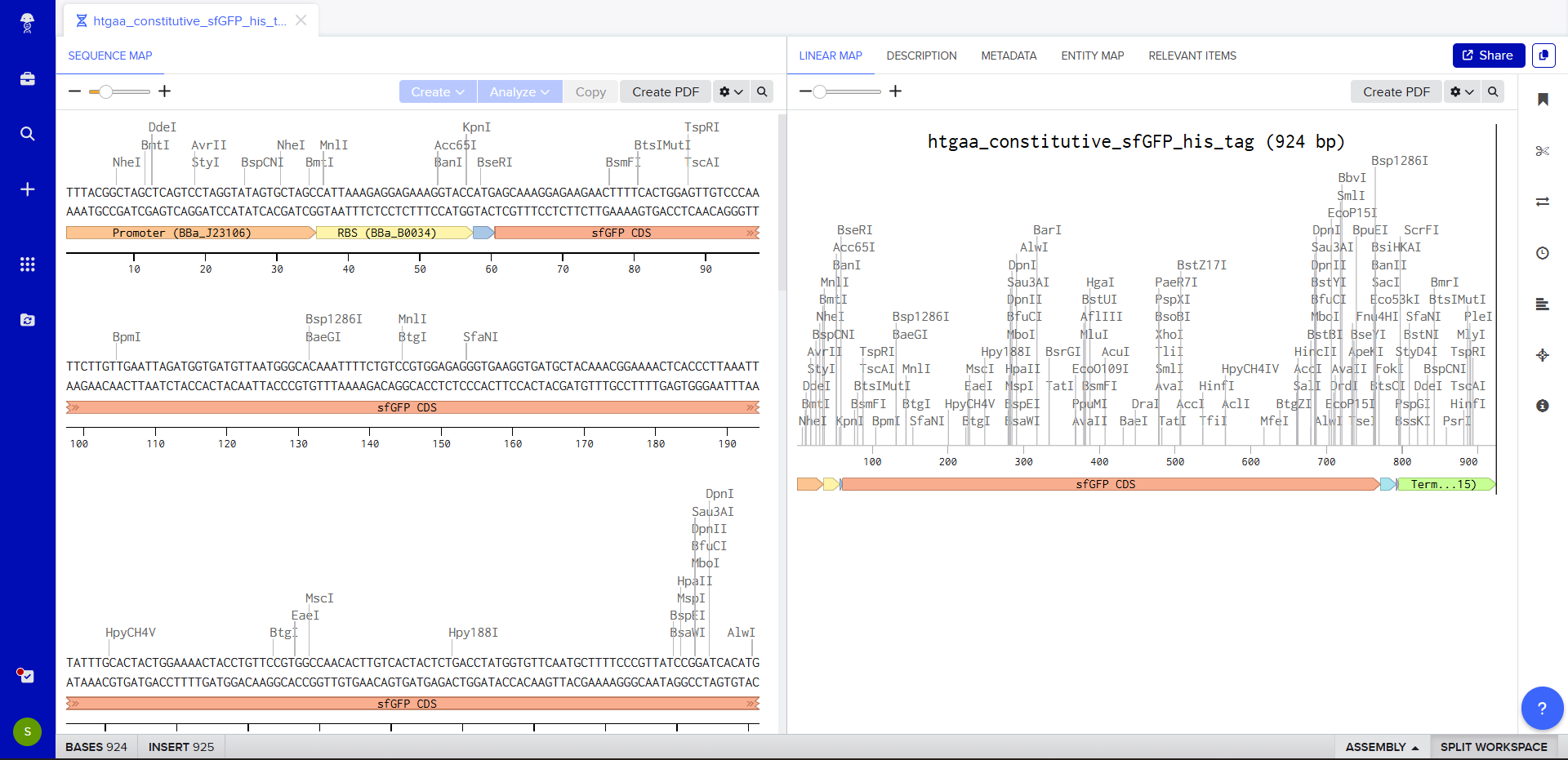
- We go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator) and paste the sequences into the Benchling file one after the other (replacing the coding sequence with the codon optimized DNA sequence of interest). Each time we add a new piece of the sequence, we make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below).
WHAT YOU SHOULD PINPOINT
Promoter (e.g. BBa_J23106)TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
RBS (e.g. BBa_B0034 with spacers for optimal expression)CATTAAAGAGGAGAAAGGTACC
Start CodonATG
Coding Sequence (your codon optimized DNA for a protein of interest, sfGFP for example)AGCAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCCGTGGAGAGGGTGAAGGTGATGCTACAAACGGAAAACTCACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCGTGGCCAACACTTGTCACTACTCTGACCTATGGTGTTCAATGCTTTTCCCGTTATCCGGATCACATGAAACGGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGCACTATATCTTTCAAAGATGACGGGACCTACAAGACGCGTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATCGTATCGAGTTAAAGGGTATTGATTTTAAAGAAGATGGAAACATTCTTGGACACAAACTCGAGTACAACTTTAACTCACACAATGTATACATCACGGCAGACAAACAAAAGAATGGAATCAAAGCTAACTTCAAAATTCGCCACAACGTTGAAGATGGTTCCGTTCAACTAGCAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCGACACAATCTGTCCTTTCGAAAGATCCCAACGAAAAGCGTGACCACATGGTCCTTCTTGAGTTTGTAACTGCTGCTGGGATTACACATGGCATGGATGAGCTCTACAAA
7x His Tag (Let’s add a 7×His tag at the C-terminus of the protein to enable protein purification from E. coli)CATCACCATCACCATCATCAC
Stop CodonTAA
Terminator (e.g. BBa_B0015)
CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
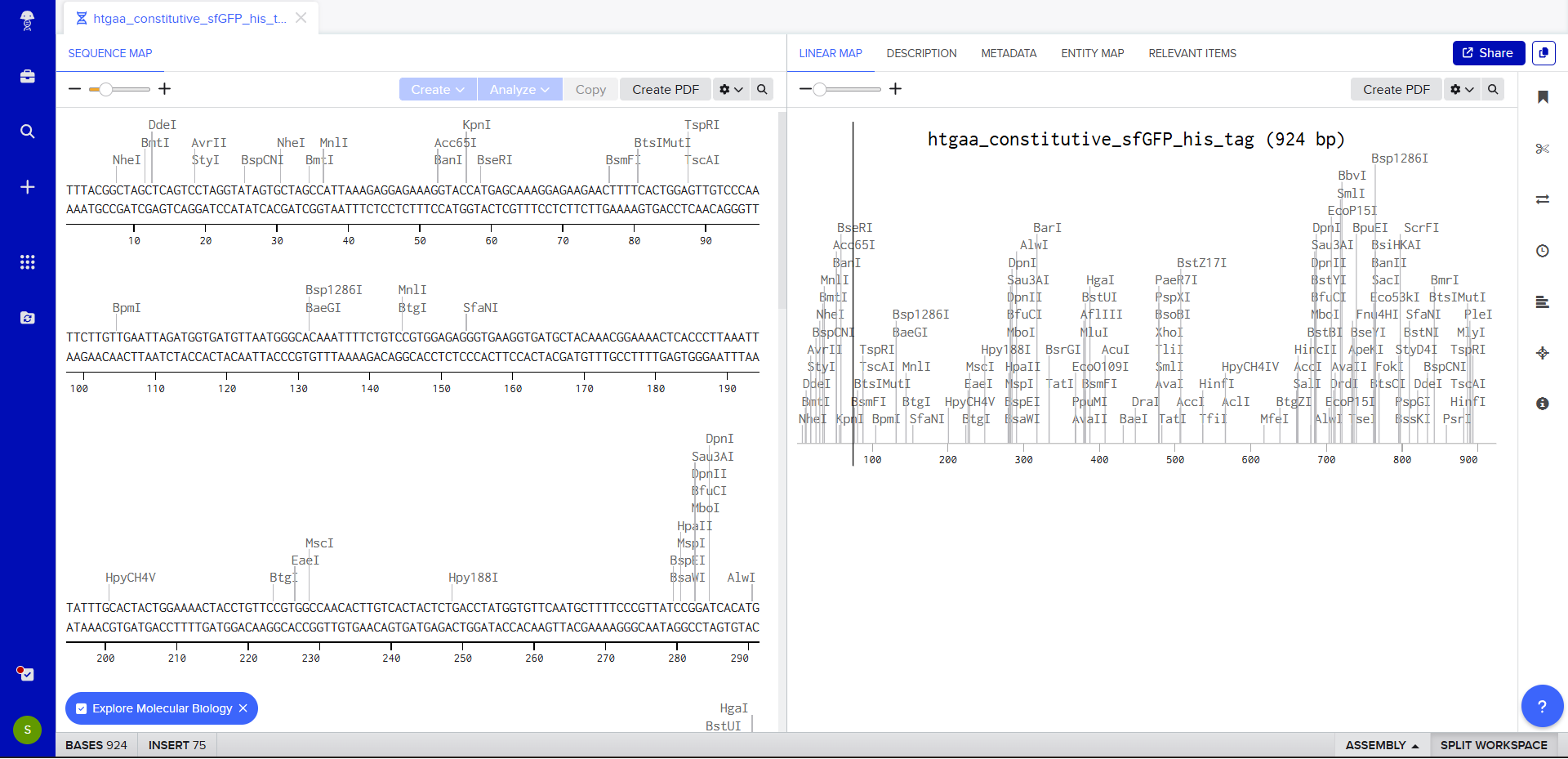
- Once this is completed, we click on Linear Map to preview the entire sequence.
Note: This is not required for this exercise, but to share the design with others, ensure that link sharing is turned on!
| 
|
|---|---|
| 
|
| 
|
The insert sequence that was built is commonly referred to as an expression cassette in molecular biology (a sequence you can drop into any vector and it’ll perform its function). We now download the FASTA file for the sequence made.
| 
|
|---|---|
| 
|
It’s helpful to visualize DNA designs using SBOL Canvas (Synthetic Biology Open Language) to convey the design. Here’s an example of what we just annotated in Benchling:
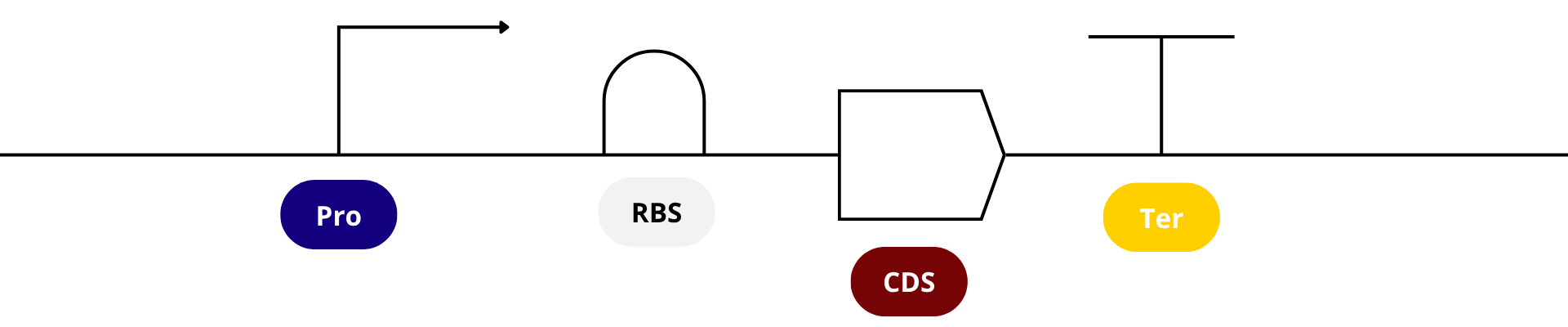
SECTION B. TWIST
Part 5: DNA Read/Write/Edit
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
For my project — Reversible Cell-Free Biosensor for ROS-Mediated Radiation Damage — we would want to sequence three categories of DNA:
| 1. The ROS-responsive promoter region |
|---|
| - Specifically, oxidative stress–responsive regulatory elements (e.g., OxyR/SoxR-regulated promoters from E. coli). |
| Why? |
| - To verify the exact sequence integrity of the promoter controlling our reporter. |
| - Small mutations in regulatory regions can drastically alter activation threshold, leakiness, or response dynamics. |
| - Since our system depends on reversible, tunable activation (not binary irreversible switching), promoter fidelity is critical for predictable behavior. |
| 2. The full genetic construct used in the TX–TL system |
| This includes: |
| - Promoter |
| - RBS |
| - Reporter gene (e.g., GFP variant) |
| - Degron tag |
| - Terminator |
| Why? |
| - To confirm assembly correctness after cloning or synthesis. |
| - To ensure no frameshifts, truncations, or rearrangements occurred. |
| - To validate that the degron sequence is intact (since reversibility depends on controlled protein degradation). |
| 3. DNA stability after ROS exposure (damage assessment) |
| - Because the biosensor operates in oxidative environments, we may also sequence recovered plasmid DNA after repeated ROS cycles. |
| Why? |
| - To assess oxidative damage accumulation. |
| - To evaluate mutation rates under stress. |
| - To determine long-term reusability limits of the system. |
This directly connects to governance and safety: understanding failure modes prevents misleading signal interpretation.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
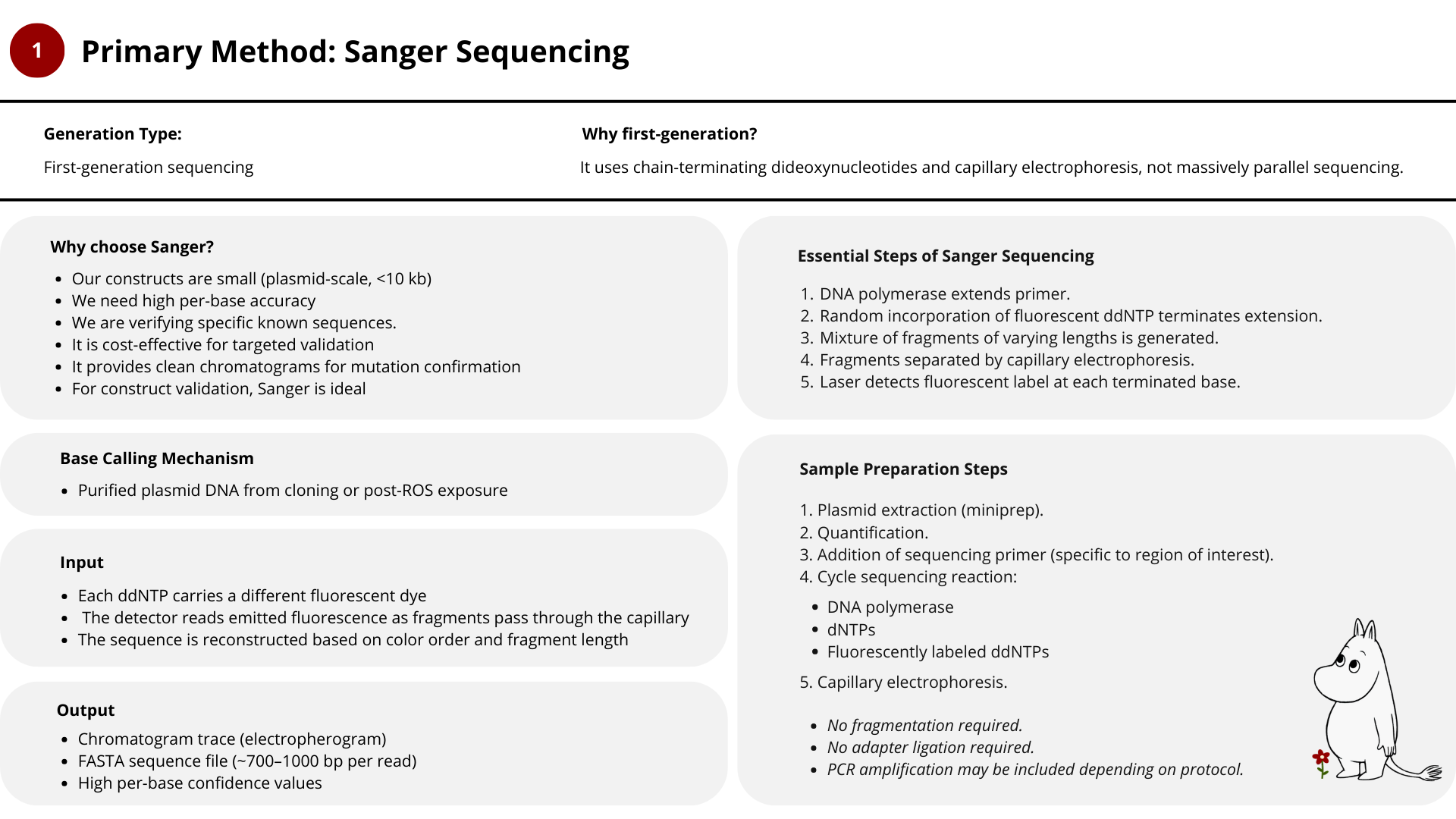
- Is your method first-, second- or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
For this project, I would use a combination of Sanger sequencing and Illumina sequencing, depending on the question being asked.
1. Sanger
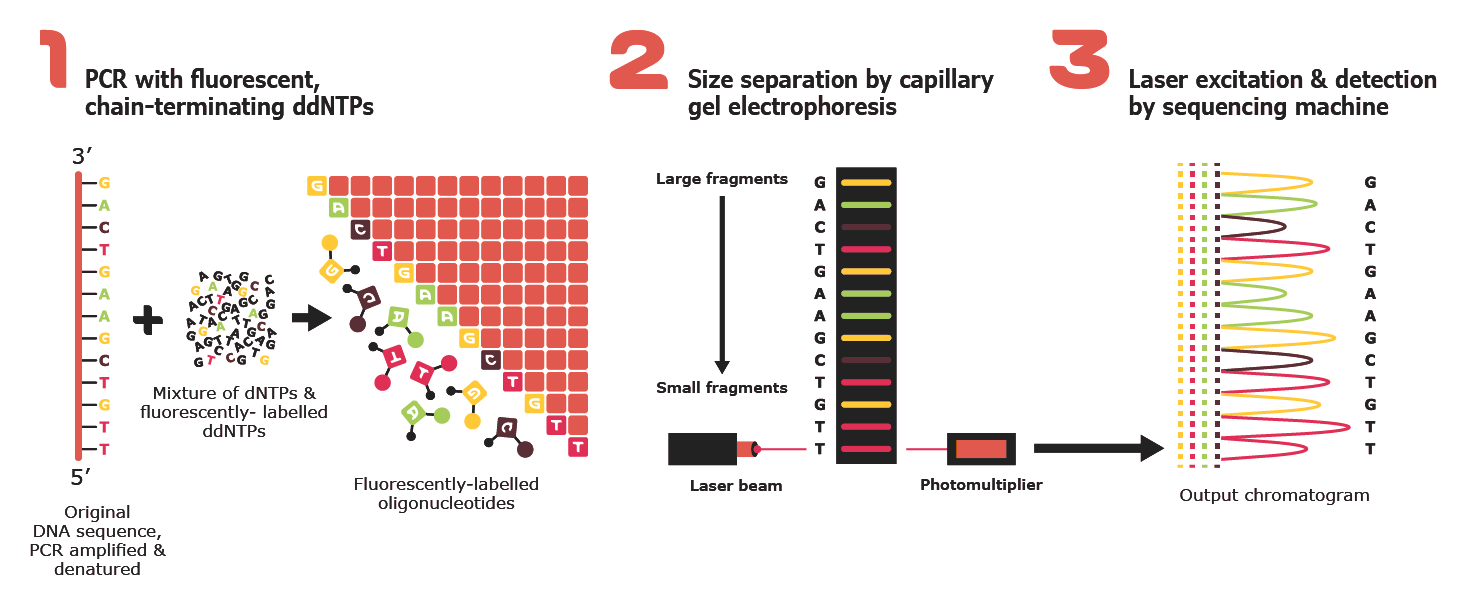
2. Illuimina

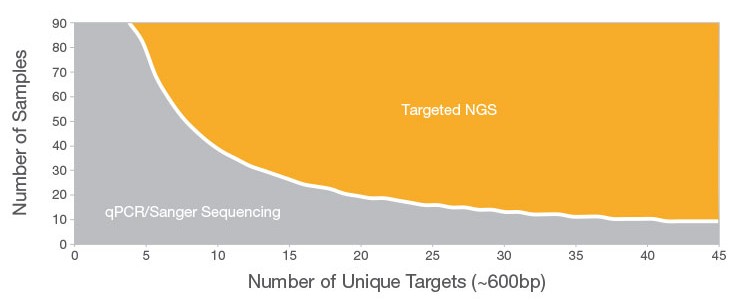
Final Justification for Technology Choice
For the reversible ROS biosensor:
- Sanger sequencing is sufficient and ideal for construct validation.
- Illumina sequencing becomes valuable when studying oxidative mutation accumulation and long-term robustness.
This sequencing strategy directly supports:
- Reliability
- Reversibility characterization
- Governance considerations
- Failure-mode understanding
- Safe system deployment
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
For the reversible ROS-mediated hydrogel biosensor, we would synthesize a minimal genetic circuit designed for oxidative stress detection and transient fluorescent output.
The construct would include:
- A ROS-responsive promoter (e.g., OxyR-regulated promoter)
- A ribosome binding site (RBS)
- A fluorescent reporter gene (e.g., sfGFP)
- A short degron tag to ensure rapid protein degradation
- A transcriptional terminator
Why synthesize this DNA?
Because:
- The promoter must be precisely tuned to oxidative stress.
- The degron must be fused correctly to ensure reversibility.
- The full construct must function in a cell-free TX–TL system.
- Synthetic DNA reduces cloning errors.
- It enables modular optimization.
We are not synthesizing a whole genome.
We are synthesizing a minimal functional sensing circuit embedded in a biomaterial.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
- What are the essential steps of your chosen sequencing methods?
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
We would use commercial gene synthesis services, such as those provided by:
Twist Bioscience
These companies use high-throughput DNA synthesis platforms based on phosphoramidite chemistry and silicon-based parallel synthesis.
Simplified process:
- Attach first base to solid surface.
- Add chemically protected nucleotide.
- Remove protective group.
- Add next nucleotide.
- Repeat cycle.
Each cycle adds ONE base. This is automated.
For longer fragments:
- Short oligos are synthesized.
- Then assembled enzymatically into longer genes.
- Verified by sequencing.
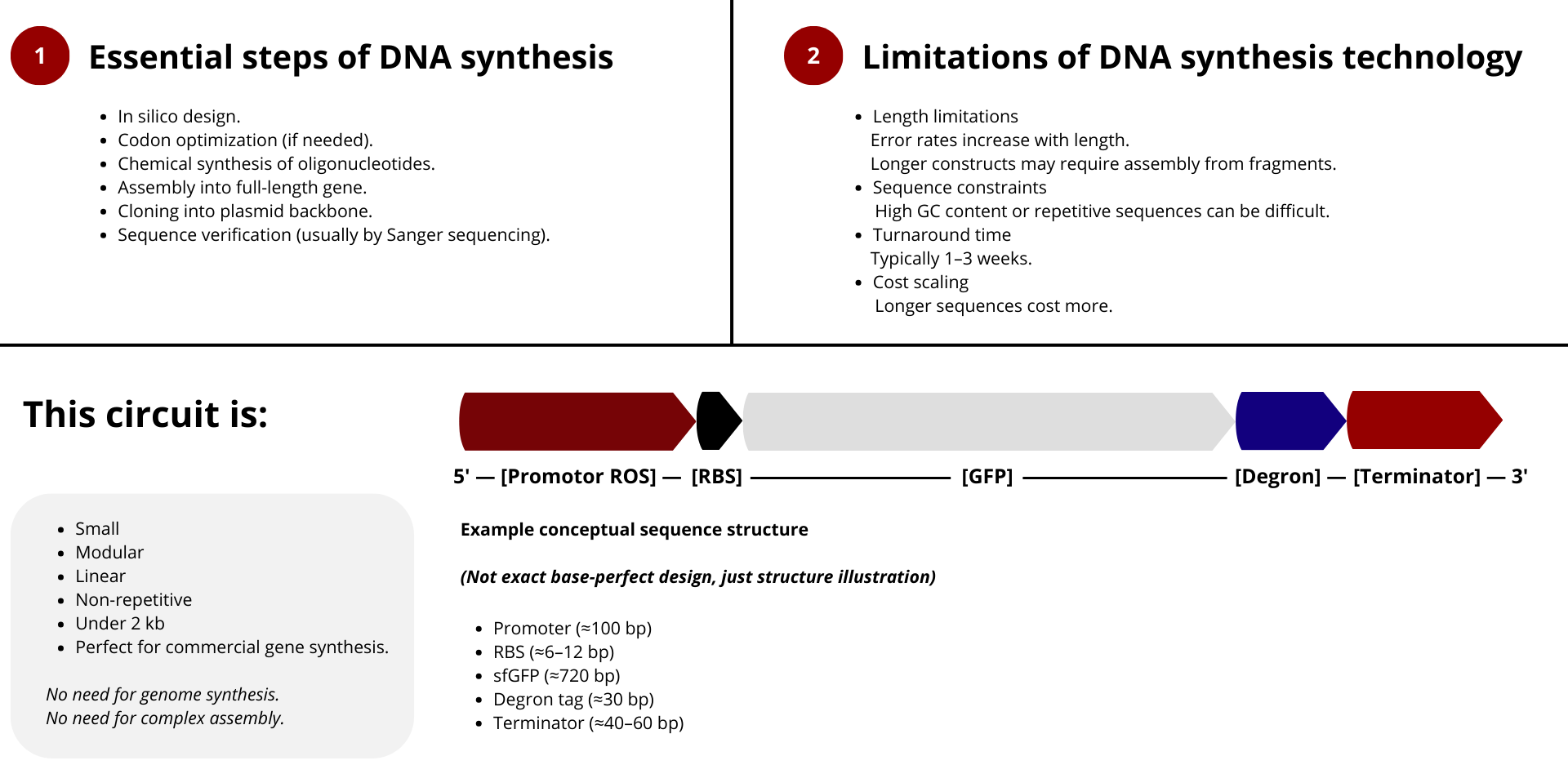
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
For this biosensor, I would edit the genetic circuit itself to optimize sensing dynamics, reversibility, and robustness.
Specifically, we would edit:

(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
- How does your technology of choice edit DNA? What are the essential steps?
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The most appropriate technology for precise edits in genetic constructs would be:
CRISPR-based editing systems.
Specifically: CRISPR-Cas9

Limitations of DNA Editing Methods
1. Off-target effects (CRISPR)
- Cas9 can cut unintended regions.
- Less relevant for small plasmids, more relevant for genomes.
2. Efficiency variability
- Not all cells incorporate edits.
- Requires screening.
3. Repair pathway dependence
- Precise edits require homologous recombination.
- Not always efficient.
4. Context sensitivity
- Changing one base can unpredictably alter promoter behavior.
- Requires iterative testing.
For this project DNA editing is not strictly required for initial system implementation. However, it would be essential for iterative optimization of promoter sensitivity, degradation kinetics, and response tuning.
Resources
- Secuenciación Sanger Pasos y método. (s.f.). Merck©. https://www.sigmaaldrich.com/MX/es/technical-documents/protocol/genomics/sequencing/sanger-sequencing
- Differences between NGS and Sanger sequencing. (s.f.). Illumina©. https://www.illumina.com/science/technology/next-generation-sequencing/beginners/advantages/ngs-vs-sanger.html
- A Simple Guide to Phosphoramidite Chemistry and How it Fits in Twist Bioscience’s Commercial Engine. Twist Bioscience. https://www.twistbioscience.com/blog/science/simple-guide-phosphoramidite-chemistry-and-how-it-fits-twist-biosciences-commercial
- CRISPR: ¿Qué es y cómo funciona?. (s.f.). genotipia. https://genotipia.com/crispr-cas/
- OpenAI. (2026). ChatGPT (GPT-5.2) [Large language model]. https://chat.openai.com/