HTGAA - Week 4: Protein Design Part I
My Homework
WEEK 4 - BIOINFORMATICS PART I
This week focuses on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Lecture (Tues, Feb 24)
Protein Design Part I
(▶️Recording)
Thras Karydis, Jon Kaufman
Recitation (Wed, Feb 25)
Protein folding
(▶️Recording)
Allan Costa
Protein Design I
Objective:
- Learn basic concepts:
- amino acid structure
- 3D protein visualization
- the variety of ML-based design tools
- Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Part A. Conceptual Questions
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
The following questions will be answer: 1, 2, 3, 4, 5, 6, 8, 9, and 10
The following questions will be left unanswer: 7 and 11
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
To estimate how many amino acid molecules are consumed in a 500 g piece of meat, we need to make reasonable biochemical assumptions.
1.1. Estimate protein content in meat
On average, raw beef contains approximately 20–25% protein by weight.
To stay conservative, we assume:
Protein content ≈ 20%
\[ 500 \, g \times 0.20 = 100 \, g \text{ protein} \]So, 500 g of meat contains approximately:
100 g of protein
1.2. Estimate number of moles of amino acids
We are told that the average amino acid has a molecular weight of approximately:
\[ 100 \, \text{Daltons} = 100 \, g/mol \]If we assume protein is fully hydrolyzed into individual amino acids, then:
\[ \frac{100 \, g}{100 \, g/mol} = 1 \, mol \]So, 100 g of amino acids corresponds to:
1 mole of amino acids
1.3. Convert moles to number of molecules
Using Avogadro’s number:
\[ 1 \, mol = 6.022 \times 10^{23} \text{ molecules} \]Therefore:
You ingest approximately 6.0 × 10²³ amino acid molecules
Final Answer
A 500 g piece of meat contains on the order of:
~ 10²⁴ amino acid molecules
(approximately one mole of amino acids)
Important Notes
- This is an order-of-magnitude estimate.
- Real proteins are polymers, so their molecular weights are much larger.
- The calculation assumes complete digestion into free amino acids.
- Water content and protein percentage vary by meat type and preparation.
Interpretation
Eating 500 g of meat means consuming roughly Avogadro-scale molecular quantities of amino acids — on the order of (10²⁴) individual molecules.
This illustrates how biological systems operate at unimaginably large molecular scales, even in everyday nutrition.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The answer is straightforward: we do not incorporate foreign organisms as whole structures — we digest them into molecular building blocks.
2.1. Digestion breaks macromolecules into basic units
Proteins in beef or fish are large, highly ordered macromolecules. During digestion:
- Stomach acid (HCl) denatures proteins.
- Proteases such as pepsin, trypsin, and chymotrypsin cleave peptide bonds.
- Proteins are hydrolyzed into short peptides and free amino acids.
By the time nutrients are absorbed in the small intestine, the original protein structures no longer exist.
We absorb:
- Amino acids
- Simple sugars
- Fatty acids
- Nucleotides
not intact tissues
2.2. Molecular identity is lost during digestion
A cow muscle protein (for example, bovine actin) is not transferred into your muscles as bovine actin. It is broken down into its constituent amino acids:
\[ \text{Protein} \rightarrow \text{Amino acids} \]Those amino acids are chemically indistinguishable from amino acids obtained from fish, plants, or synthesized endogenously.
At the molecular level:
An amino acid is just an amino acid — it carries no “species identity.”
2.3. Your genome determines what you become
Once absorbed, amino acids enter your metabolic pool. Your ribosomes then synthesize proteins according to:
\[ \text{DNA} \rightarrow \text{RNA} \rightarrow \text{Protein} \]Your DNA sequence encodes human proteins, not cow or fish proteins.
Therefore:
- You rebuild human actin.
- You rebuild human collagen.
- You rebuild human enzymes.
Your phenotype is determined by your genome, not by the origin of your nutrients.
2.4. Information vs. Matter
This question highlights a fundamental biological principle:
Biological identity is determined by information, not raw material.
Matter (carbon, nitrogen, oxygen, amino acids) is universal.
Biological structure depends on how that matter is organized, and organization is encoded in DNA.
Final Answer
Humans do not become cows or fish after eating them because digestion reduces food to molecular building blocks. These building blocks are then reassembled according to human genetic instructions.
We recycle matter — but we do not inherit structural identity from what we eat.
3. Why are there only 20 natural amino acids?
Indeed, why are there only 20 amino acids when the triplet genetic code has 64 codons available? Similarly, could the system work effectively with less than 20? The existence of 20 canonical amino acids is not a chemical inevitability — it is the result of evolutionary optimization. There is no fundamental law of physics that limits proteins to 20 amino acids. Instead, the number reflects a balance between chemical diversity, translational fidelity, and evolutionary stability.
3.1. The genetic code constrains the set
Proteins are encoded by triplet codons:
\[ 4^3 = 64 \text{ possible codons} \]Out of these:
- 61 encode amino acids
- 3 are stop codons
The canonical genetic code maps these 61 codons to 20 amino acids. This mapping is highly redundant (degenerate), which increases robustness against mutations.
Expanding the number of amino acids would require:
- New tRNAs
- New aminoacyl-tRNA synthetases
- Rewiring of codon assignments
This is evolutionarily costly.
3.2. Chemical sufficiency
The 20 amino acids provide a remarkably broad range of chemical functionality:
- Nonpolar (hydrophobic packing)
- Polar uncharged (hydrogen bonding)
- Charged (electrostatics)
- Aromatic (π interactions)
- Special cases (glycine flexibility, proline rigidity, cysteine disulfide bonding)
With just 20 building blocks, proteins can:
- Fold into stable 3D structures
- Catalyze diverse chemical reactions
- Form dynamic assemblies
Adding many more amino acids would yield diminishing functional returns.
3.3. Evolutionary “freeze” of the code
Once the genetic code became established in early life, it became extremely difficult to change.
This is known as the frozen accident hypothesis:
Once organisms shared a common genetic code, large-scale changes would be lethal.
Thus, the 20 amino acids became locked in by evolutionary history.
3.4. Are there really only 20?
Interestingly, modern biology slightly exceeds 20:
- Selenocysteine (21st amino acid)
- Pyrrolysine (22nd amino acid)
These are incorporated via special recoding mechanisms.
Additionally, synthetic biology has engineered organisms that incorporate noncanonical amino acids, proving that 20 is not a hard biochemical limit — just the natural evolutionary standard.
Final Answer
There are 20 natural amino acids because evolution selected a chemically sufficient, robust, and efficient set early in the history of life.
The genetic code then became evolutionarily fixed, making large-scale expansion unlikely. The number 20 reflects evolutionary optimization — not chemical necessity.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes. Non-natural (noncanonical) amino acids can be synthesized chemically and even incorporated into proteins using engineered translation systems.
There is no chemical rule limiting amino acids to the 20 canonical ones. The only strict requirement for incorporation into proteins is that the molecule must:
- Contain an α-amino group
- Contain an α-carboxyl group
- Be compatible with ribosomal geometry
- Be recognized by a tRNA / aminoacyl-tRNA synthetase pair
Modern synthetic biology has successfully expanded the genetic code to include dozens of artificial amino acids.
4.1 Design some new amino acids
4.1.1. Design Principles
When designing a new amino acid, we must consider:
- Side-chain size and steric compatibility
- Polarity and hydrogen bonding capacity
- Electronic effects
- Stability under physiological conditions
- Synthetic accessibility
A) Fluorinated Hydrophobic Amino Acid
Structure Concept:
Replace a methyl group in leucine with a trifluoromethyl group.
Side chain: \[ -CH_2-CH(CF_3)_2 \]Purpose:
- Increase hydrophobicity
- Alter packing interactions
- Increase metabolic stability
Fluorinated residues are useful for:
- Stabilizing protein cores
- Modifying membrane interactions
- 19F NMR tracking
B) Photo-Crosslinking Amino Acid
Structure Concept:
Attach a diazirine group to a phenylalanine-like ring.
Side chain: \[ -CH_2-phenyl-diazrine \]Purpose:
- UV-activated covalent crosslinking
- Study protein–protein interactions
- Capture transient binding events
This would allow light-controlled structural locking of proteins.
C) Redox-Active Aromatic Amino Acid
Structure Concept:
Modify tyrosine to include a quinone-like moiety.
Side chain:
\[ -CH_2-aromatic-quinone \]Purpose:
- Electron transfer capability
- Catalysis in synthetic enzymes
- Bioelectronic interfaces
This could enhance long-range electron transport in engineered proteins.
Are These Realistic?
Yes. Variants of these ideas already exist in synthetic biology:
- Fluorinated amino acids
- Photo-reactive amino acids
- Click-chemistry compatible residues
- Redox-active artificial cofactors
Genetic code expansion techniques allow site-specific incorporation using engineered:
- Orthogonal tRNA
- Engineered aminoacyl-tRNA synthetase
- Reassigned stop codons (often UAG)
Final Answer
Yes, non-natural amino acids can be synthesized and incorporated into proteins. The natural 20 amino acids represent an evolutionary standard, not a chemical limit.
By modifying side chains, we can design amino acids with enhanced hydrophobicity, photo-reactivity, redox properties, or catalytic potential — dramatically expanding the functional landscape of proteins.
5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids did not require life to exist. They can form through purely abiotic chemical processes under the right physical conditions. Before enzymes evolved, amino acids were likely synthesized through prebiotic chemistry on early Earth — and possibly delivered from space.
5.1. Prebiotic Atmospheric Chemistry
In 1953, Stanley Miller and Harold Urey demonstrated that amino acids can form spontaneously from simple gases when energy is supplied.
They simulated early Earth conditions using:
- Methane (CH₄)
- Ammonia (NH₃)
- Hydrogen (H₂)
- Water vapor (H₂O)
- Electrical sparks (lightning)
After several days, the system produced amino acids such as:
- Glycine
- Alanine
- Aspartic acid
This experiment showed that amino acids can emerge from non-living chemistry.
Reaction principle (simplified):
\[ \text{Simple gases} + \text{energy} \rightarrow \text{Organic molecules} \]5.2. Hydrothermal Vent Chemistry
Another hypothesis suggests that amino acids formed near deep-sea hydrothermal vents.
These environments provide:
- Mineral catalysts (iron, nickel sulfides)
- Redox gradients
- Thermal energy
- High pressure
Mineral surfaces may have catalyzed the formation of organic molecules and concentrated them locally.
5.3. Extraterrestrial Delivery
Amino acids have been detected in carbonaceous meteorites, such as the Murchison meteorite.
These findings suggest that:
- Amino acids can form in interstellar space
- They can survive planetary accretion
- Early Earth may have received organic molecules via meteorite bombardment
Thus, part of Earth’s prebiotic inventory may have been extraterrestrial.
5.4. No Enzymes Required
Modern organisms synthesize amino acids using enzyme-catalyzed pathways. However, enzymes are highly evolved catalysts.
Before life:
- Chemistry was driven by thermodynamics and energy input
- Catalysis may have been mineral-based
- Reaction networks were simpler but chemically plausible
Life did not invent amino acids — it inherited them from chemistry.
Final Answer
Amino acids likely originated through abiotic chemical reactions on early Earth (e.g., atmospheric discharge or hydrothermal systems) and possibly through extraterrestrial synthesis. They existed before enzymes because their formation does not require biological catalysis — only appropriate chemical conditions and energy sources.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
An α-helix formed from D-amino acids would be left-handed.
6.1. Chirality Determines Helical Handedness
Natural proteins are built from L-amino acids.
When L-amino acids adopt an α-helical conformation, they form a:
Right-handed α-helix
This is the most energetically favorable geometry due to:
- Backbone bond angles (φ and ψ)
- Steric constraints
- Optimal hydrogen bonding alignment
6.2. Mirror Symmetry Argument
D-amino acids are the mirror images of L-amino acids.
Because chirality inverts stereochemistry at the α-carbon, the entire conformational energy landscape is mirrored.
Therefore:
- L-amino acids → right-handed α-helix
- D-amino acids → left-handed α-helix
The structures are mirror images of each other.
6.3. Hydrogen Bond Geometry
The α-helix is stabilized by hydrogen bonds:
\[ C=O_{(i)} \rightarrow H-N_{(i+4)} \]The spatial orientation required for optimal hydrogen bonding depends on backbone stereochemistry.
Switching from L to D reverses:
- Dihedral angle preferences
- Side-chain orientation
- Overall helical twist direction
6.4. Energetics
For L-amino acids:
- Right-handed helices are lower in energy.
- Left-handed helices are sterically disfavored.
For D-amino acids:
- The energetic preference is inverted.
Thus, a D-polypeptide naturally favors a left-handed α-helix.
Final Answer
An α-helix composed entirely of D-amino acids would adopt a left-handed conformation, because reversing chirality at the α-carbon mirrors the backbone geometry and inverts the preferred helical handedness.
8. Why are most molecular helices right-handed?
Most molecular helices in biology are right-handed because life is built almost exclusively from L-amino acids and D-sugars. Molecular chirality determines the preferred helical geometry. Right-handed helices are not universally required by physics — they are a consequence of stereochemistry and evolutionary selection.
8.1. Chirality Bias in Biology
Biological systems exhibit homochirality:
- Proteins are built from L-amino acids.
- Nucleic acids contain D-ribose or D-deoxyribose.
Because helices emerge from repeating chiral building blocks, their handedness is dictated by the stereochemistry of those monomers.
For example:
- L-amino acids → right-handed α-helices
- D-sugars → right-handed DNA double helix (B-DNA)
If chirality were inverted, handedness would invert.
8.2. Energetic Favorability
Helical structures form when:
- Backbone dihedral angles minimize steric clashes
- Hydrogen bonds align optimally
- Side chains pack efficiently
For L-amino acids, the lowest-energy α-helical conformation is right-handed. Left-handed helices are possible but typically sterically disfavored in L-polypeptides. Thus, the dominance of right-handed helices reflects energetic optimization under stereochemical constraints.
8.3. Repeating Geometry and Twist
A helix arises from repeating units with constrained bond angles.
Because bond rotations are not symmetric in chiral molecules, the accumulation of small angular preferences results in a macroscopic twist.
This is an emergent geometric property:
\[ \text{Local chirality} \rightarrow \text{Global helical handedness} \]8.4. Is Right-Handedness Universal?
No.
- Polymers built from D-amino acids form left-handed helices.
- Synthetic achiral polymers can form either handedness.
- Certain protein segments (e.g., polyproline II helices) may adopt left-handed conformations.
Thus, right-handed dominance in biology reflects molecular asymmetry, not universal physical law.
Final Answer
Most molecular helices in biology are right-handed because they are built from chiral building blocks (L-amino acids and D-sugars) whose stereochemistry favors right-handed twist geometries. Helical handedness emerges from the accumulation of local stereochemical constraints into a global structural bias.
9. Why do β-sheets tend to aggregate?
- What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because their structure exposes hydrogen-bonding backbone groups and often hydrophobic side chains, making intermolecular association energetically favorable.
The driving force is primarily:
- Backbone hydrogen bonding
- The hydrophobic effect
- Reduction of solvent-exposed surface area
- Overall free energy minimization
9.1. Backbone Hydrogen Bonding Is Not Fully Satisfied
In an isolated or partially unfolded polypeptide:
- Carbonyl (C=O) groups
- Amide (N–H) groups
are capable of forming hydrogen bonds. If these groups are not satisfied intramolecularly, they seek partners intermolecularly.
When multiple β-strands align:
\[ C=O_{(i)} \leftrightarrow H-N_{(j)} \]They form extended hydrogen-bond networks between molecules. This makes β-sheets particularly prone to forming intermolecular structures.
9.2. The Extended Geometry of β-Strands
β-strands are relatively:
- Extended
- Planar
- Repetitive
This geometry allows:
- Easy stacking
- Sheet-to-sheet association
- Formation of fibrillar structures
Unlike α-helices (which are internally hydrogen-bonded), β-strands expose bonding potential along their length.
9.3. Hydrophobic Effect
Many β-sheet–forming sequences contain hydrophobic residues.
When strands aggregate:
- Hydrophobic side chains become buried
- Ordered water molecules are released into bulk solvent
- Solvent entropy increases
This contributes favorably to:
\[ \Delta G = \Delta H - T\Delta S \]The increase in solvent entropy (ΔS > 0) often drives aggregation.
9.4. Structural Complementarity
β-sheets allow:
- Tight side-chain interdigitation
- Steric zipper formation
- Highly ordered packing
This geometric complementarity stabilizes aggregates such as amyloid fibrils.
9.5. Thermodynamic Perspective
Aggregation is favored when:
\[ \Delta G_{aggregation} < 0 \]This occurs due to:
- Enthalpic gain from hydrogen bonding
- Entropic gain from water release
- Reduced solvent-exposed surface area
Thus, β-sheet aggregation is often thermodynamically favorable, especially at high concentration or under partially denaturing conditions.
Final Answer
β-sheets tend to aggregate because their extended backbone structure allows extensive intermolecular hydrogen bonding and efficient hydrophobic packing. The primary driving forces are backbone hydrogen bonding and the hydrophobic effect, which together lower the system’s free energy and stabilize ordered aggregates.
10. Why do many amyloid diseases form β-sheets?
- Can you use amyloid β-sheets as materials?
Many amyloid diseases are associated with β-sheet formation because β-sheets provide a structurally stable, energetically favorable architecture for protein aggregation. The same physical principles that stabilize β-sheets in normal proteins can drive pathological self-assembly under destabilizing conditions.
10.1. Misfolding Exposes Aggregation-Prone Regions
Many proteins contain segments with high β-sheet propensity.
Under normal conditions:
- Proteins fold into native conformations
- Aggregation-prone regions are buried
However, mutations, oxidative stress, or aging can:
- Destabilize native folds
- Expose hydrophobic and hydrogen-bonding surfaces
Once exposed, these regions can align into intermolecular β-sheets.
10.2. Cross-β Architecture
Amyloid fibrils share a characteristic structural motif:
- β-strands run perpendicular to the fibril axis
- Hydrogen bonds run parallel to the fibril axis
This “cross-β” structure creates:
- Extensive hydrogen-bond networks
- High mechanical stability
- Repetitive, ordered packing
Because backbone hydrogen bonds are strong and directional, β-sheets form highly stable fibrillar assemblies.
10.3. Thermodynamic Driving Forces
Amyloid formation is driven by:
- Backbone hydrogen bonding
- Hydrophobic packing
- Release of ordered water (entropy gain)
- Reduction of exposed surface area
Thus, amyloid fibrils often represent a deep thermodynamic minimum. In some cases, the amyloid state may be more stable than the native fold.
10.4. Why So Many Diseases?
Examples include:
- Alzheimer’s disease
- Parkinson’s disease
- Huntington’s disease
- Prion diseases
In each case, a normally soluble protein adopts an aggregation-prone β-sheet–rich structure. Because β-sheets allow extensive intermolecular stabilization, once nucleation occurs, fibril growth can become self-propagating.
10.5. Can Amyloid β-Sheets Be Used as Materials?
Yes. Although pathological in some contexts, amyloid fibrils have remarkable material properties:
- High tensile strength
- Nanometer-scale precision
- Self-assembly capability
- Chemical robustness
Potential applications include:
- Biomaterials and scaffolds
- Nanowires
- Drug delivery systems
- Tissue engineering frameworks
- Bioelectronic interfaces
Some organisms naturally use functional amyloids (e.g., bacterial biofilms), demonstrating that amyloid structures are not inherently pathological.
Final Answer
Many amyloid diseases form β-sheets because the β-sheet architecture allows extensive intermolecular hydrogen bonding and hydrophobic packing, creating highly stable cross-β fibrils. While pathological in neurodegenerative diseases, amyloid β-sheet assemblies can also be harnessed as robust, self-assembling nanomaterials in biotechnology and materials science.
Part B: Protein Analysis and Visualization
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
1. Briefly describe the protein you selected and why you selected it.
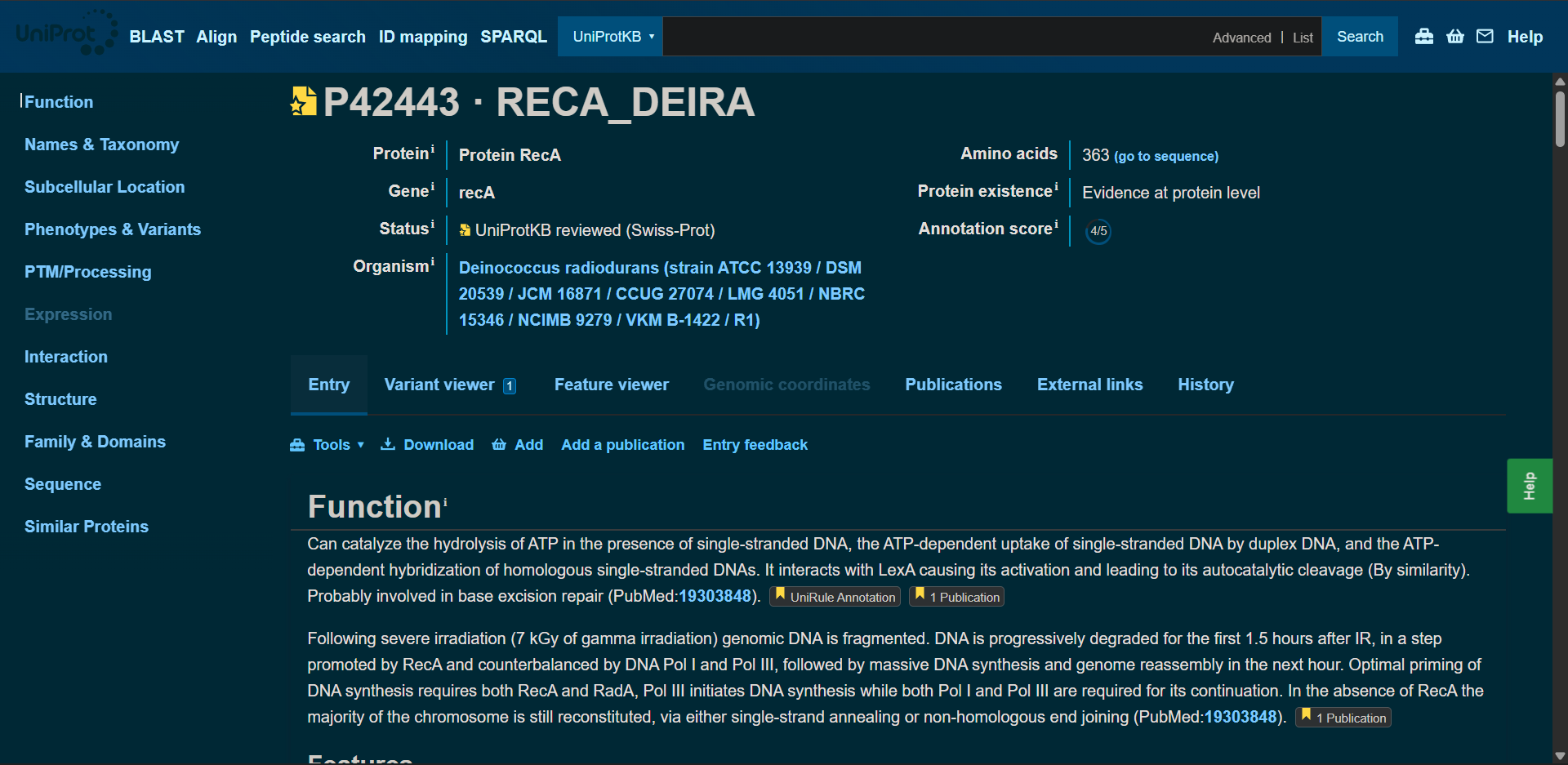
I selected the protein RecA from the extremophilic bacterium Deinococcus radiodurans. RecA is a DNA recombination and repair protein that plays a central role in homologous recombination and in the repair of double-strand DNA breaks.
D. radiodurans is known for its extraordinary resistance to ionizing radiation, desiccation, and other extreme environmental stresses. It can survive radiation levels thousands of times higher than those lethal to humans. RecA is essential for this remarkable resilience, as it facilitates DNA strand exchange and genome reassembly after severe DNA fragmentation.
I selected this protein because of its strong relevance to space biology and astrobiology. Radiation is one of the main challenges for life beyond Earth, and understanding the molecular mechanisms that enable DNA repair under extreme radiation conditions provides insight into how life might survive in extraterrestrial environments such as Mars. Additionally, RecA belongs to a highly conserved protein family, making it ideal for evolutionary and structural analysis.
2. Identify the amino acid sequence of your protein.
2.1. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
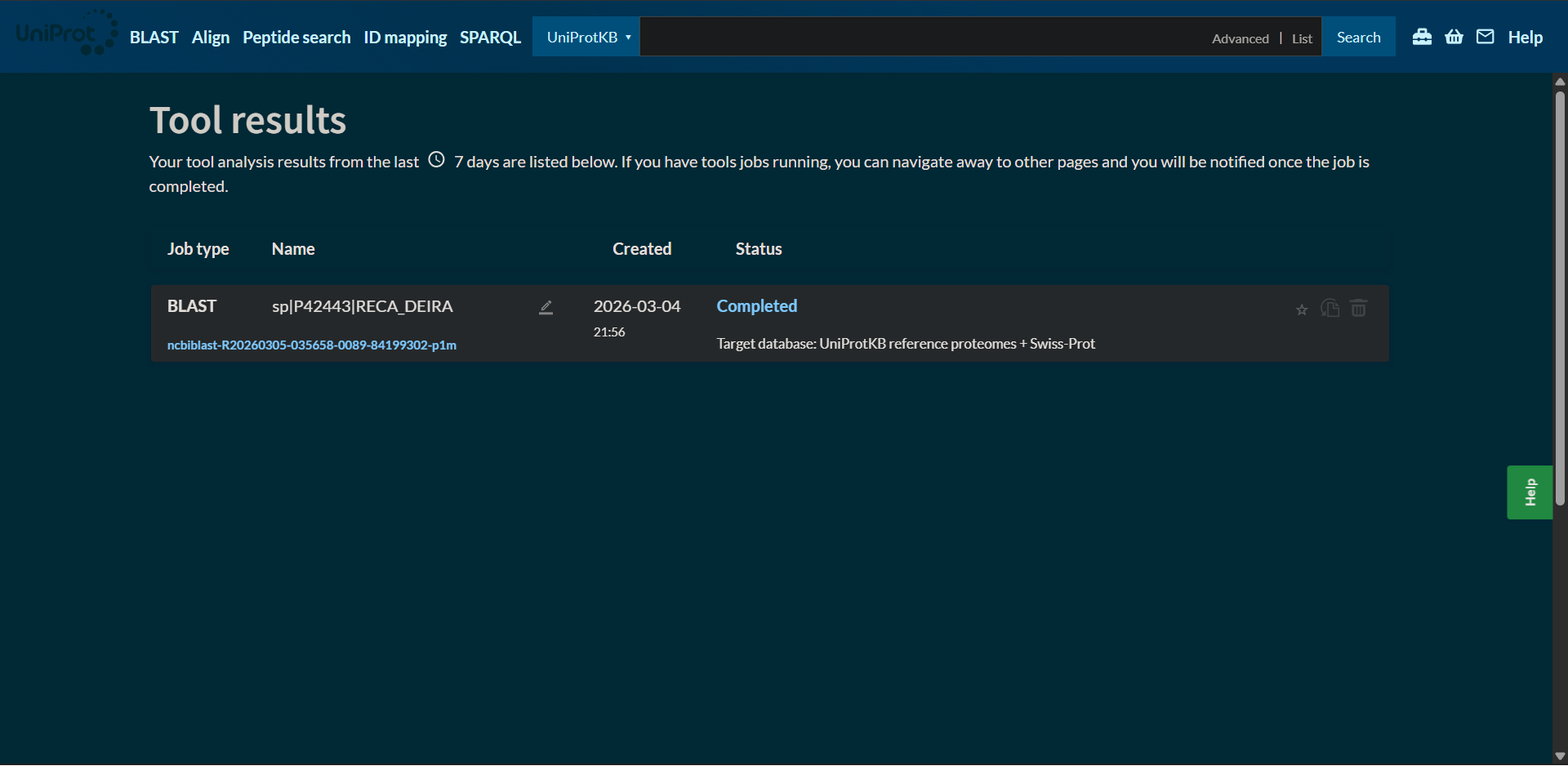
2.2. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
2.3. Does your protein belong to any protein family?
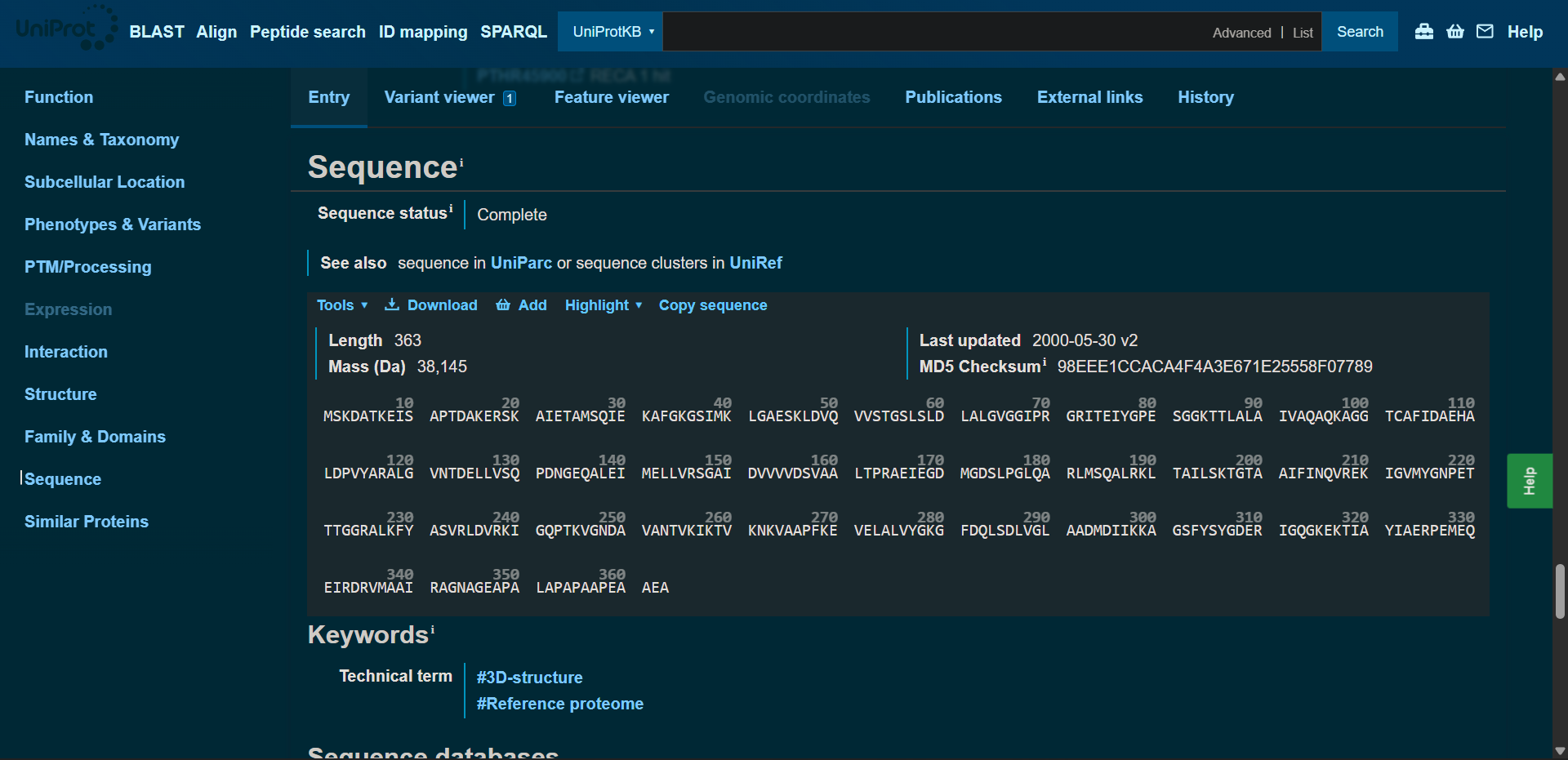
The amino acid sequence was obtainted from UniProt:
| 
|
|---|
RecA - Amino acid sequence
sp|P42443|RECA_DEIRA Protein RecA OS=Deinococcus radiodurans (strain ATCC 13939 / DSM 20539 / JCM 16871 / CCUG 27074 / LMG 4051 / NBRC 15346 / NCIMB 9279 / VKM B-1422 / R1) OX=243230 GN=recA PE=1 SV=2
MSKDATKEISAPTDAKERSKAIETAMSQIEKAFGKGSIMKLGAESKLDVQVVSTGSLSLDLALGVGGIPRGRITEIYGPESGGKTTLALAIVAQAQKAGGTCAFIDAEHALDPVYARALGVNTDELLVSQPDNGEQALEIMELLVRSGAIDVVVVDSVAALTPRAEIEGDMGDSLPGLQARLMSQALRKLTAILSKTGTAAIFINQVREKIGVMYGNPETTTGGRALKFYASVRLDVRKIGQPTKVGNDAVANTVKIKTVKNKVAAPFKEVELALVYGKGFDQLSDLVGLAADMDIIKKAGSFYSYGDERIGQGKEKTIAYIAERPEMEQEIRDRVMAAIRAGNAGEAPALAPAPAAPEAAEA
Protein lenght: 363 amino acids, wich aligns with the data shown at the UniProt site
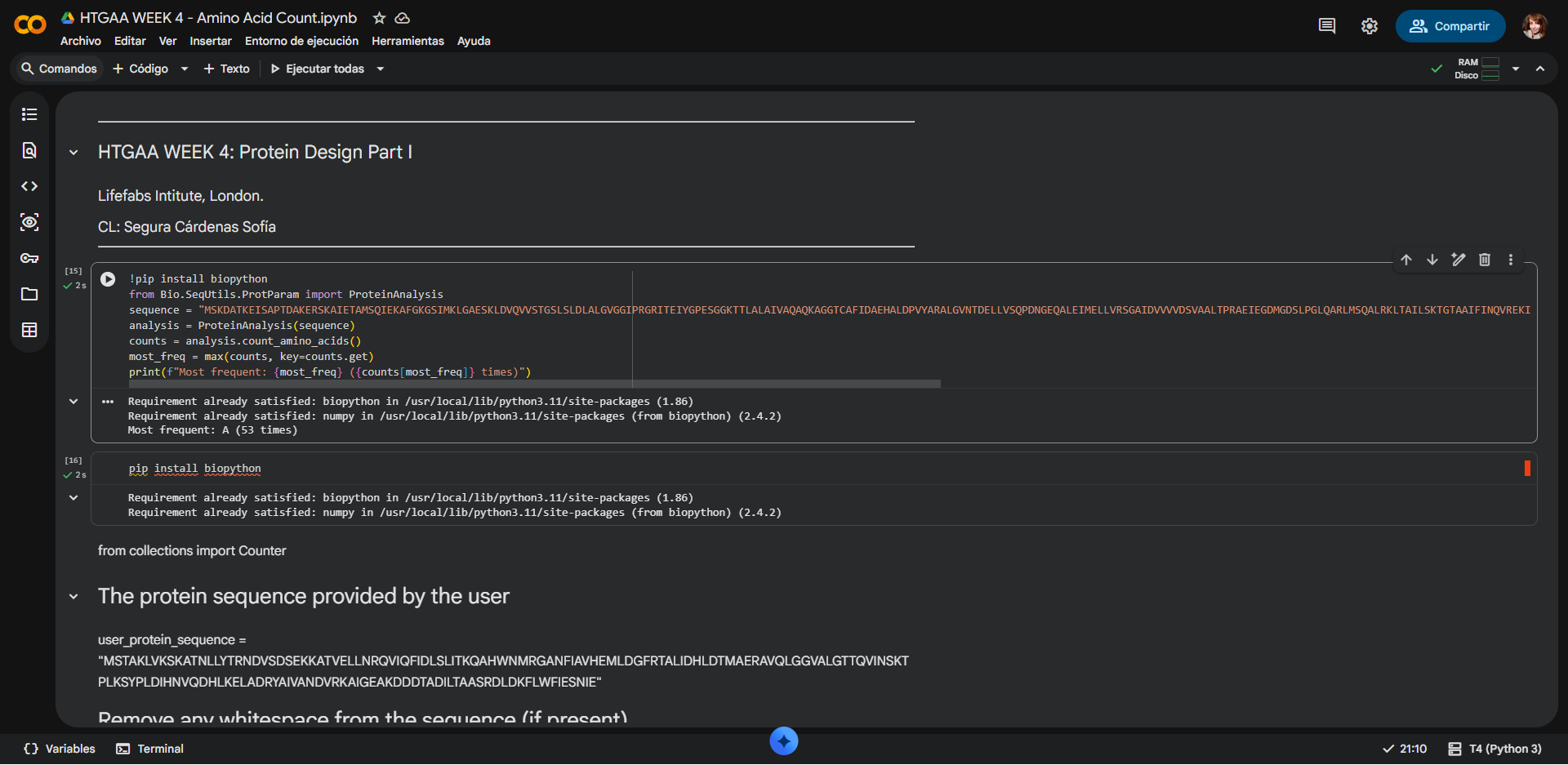
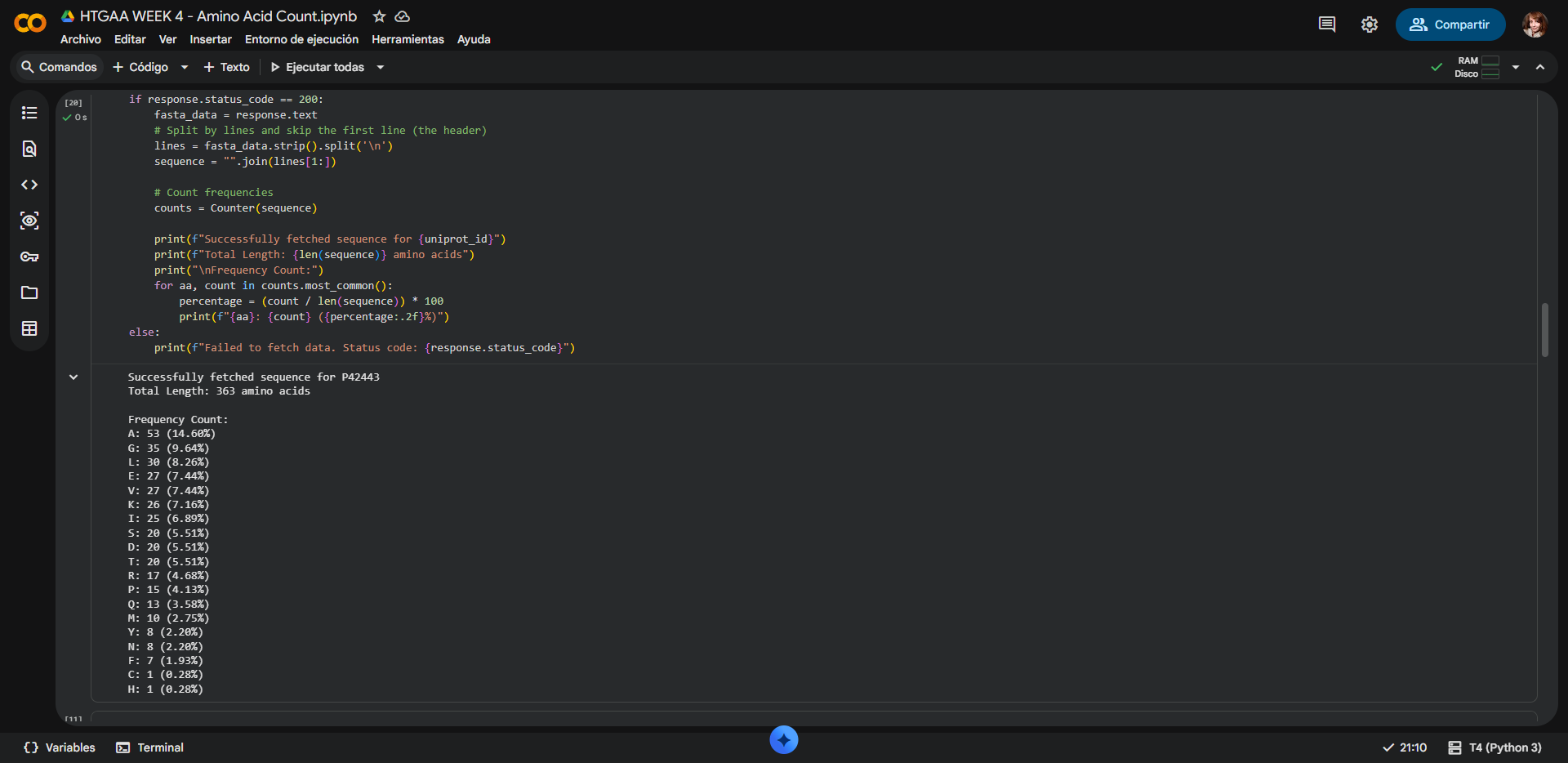
Most frecuent amino acid: Alanine (Ala, A), with a Frequency Count of 53 residues (14.60%)
Counting result by Google Colab notebook:
| 
|
|---|
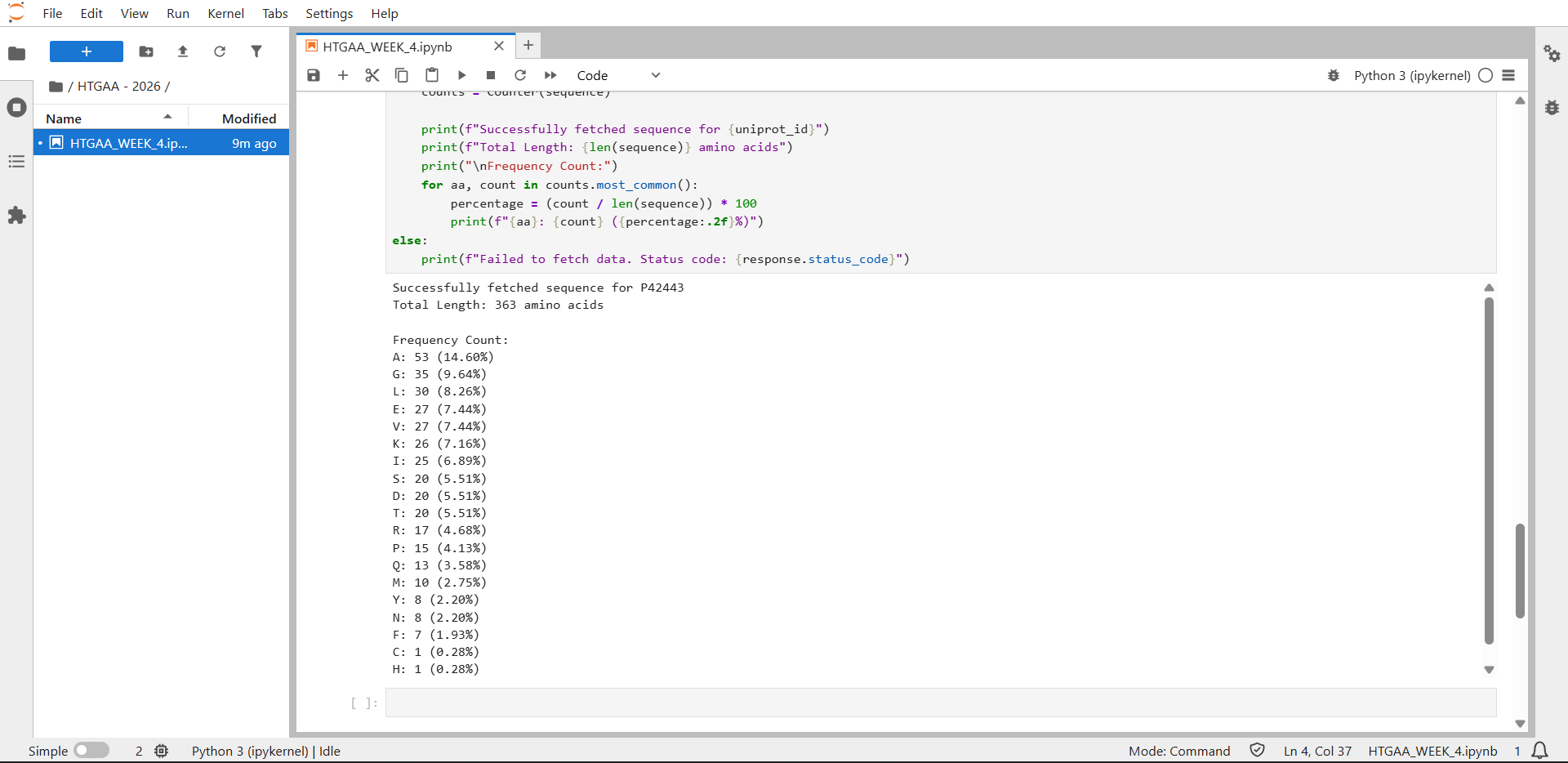
Counting result by JupyterLab:
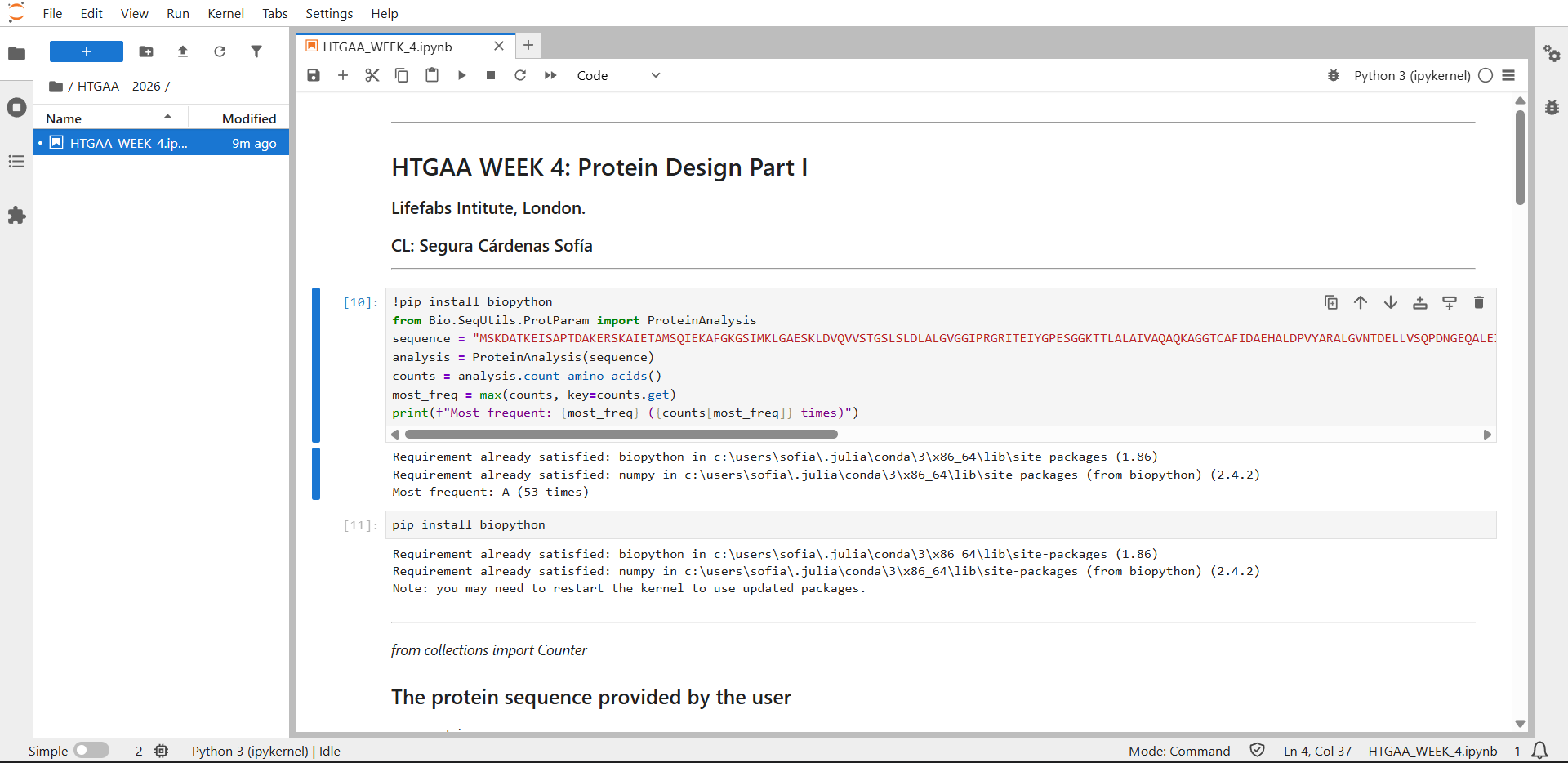
| 
|
|---|
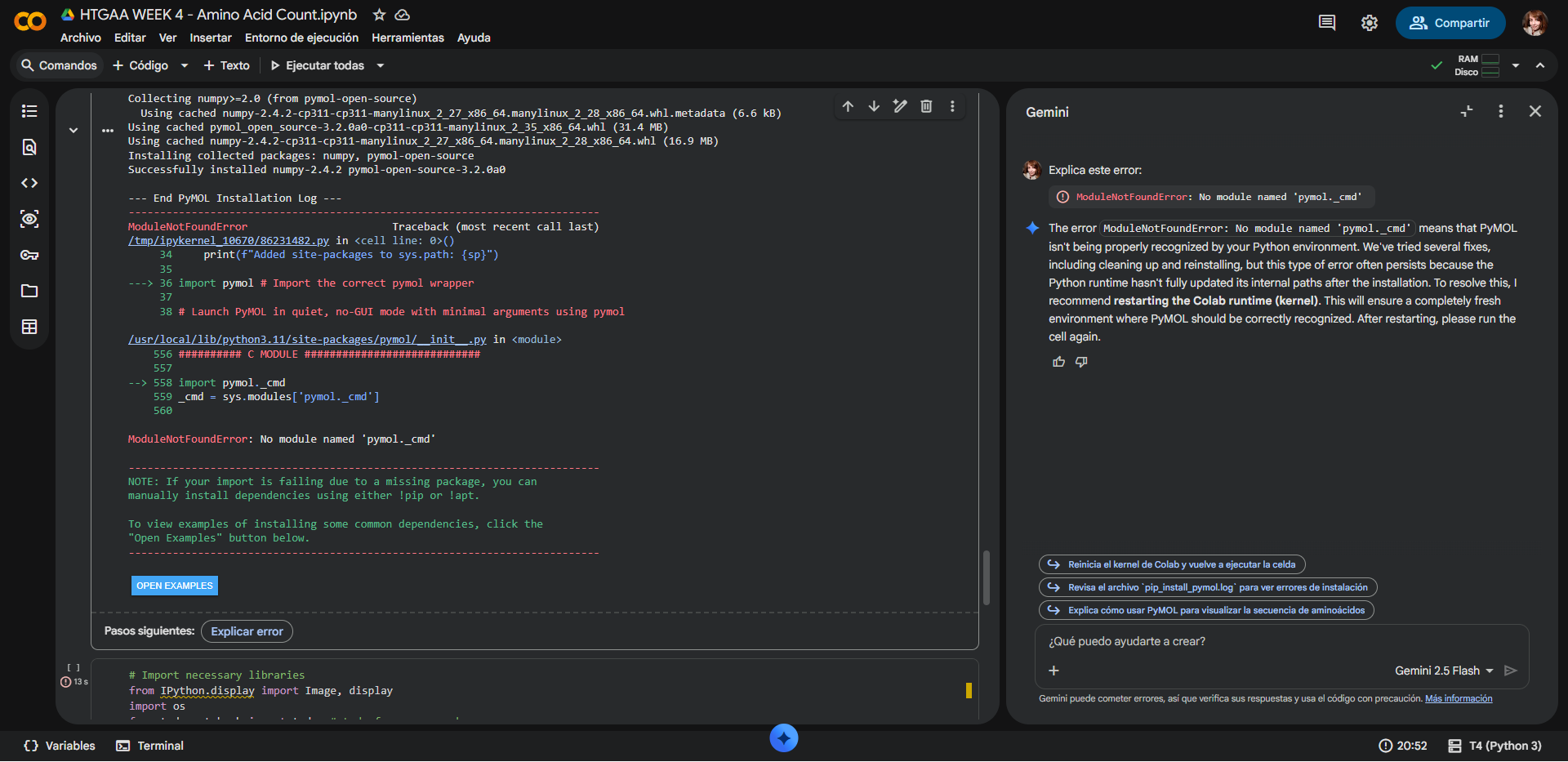
There was a small error on the second half of the Colab code, but the first part runs without issues. Even with assistance from Gemini AI it was not posible for it to run correctly:
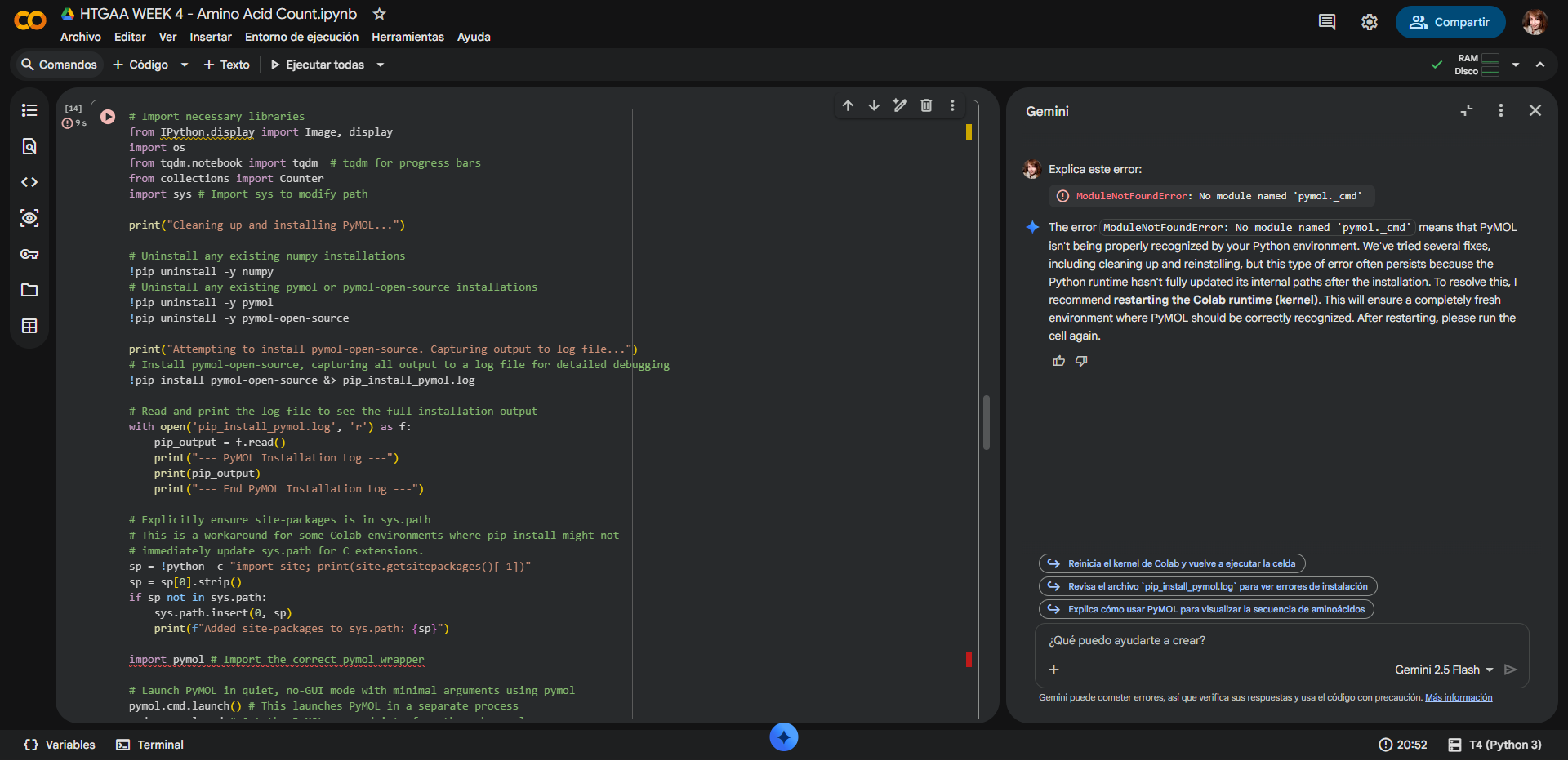
| 
|
|---|
Access to the Google Colab notebook used to count the frequency of amino acids: Click here
Protein sequence homologs: 250 hits found with BLAST
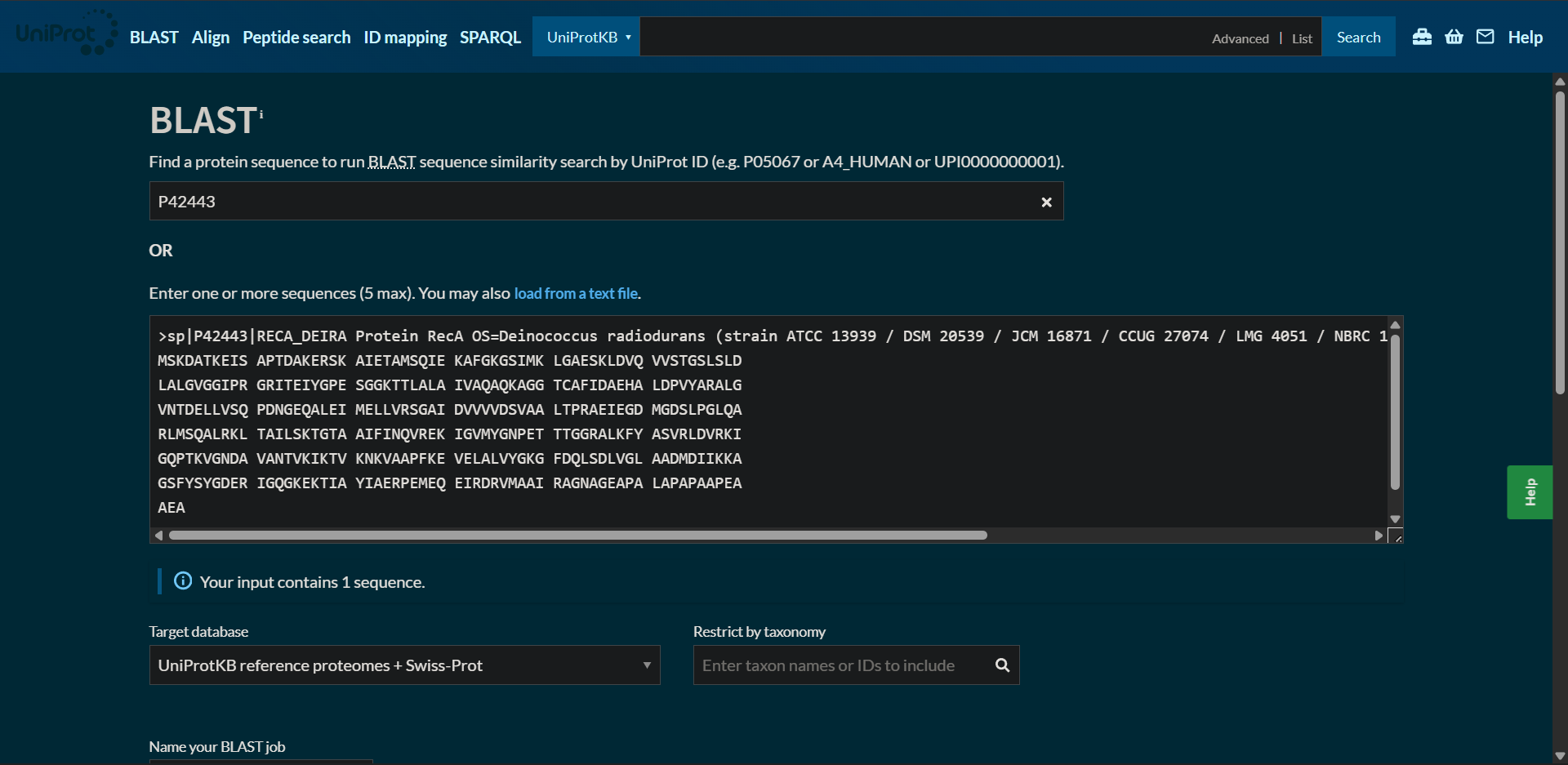
| 
|
|---|---|
| 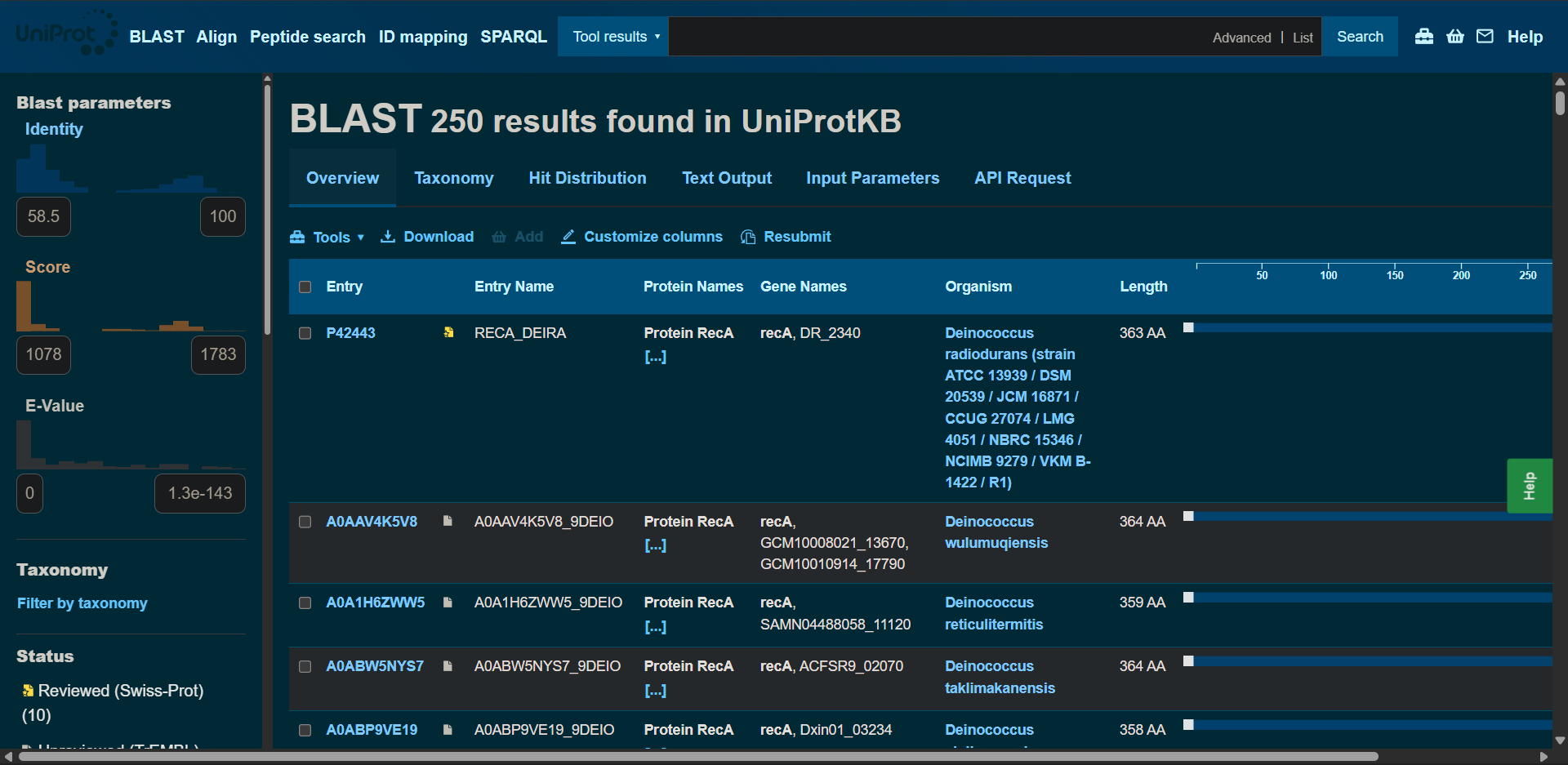
|
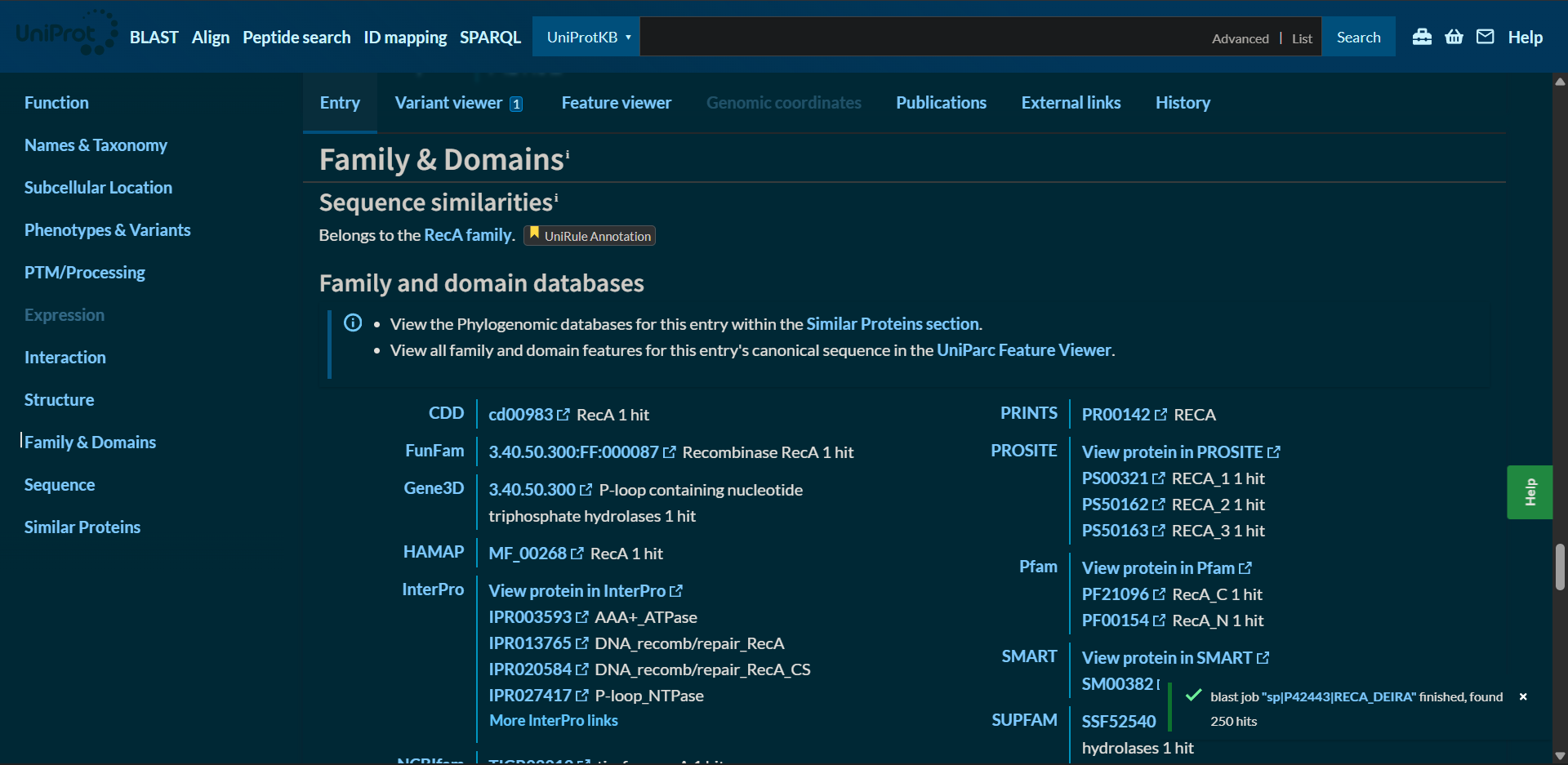
Protein affiliation (family): it belongs to the RecA family
According to UniProt and InterPro classification, RecA belongs to the RecA/Rad51 protein family. This family includes bacterial RecA, archaeal RadA, and eukaryotic Rad51 proteins. These proteins share a conserved ATPase domain of the P-loop NTP-binding superfamily and play essential roles in homologous recombination and DNA repair.
3. Identify the structure page of your protein in RCSB
3.1. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
3.2. Are there any other molecules in the solved structure apart from protein?
3.3. Does your protein belong to any structure classification family?
Structure released
- Deposited: 2004-10-08
- Released: 2004-12-21
- Deposition Author(s): Bell, C.E., Rajan, R. (2004) J Mol Biology 344: 951-963.
- Title: Crystal structure of RecA from Deinococcus radiodurans: insights into the structural basis of extreme radioresistance.
- DOI: https://doi.org/10.1016/j.jmb.2004.09.087
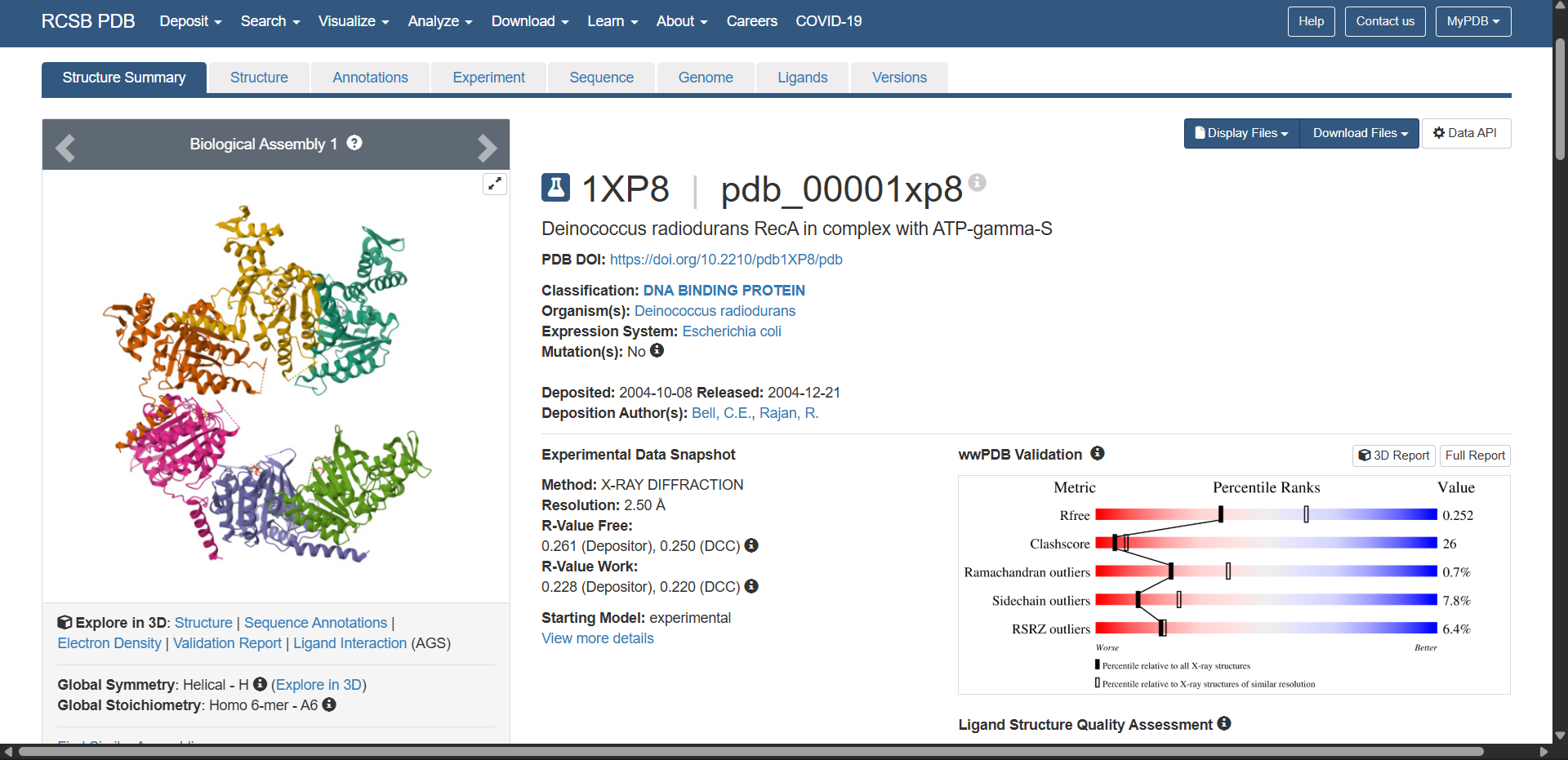
- Quality: Resolution of 2.50 Å
This is considered a good quality crystal structure. In X-ray crystallography, the resolution indicates the level of structural detail observed in the electron density map. Lower values correspond to higher structural precision.
- Molecules in the structure:
Yes. According to the RCSB entry, the structure contains additional molecules besides the RecA protein. These include:
- ADP (adenosine diphosphate) — a nucleotide bound to the ATPase active site
- Magnesium ion (Mg²⁺) — a cofactor required for nucleotide binding and ATP hydrolysis
- Water molecules (HOH) — commonly observed in crystal structures
These molecules are functionally relevant because RecA is an ATPase, and nucleotide binding plays an important role in its mechanism during DNA repair and homologous recombination.
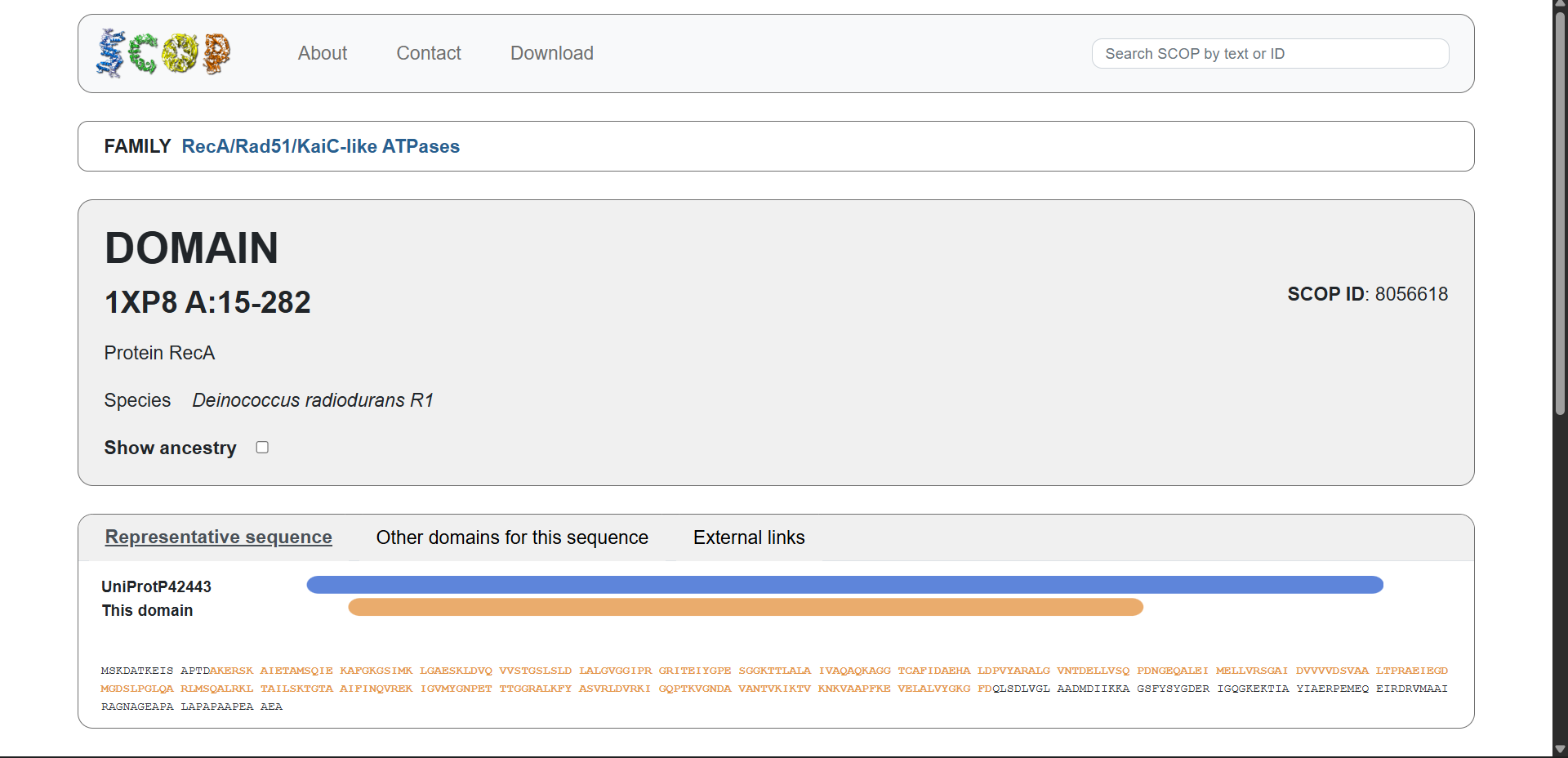
Protein affiliation:
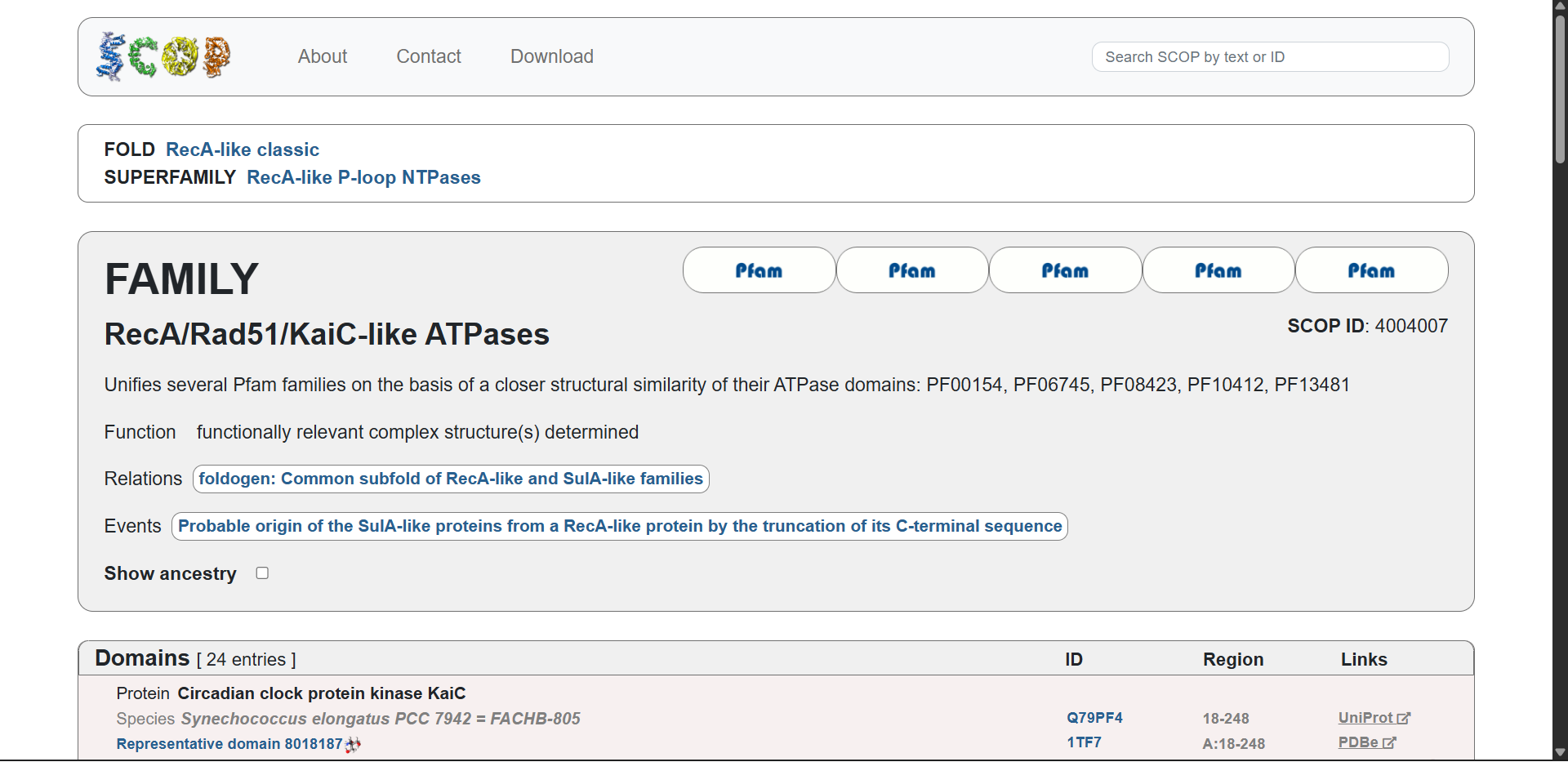
Yes. According to the SCOP structural classification database, the protein belongs to the following hierarchy:
- Fold:
RecA-like classic - Superfamily:
RecA-like P-loop NTPases - Family:
RecA/Rad51/KaiC-like ATPases - SCOP ID:
4004007
This classification groups proteins with similar three-dimensional folds and ATPase domains, even if their sequences differ. Members of this superfamily share a conserved P-loop NTP-binding domain involved in nucleotide binding and hydrolysis.
| 
|
|---|
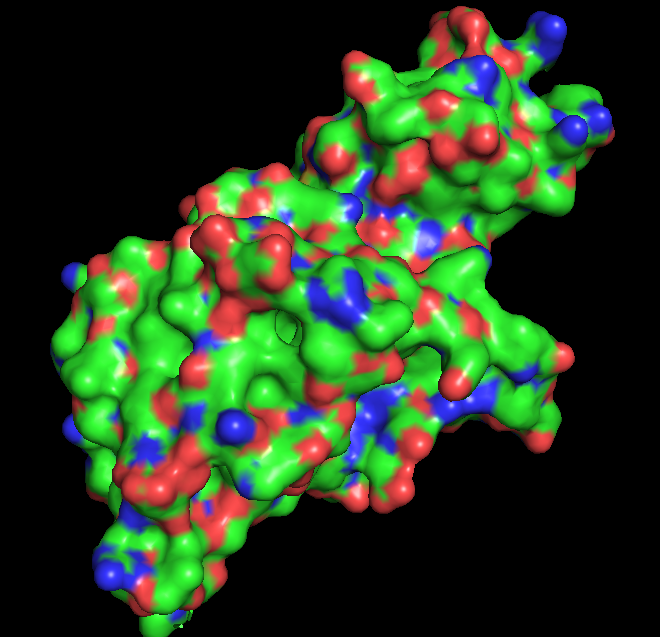
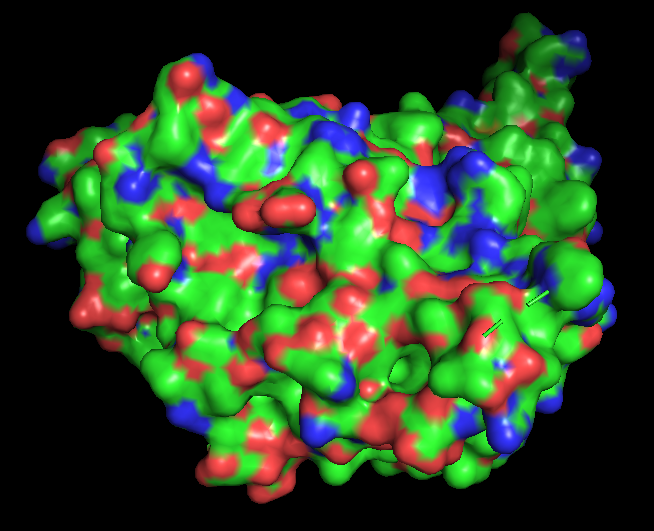
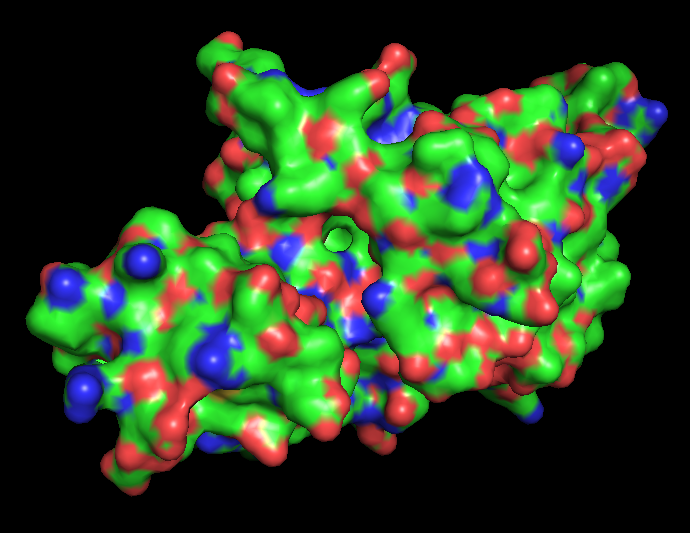
4. Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
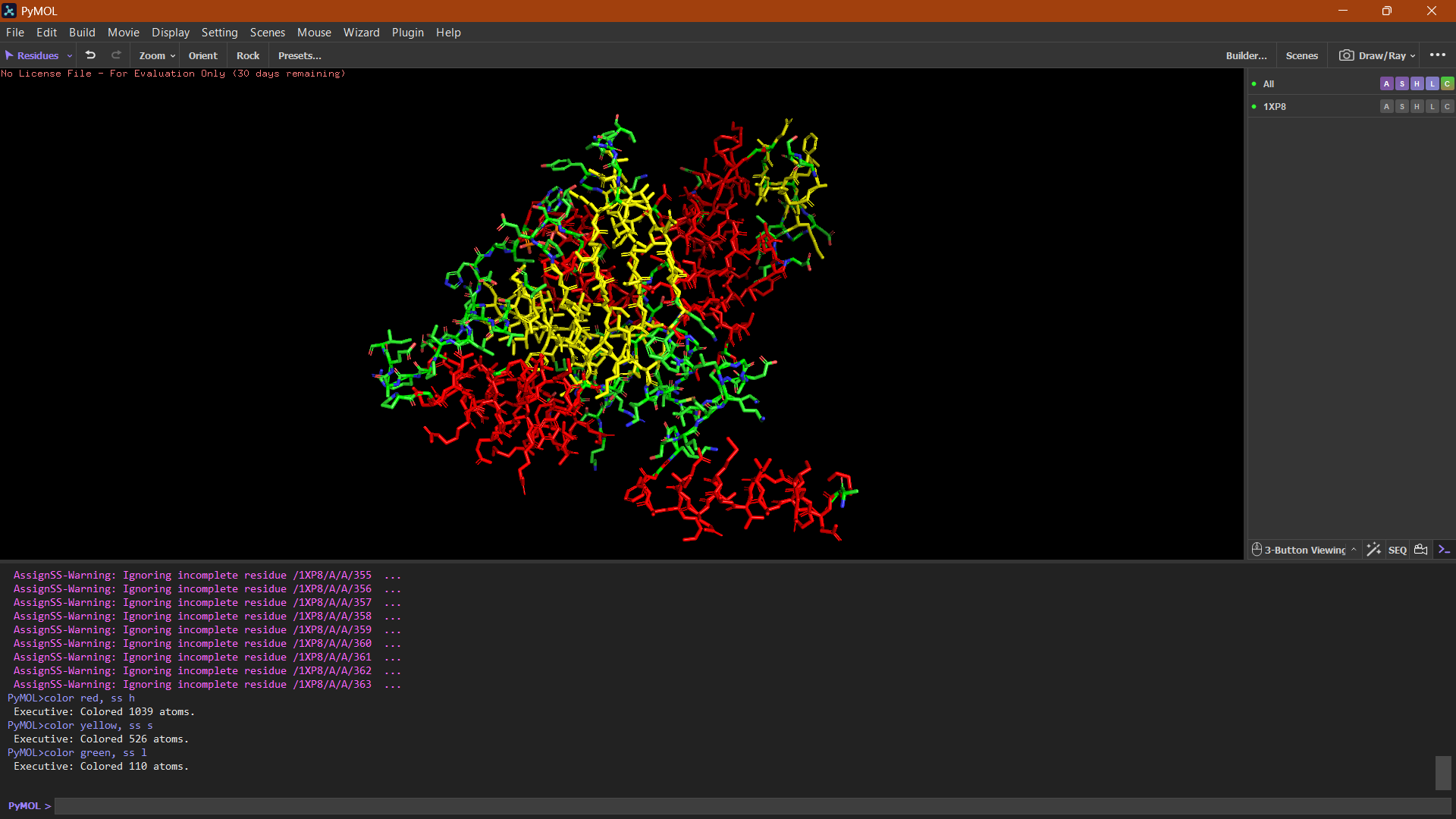
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
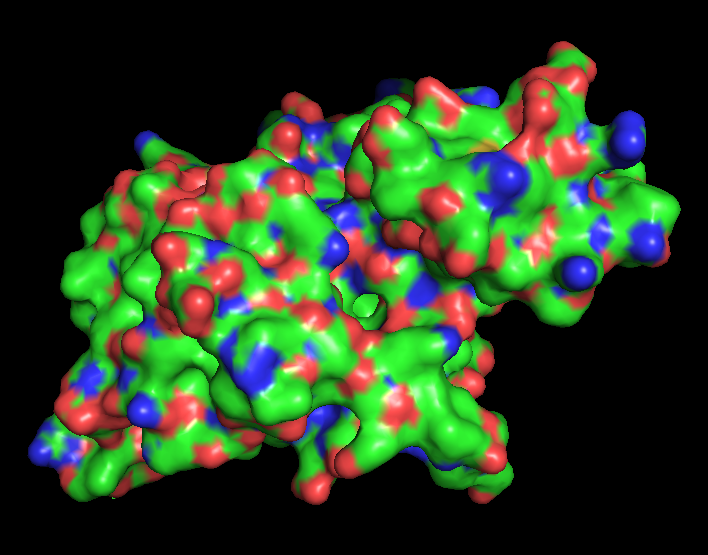
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
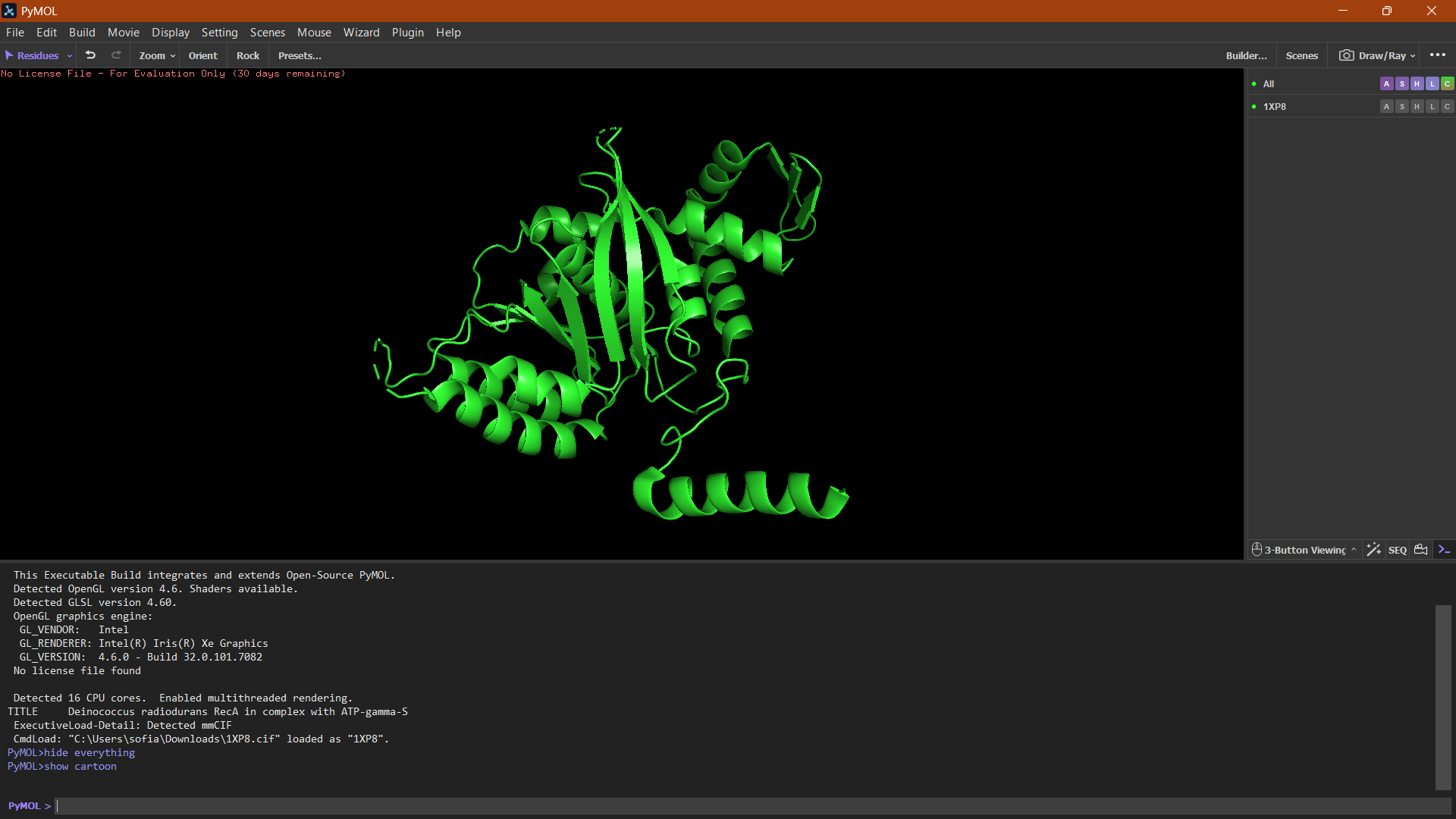
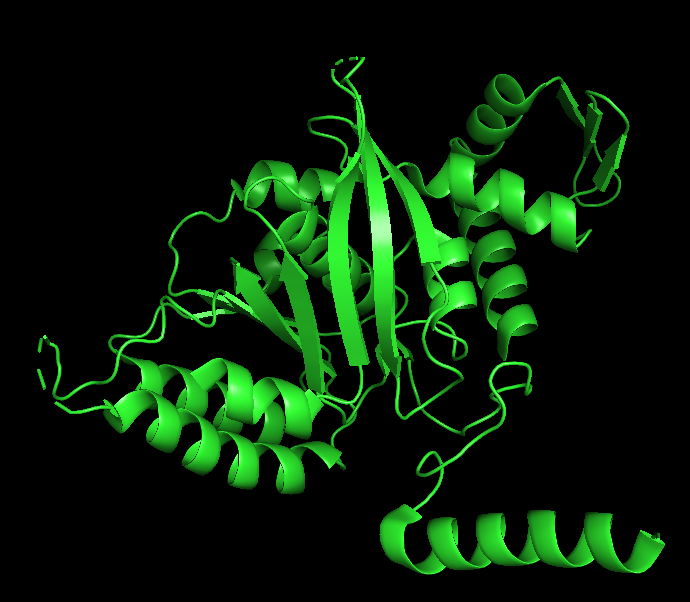
Protein visualization in PyMOL:
Cartoon
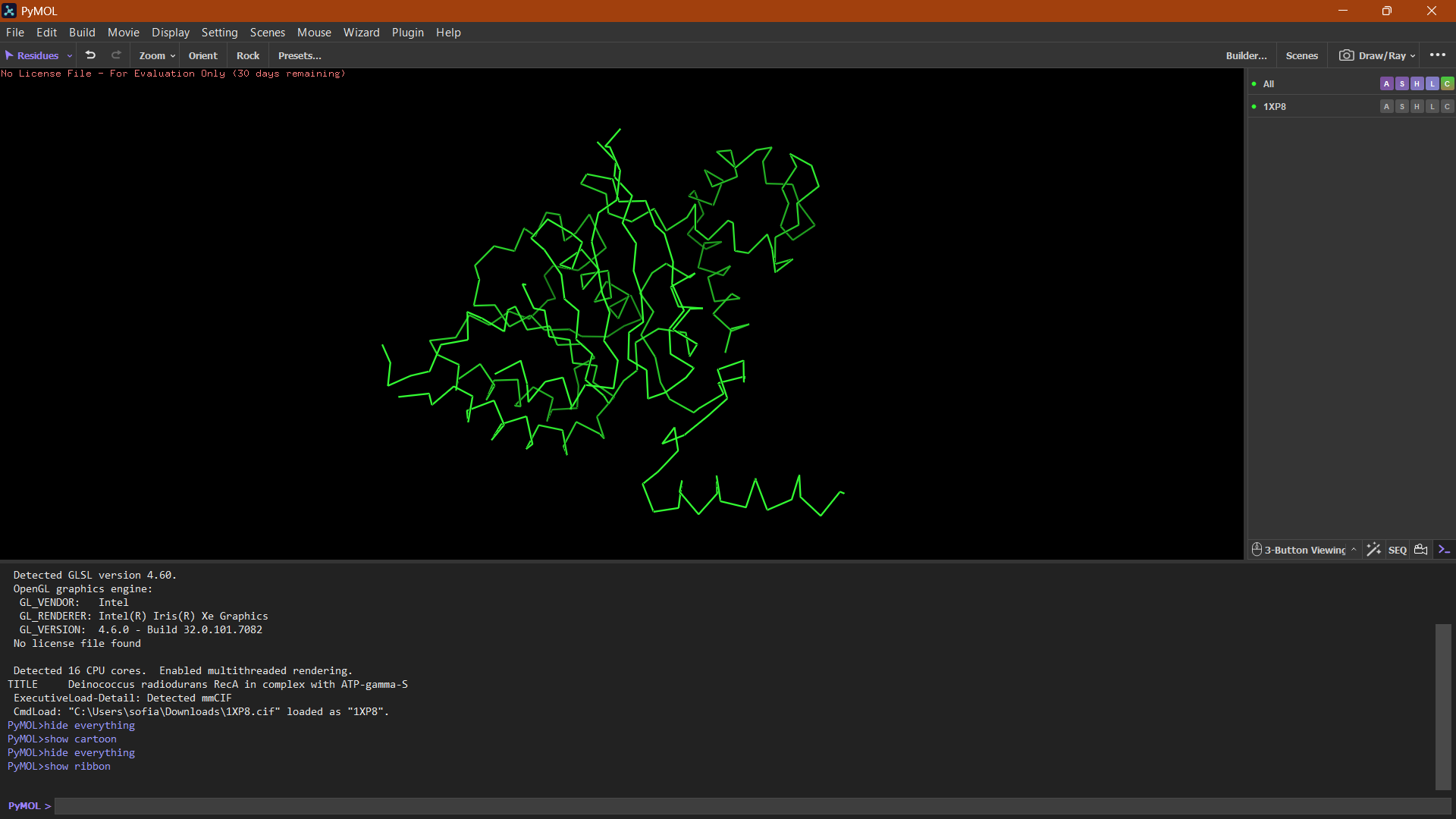
Ribbon

Ball and Stick
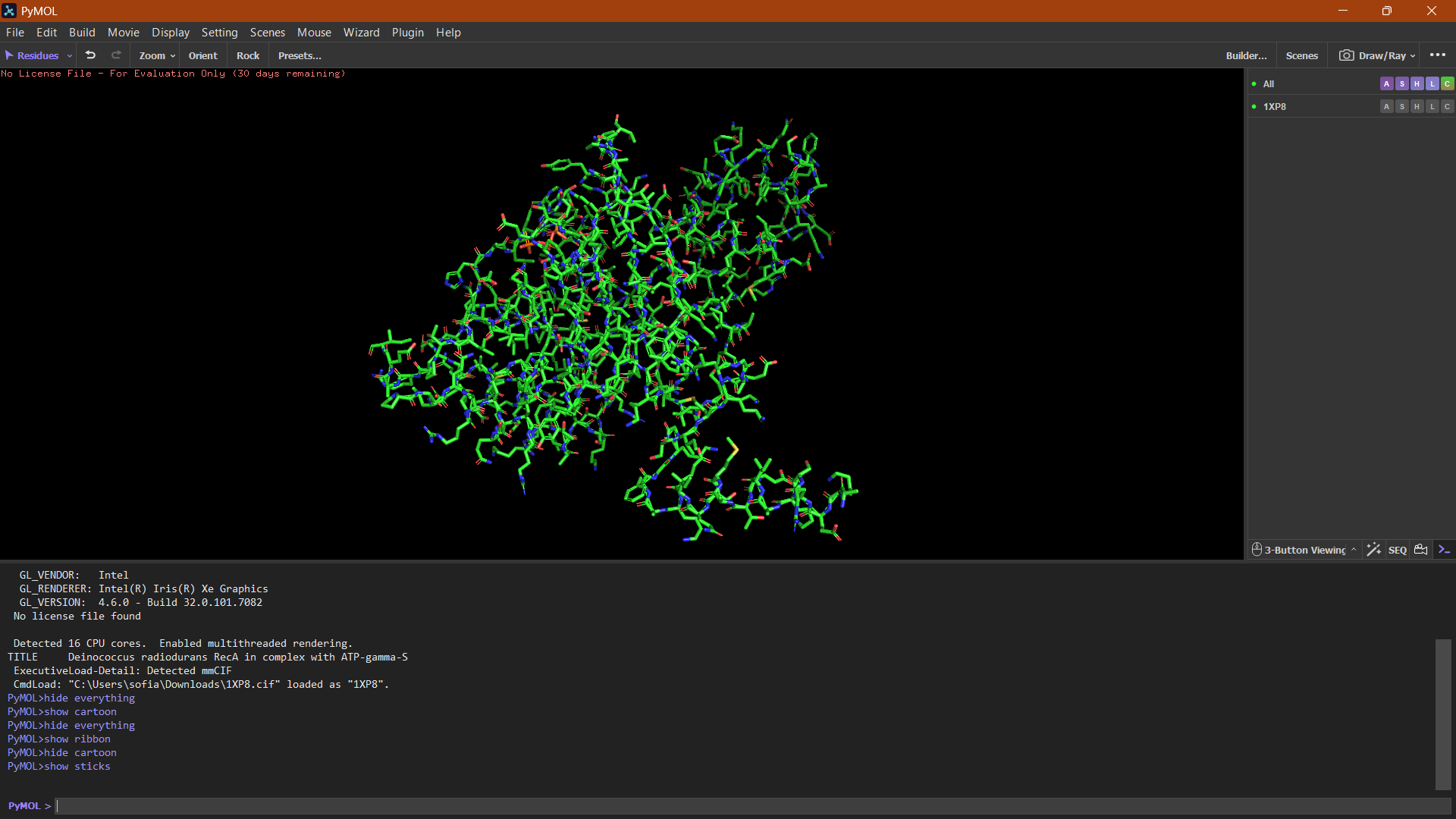

Helices or Sheets?
🔴 α-helices
🟡 β-sheets
🟢 loops or flexible regions
Cartoon:
The cartoon representation highlights the secondary structure elements of the protein. The structure contains several α-helices and a smaller number of β-sheets arranged near the center of the protein. Overall, α-helices appear to be more abundant than β-sheets.
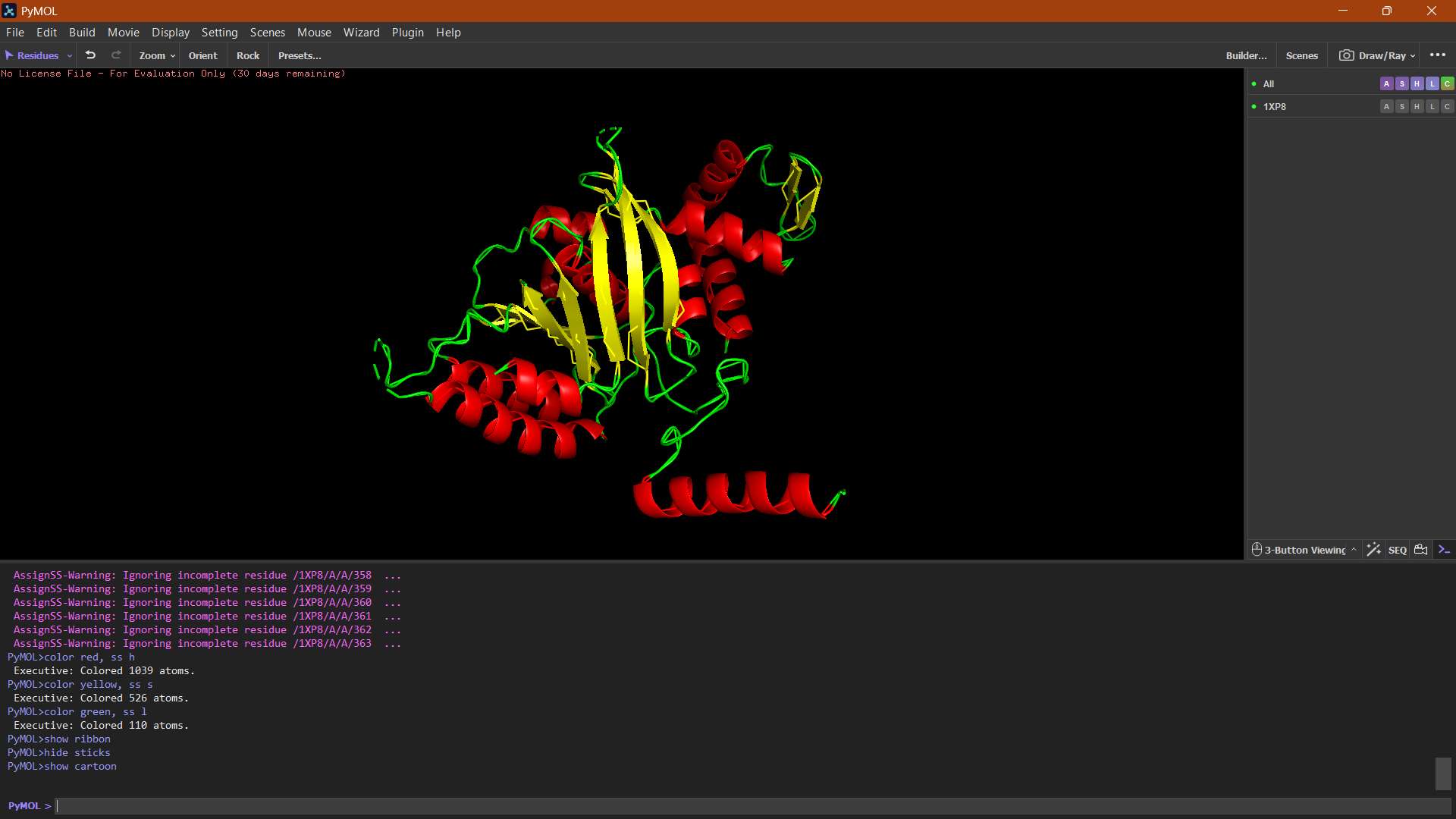
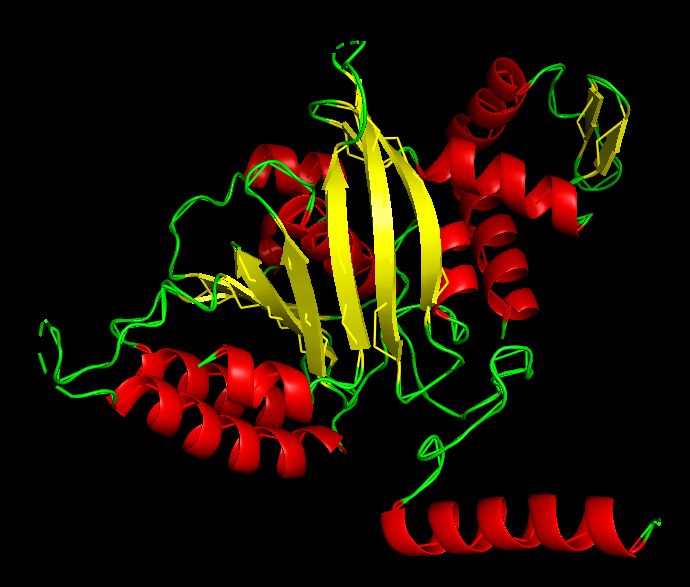
Ribbon:
The ribbon representation reveals the overall fold of the protein. The structure consists of a central β-sheet region surrounded by α-helices, which is characteristic of the RecA-like fold found in ATP-binding proteins.

Ball and Stick:
The ball-and-stick representation shows the detailed atomic arrangement of the amino acid residues. Hydrophobic residues appear mostly buried within the protein core, whereas hydrophilic residues are more exposed on the surface, which is typical for soluble cytoplasmic proteins.
Hydrophobic vs Hydrophilic
Hydrophobic residues tend to cluster within the interior of the protein structure, while hydrophilic and charged residues are more exposed on the protein surface, which is typical for soluble proteins interacting with the aqueous cytoplasm.
🔵 Hydrophilic residues (positively charged)
🔴 Negatively charged residues
Binding pockets
Surface representation of the protein reveals several small cavities distributed across the structure. These cavities likely correspond to potential ligand-binding pockets. In RecA proteins, such pockets are typically involved in nucleotide binding (ATP/ADP), which is required for DNA repair activity.
| 
| 
| 
|
|---|
Part C. Using ML-Based Protein Design Tools
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Google Colab notebook used: HTGAA_W4_Protein_Design_2026_Sofía_Segura.ipynb
C1. Protein Language Modeling
C1.1. Deep mutational scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
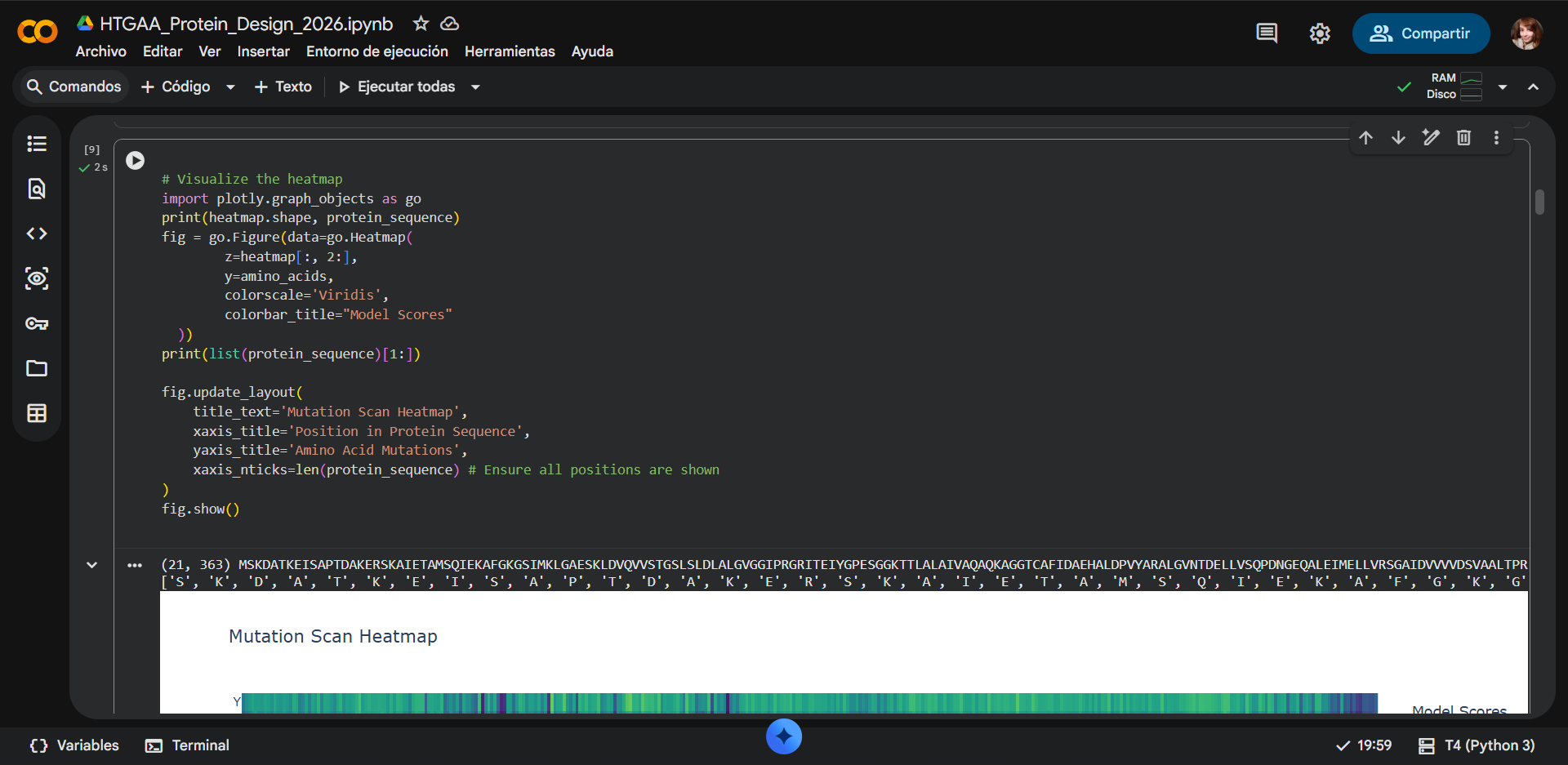
The deep mutational scan generated using the protein language model ESM-2 evaluates how likely each amino-acid substitution is at every position in the protein sequence of RecA.
In the Heatmap:
- The x-axis represents the position of residues in the protein sequence.
- The y-axis represents the amino acid substituted at that position.
- Each cell corresponds to a single mutation.
The color scale represents the model score, which reflects how compatible a mutation is with the learned evolutionary patterns of proteins.
| Color | Score Range | Interpretation |
|---|---|---|
| 🟡 Yellow | Positive (>0) | Favorable or tolerated mutation |
| 🟢 Green | Around 0 | Neutral mutation |
| 🔵 Blue | Moderately negative | Likely destabilizing mutation |
| 🟣 Purple | Very negative (< -8) | Strongly deleterious mutation |
Mutations with strongly negative scores are predicted to be highly disruptive to the protein structure or function.
| 
| 
|
|---|
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
One of the most noticeable patterns in the heatmap is the presence of vertical dark bands across several sequence positions. These vertical bands indicate positions that are highly sensitive to mutation, where almost any substitution results in a strongly negative score. This pattern suggests that these residues are evolutionarily conserved and structurally or functionally important.
In proteins such as RecA, highly conserved residues often correspond to:
- Catalytic residues
- ATP-binding residues
- Residues located in the structural core of the protein
Mutations at these positions are therefore predicted to disrupt protein folding or functional activity.
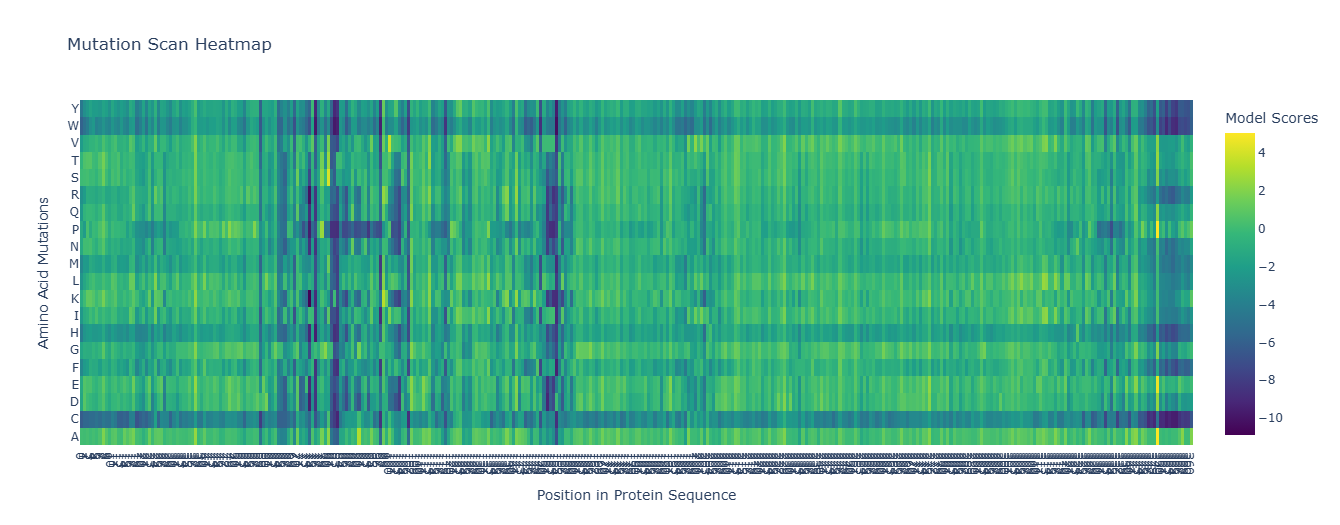
Strongly unfavorable mutations
a) Cysteine mutations
A particularly prominent pattern in the heatmap corresponds to mutations to cysteine (C), which frequently show very negative scores across many sequence positions. Introducing cysteine residues can be problematic for several reasons:
- Disulfide bond formation: Cysteine residues can form disulfide bonds, which may introduce unintended cross-links that disrupt the protein’s native structure.
- Structural constraints: Cysteine has a reactive thiol group that may interfere with local interactions within the protein core.
- Protein environment mismatch: In cytosolic proteins such as RecA, cysteines are relatively rare and often occur only at specific functional sites.
Because of these factors, many cysteine substitutions are predicted to be structurally destabilizing, which explains the strong negative scores observed in the heatmap.
b) Tryptophan mutations
Another notable pattern is the strong negative scores observed for mutations to tryptophan (W) at many positions. Tryptophan is the largest amino acid, and its bulky aromatic side chain can disrupt tightly packed regions of the protein structure. When introduced at positions that cannot accommodate large residues, it may:
- Create steric clashes
- Disturb secondary structure packing
- Destabilize the hydrophobic core
As a result, many tryptophan substitutions receive very negative model scores, indicating that these mutations are likely to be deleterious.
Favorable or tolerated mutations
Some amino acids show mostly neutral or favorable scores across the sequence. One example in the heatmap is serine (S), which appears largely green and occasionally yellow. Serine substitutions are often tolerated because:
- it is small in size
- it is polar but not strongly charged
- it can participate in hydrogen bonding
- it does not introduce major steric clashes
Because of these properties, serine can frequently replace other small or polar residues without significantly disrupting the protein structure.
Neutral mutations
Neutral mutations (shown in green) typically occur when the substituted amino acid has similar physicochemical properties to the original residue. Examples include substitutions between:
- Hydrophobic residues (e.g., V → I)
- Polar residues (e.g., S → T)
- Similarly sized amino acids
These mutations tend to preserve the overall structural stability and local interactions of the protein.
C1.2. Latent space analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
- Analyze the different formed neighborhoods: do they approximate similar proteins?
- Place your protein in the resulting map and explain its position and similarity to its neighbors.
The following dimensionality reduction technique preserves local similarity relationships, allowing sequences with similar structural or evolutionary features to cluster together in the resulting latent space.
NOTE
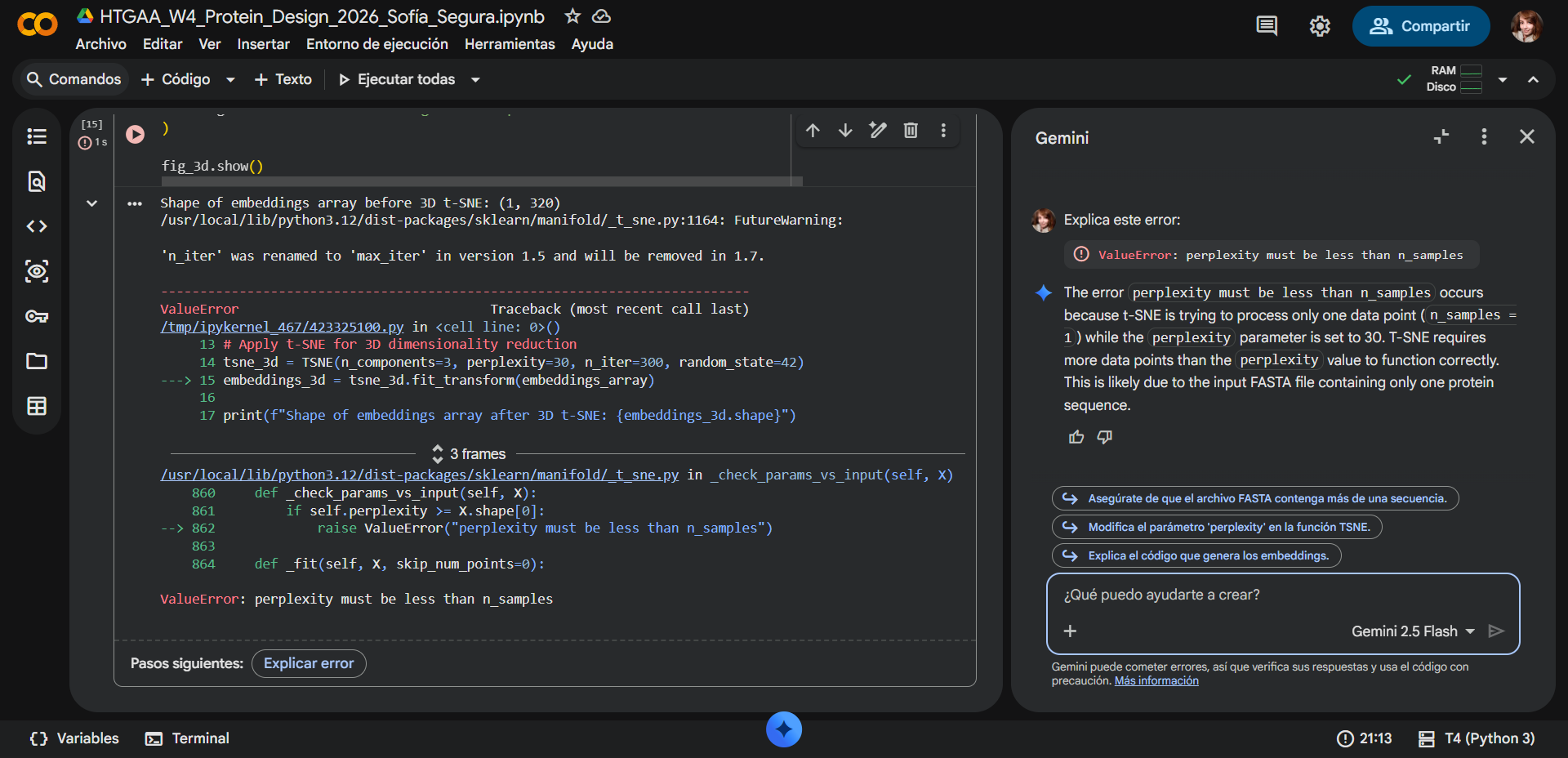
Some adjusments were made. During the latent space analysis, an error occurred while applying the t-SNE dimensionality reduction using
scikit-learn. The program returned the messageValueError: perplexity must be less than n_samples. This error arose because the input dataset initially contained only a single protein sequence corresponding to RecA from Deinococcus radiodurans. The t-SNE algorithm requires multiple samples to estimate neighborhood relationships between points, and theperplexityparameter (set to 30) must always be smaller than the number of samples in the dataset. Because only one sequence was provided, the algorithm could not compute the embedding. The issue was resolved with assistance from Gemini, which identified that theFASTAinput needed multiple sequences. The original FASTA link was therefore replaced with a dataset containing 50 homologous protein sequences related to RecA, allowing the model to generate valid embeddings and complete the latent space analysis.
With error
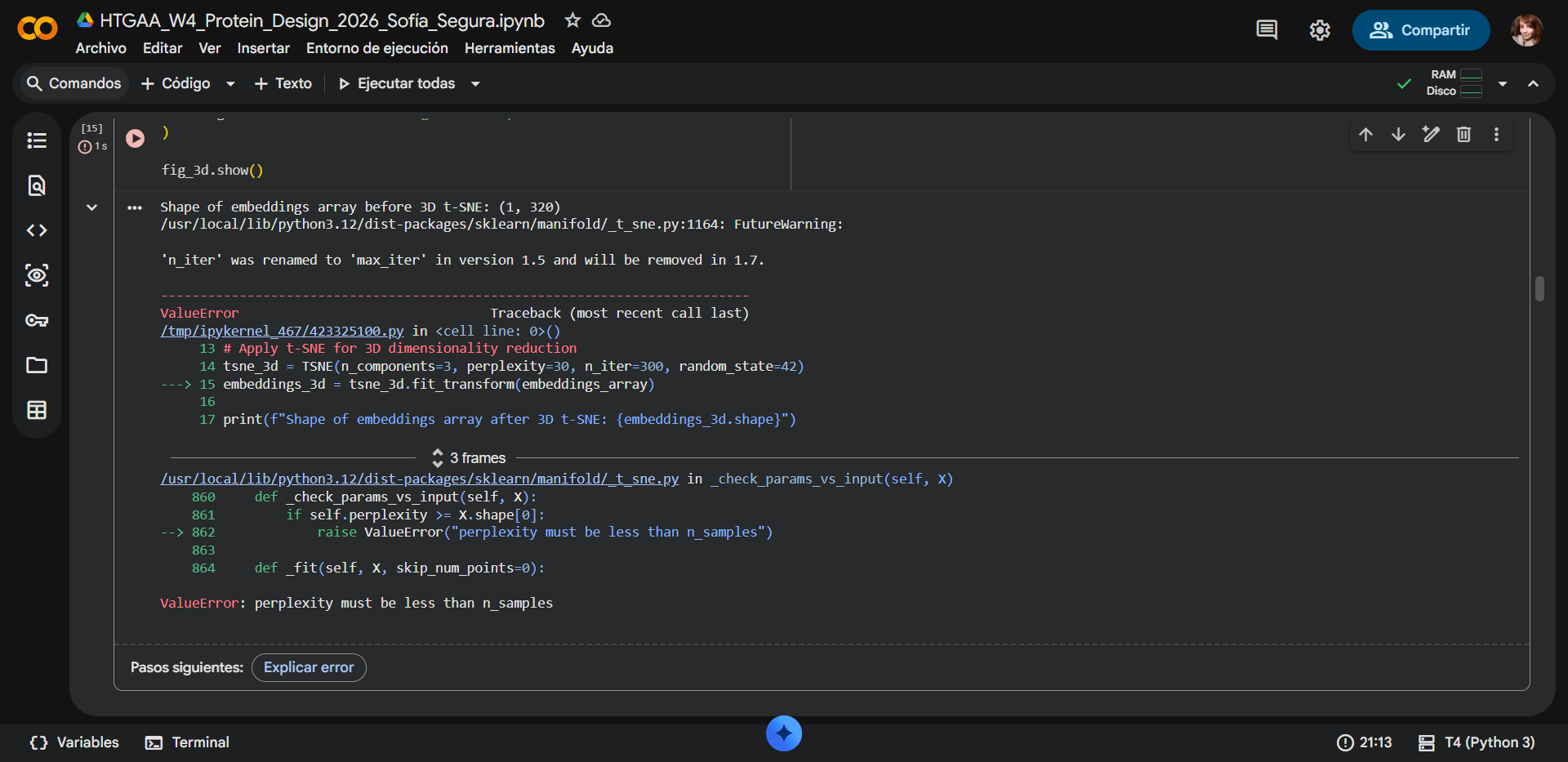
| 
|
|---|
Fixed
| 
| 
|
|---|
Final take
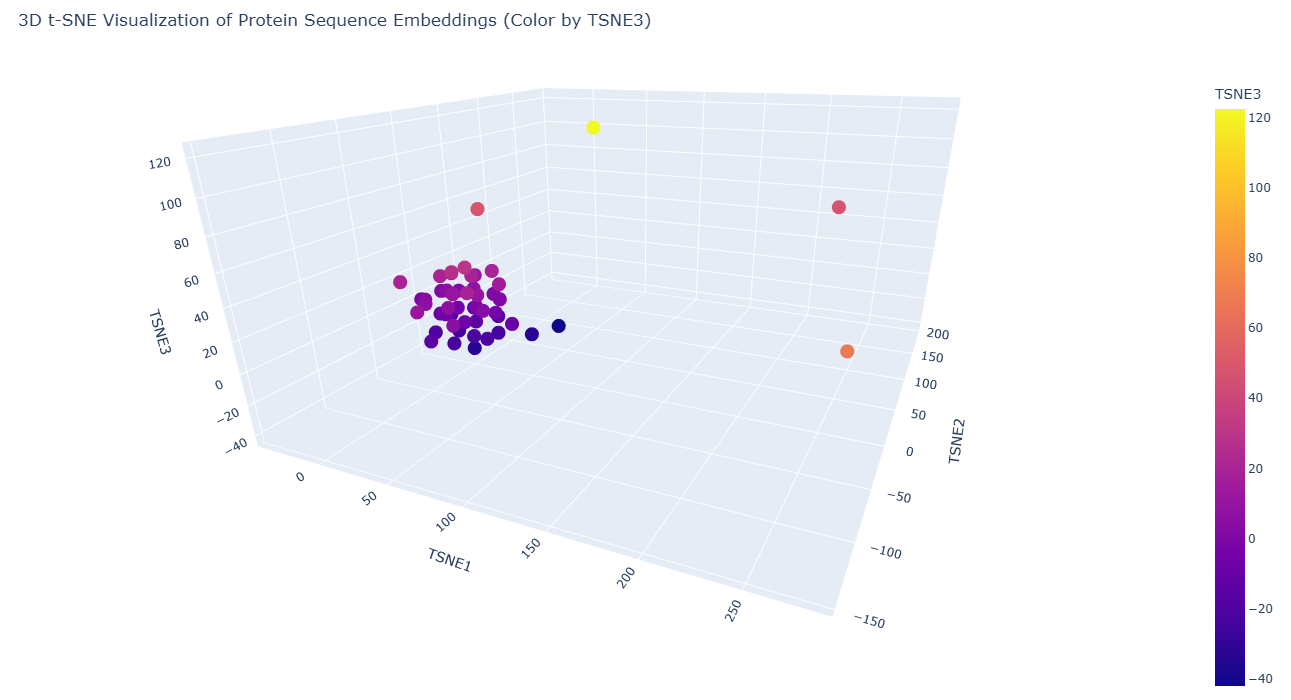
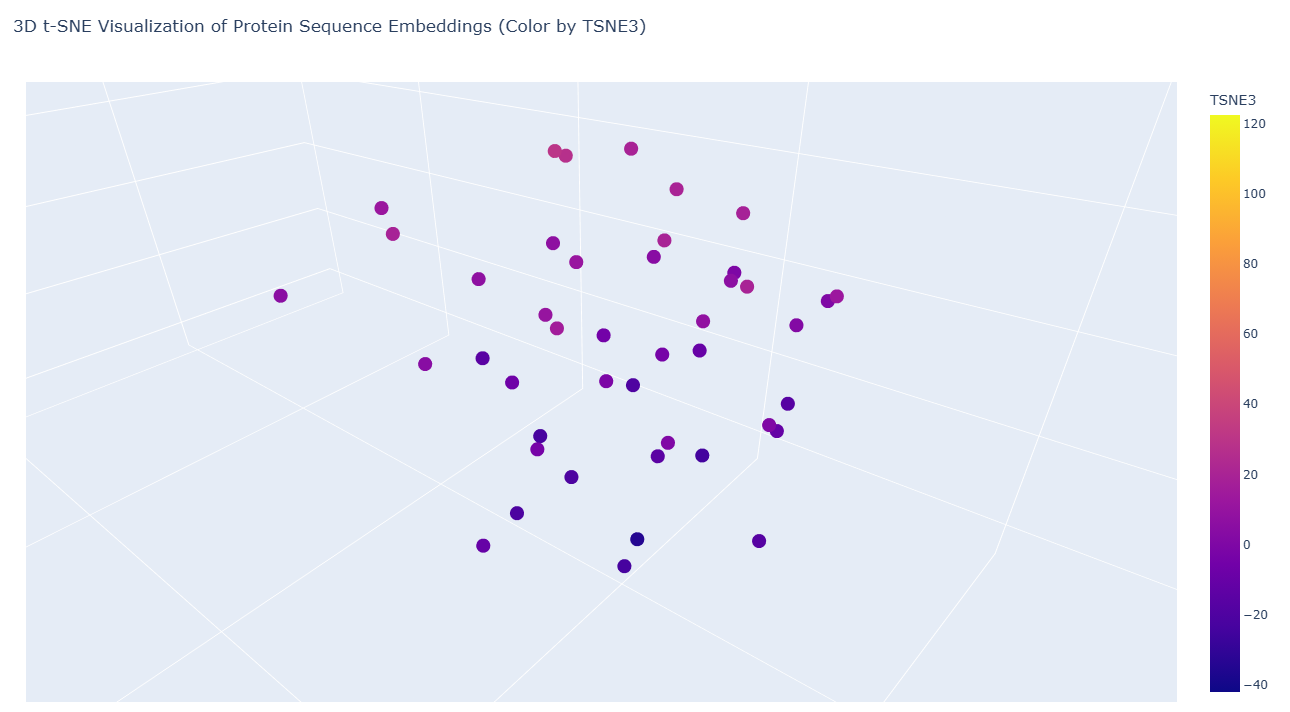
| 
|
|---|
The following color encoding helps visualize how proteins are distributed along the third dimension of the latent space and highlights subtle structural relationships that may not be obvious from spatial position alone.
| Color | TSNE3 Value | Interpretation |
|---|---|---|
| 🔵 Dark purple | Low values (negative) | Proteins positioned in lower regions of the latent dimension |
| 🟣 Magenta / 🩷 pink | Intermediate values | Proteins occupying middle regions of the embedding |
| 🟠 Orange / 🟡 yellow | High values (positive) | Proteins separated along the third dimension |
NOTE: The color scale represents the TSNE3 coordinate and does not indicate protein quality or functional superiority. Instead, it simply reflects the relative position of proteins along the third dimension of the embedding space.
a) Neighborhood structure in the embedding space
The resulting 3D visualization reveals a clear clustering pattern, where the majority of sequences form a dense neighborhood in the latent space. This clustering indicates that many sequences in the dataset share similar sequence patterns, structural motifs, or evolutionary relationships, and likely belong to related protein families or structural classes. Protein language models capture evolutionary constraints during training, meaning sequences with similar functional or structural properties tend to occupy nearby regions in the embedding space.
Within the visualization, most sequences form a compact cluster, suggesting they share significant sequence similarity and may belong to related recombination or DNA-binding protein families.
b) Outlier sequences
A small number of sequences appear separated from the main cluster, forming outliers in the latent space. These outliers may correspond to proteins that:
- Contain significant sequence divergence
- Belong to more distant homologous families
- Contain additional domains or structural insertions
Protein language models often place functionally related but evolutionarily distant proteins in nearby regions, but sequences that diverge significantly may appear as isolated points in the reduced dimensional space.
Placement of the selected protein
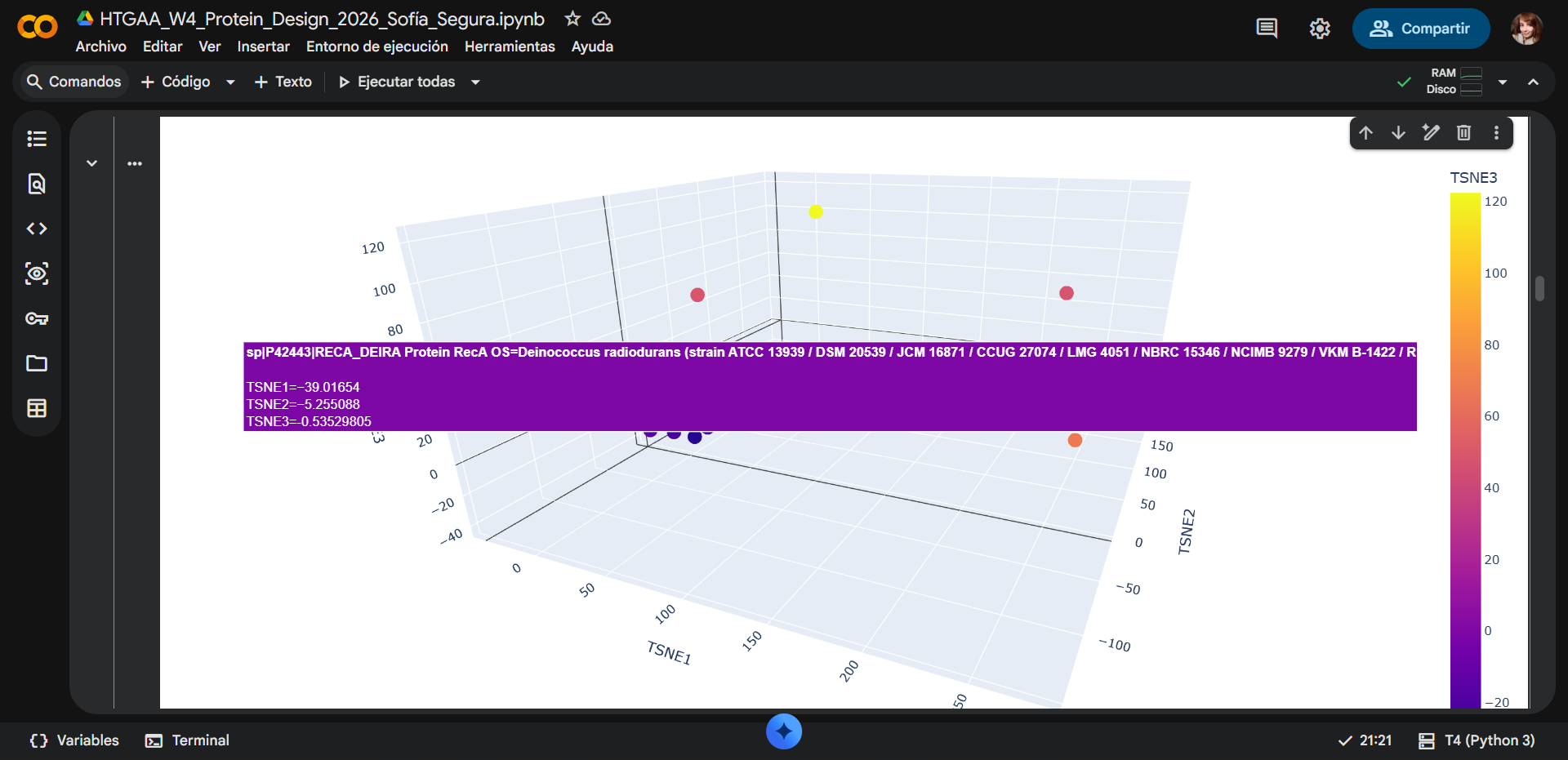
RecA from Deinococcus radiodurans, appears within the main cluster of sequences in the embedding space. Its coordinates (TSNE1 ≈ −39, TSNE2 ≈ −5, TSNE3 ≈ −0.5) place it close to several other proteins in the dataset. The surrounding points share similar color and spatial proximity, indicating that these proteins have similar embedding representations.
C2. Protein Folding
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
| 
| 
|
|---|
ESMFOLDING RESULTS
| 
|
|---|
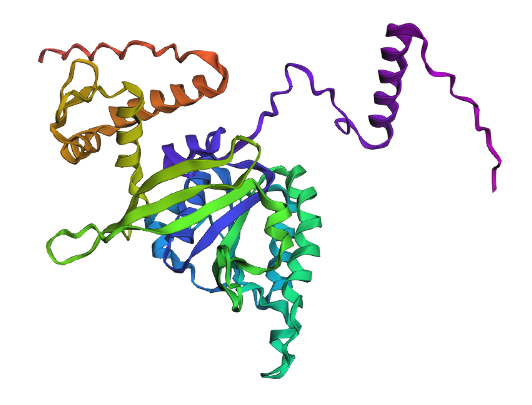
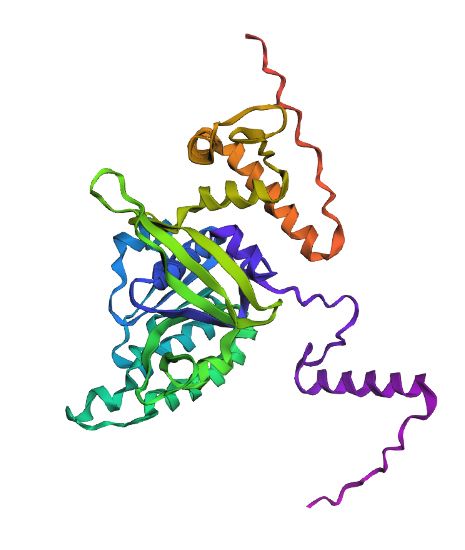
| 
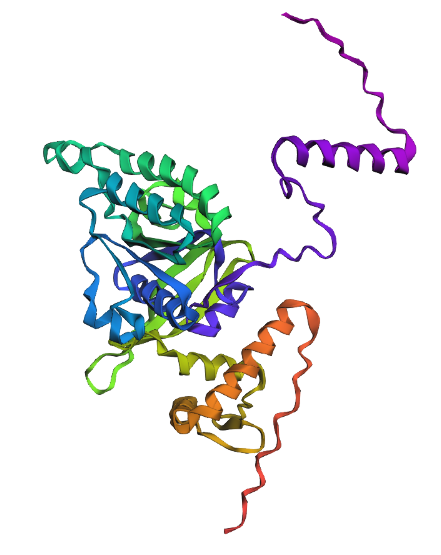
| 
|
|---|
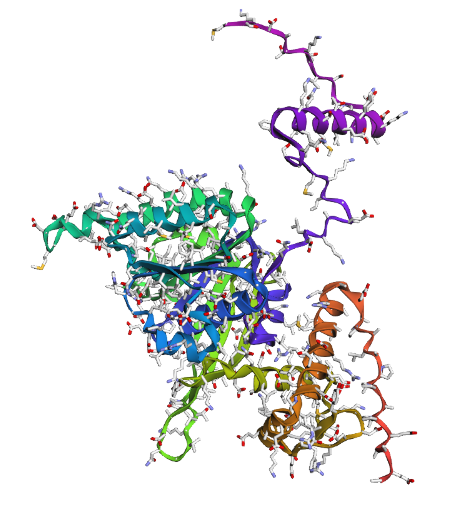
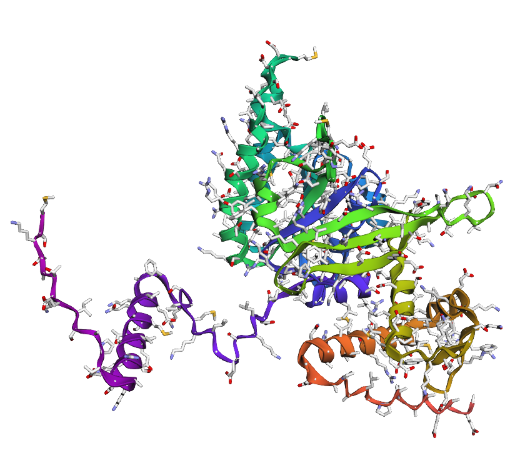
Displays
a) Sidechain
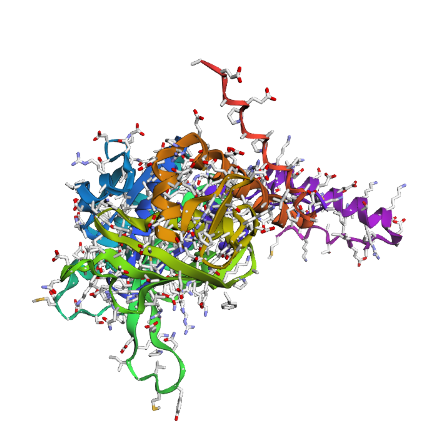
| 
| 
|
|---|
b) Mainchain
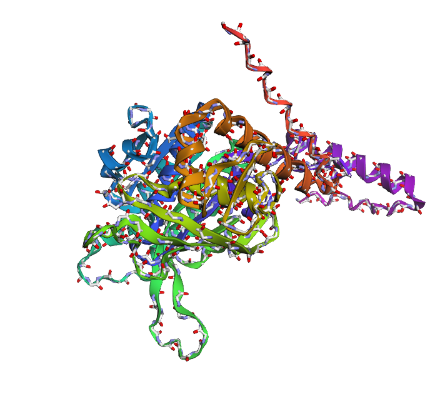
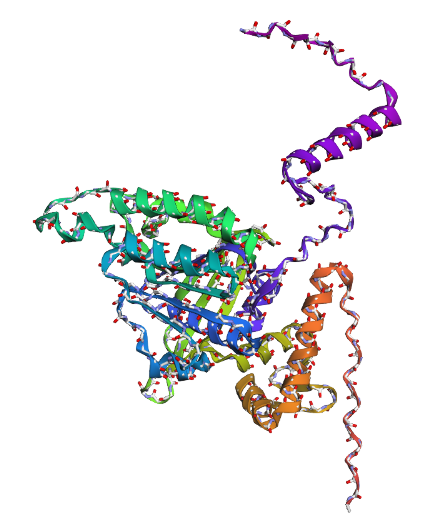
| 
| 
|
|---|
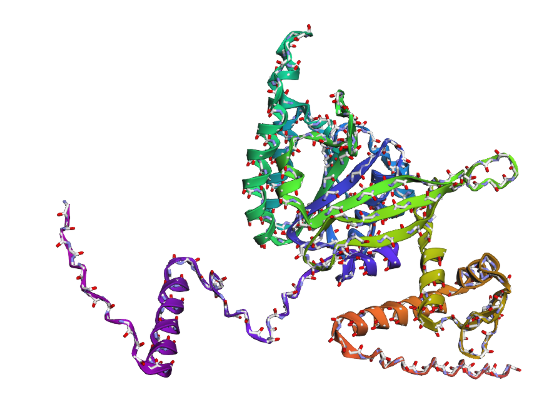
c) Sidechain + Mainchain
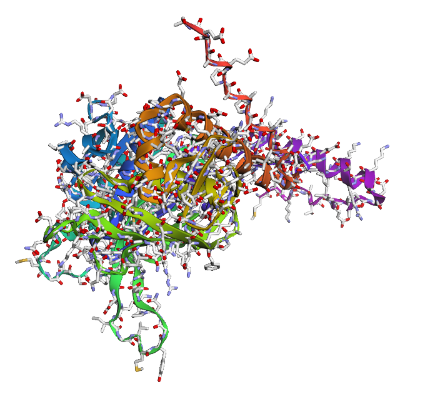
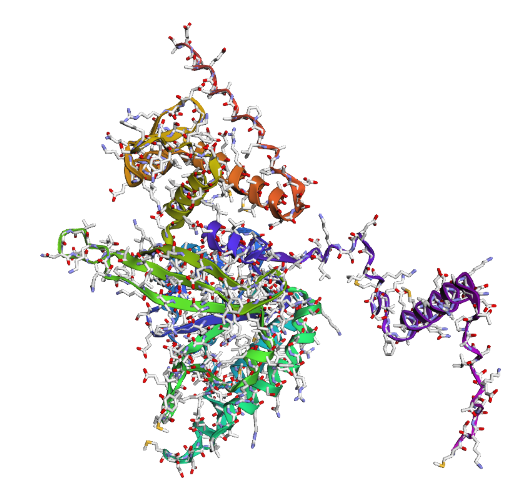
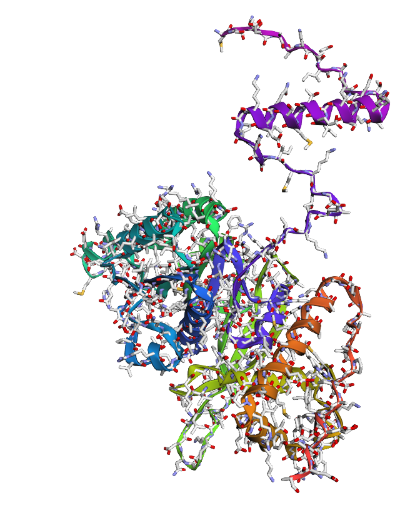
| 
| 
|
|---|
Comparison
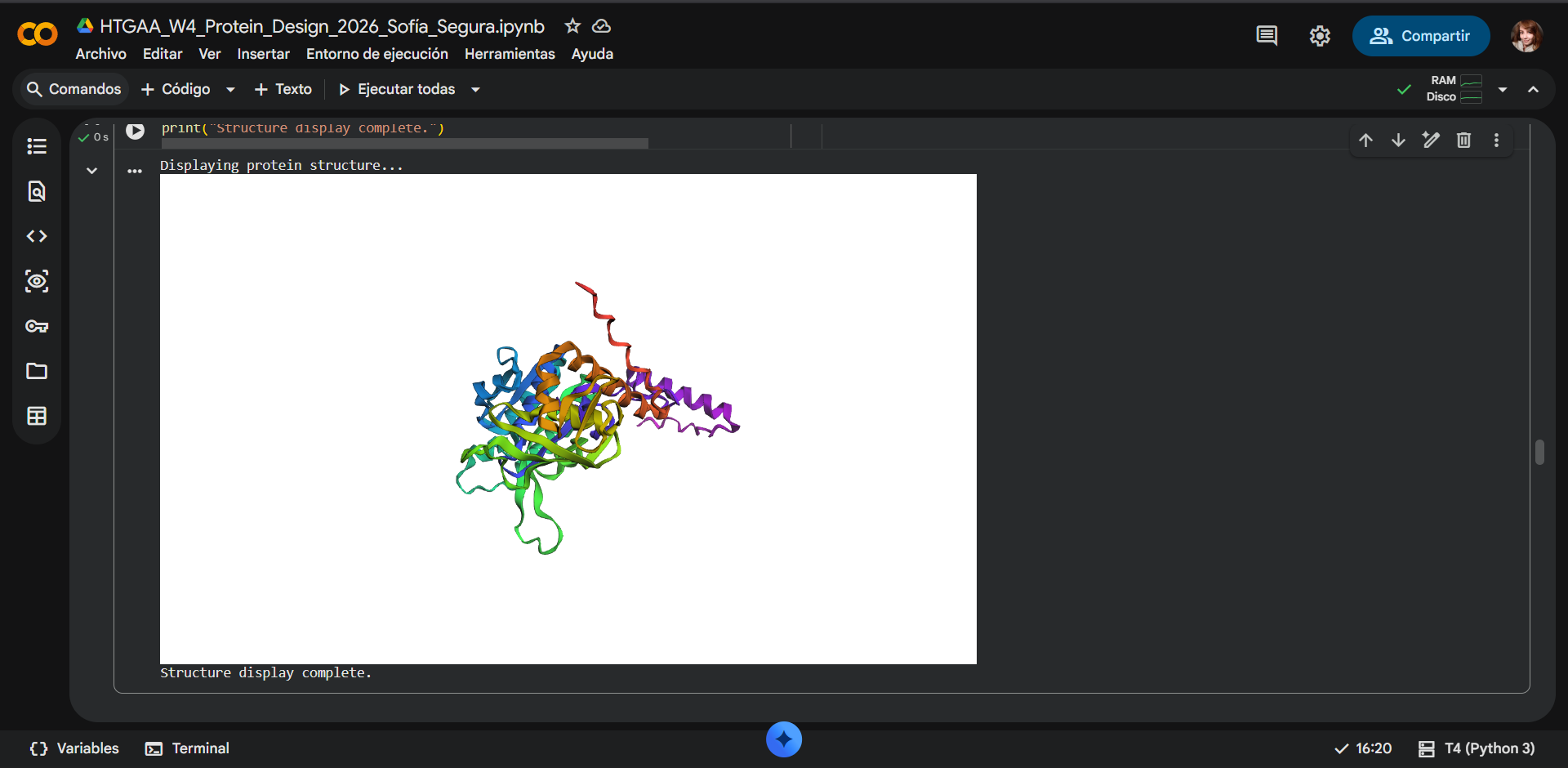
Overall, they do look similar 😃! The structure predicted with ESMFold closely resembles the monomeric fold of the RecA proteins reported in experimental structures and AlphaFold models on UniProt. The predicted structure contains the characteristic α/β core domain and several α-helices typical of RecA family proteins. However, it does not reproduce the circular oligomeric structure observed in some UniProt models (PDB 1XP8), because those structures represent multimeric assemblies composed of several RecA subunits. ESMFold predicts the structure of a single polypeptide chain (monomer), which explains why the predicted structure resembles monomeric crystal structures such as 2ofo.1 rather than the oligomeric filament assemblies (1xp8.1.F).
SWISS-MODEL: SMTL ID : 2ofo.1
Average Model Confidence (QMEANDisCo): 0.77 ± 0.05
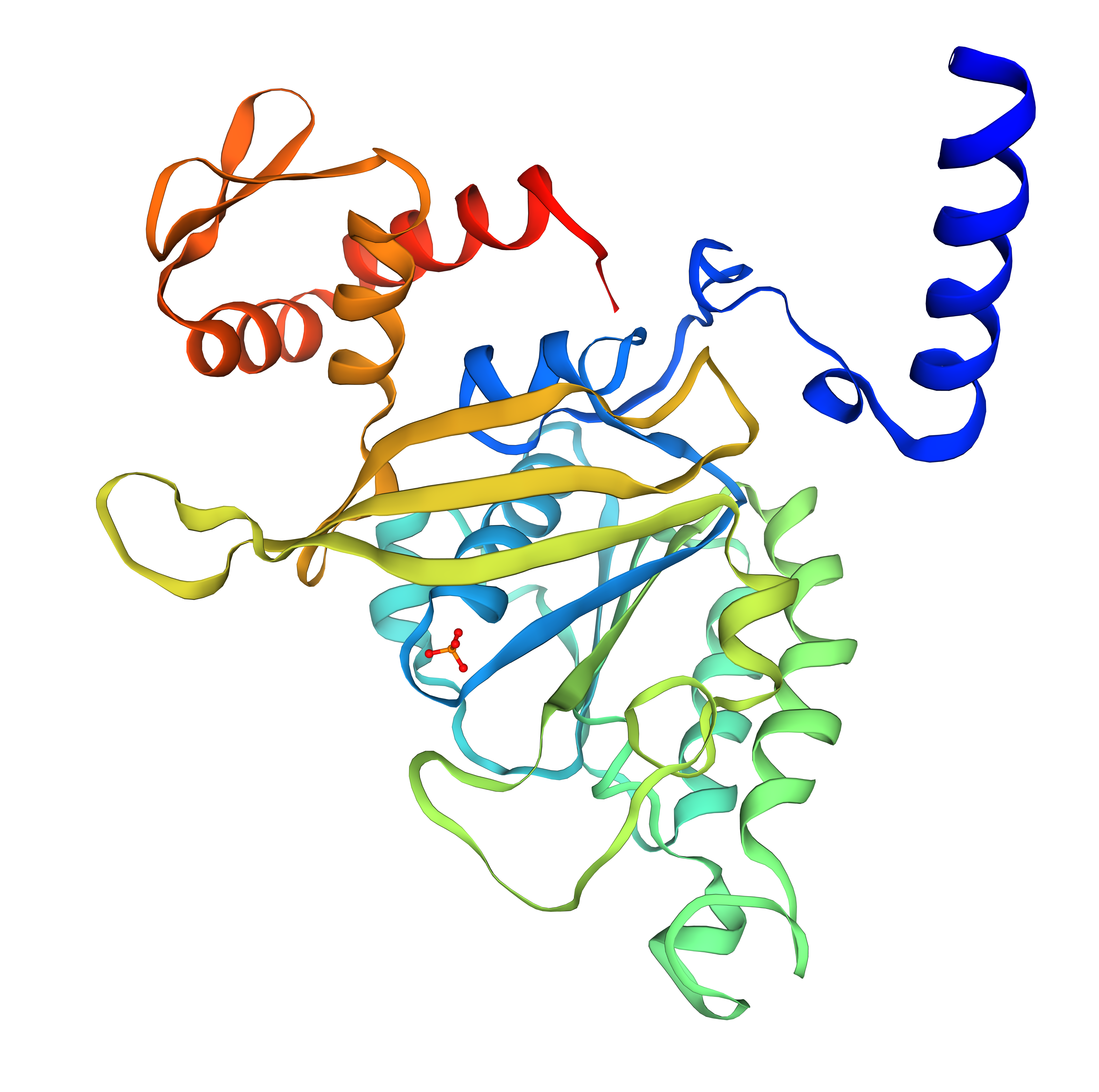

SWISS-MODEL: AlphaFold Model AF-P42443-F1
Average Model Confidence (pLDDT): 88.88
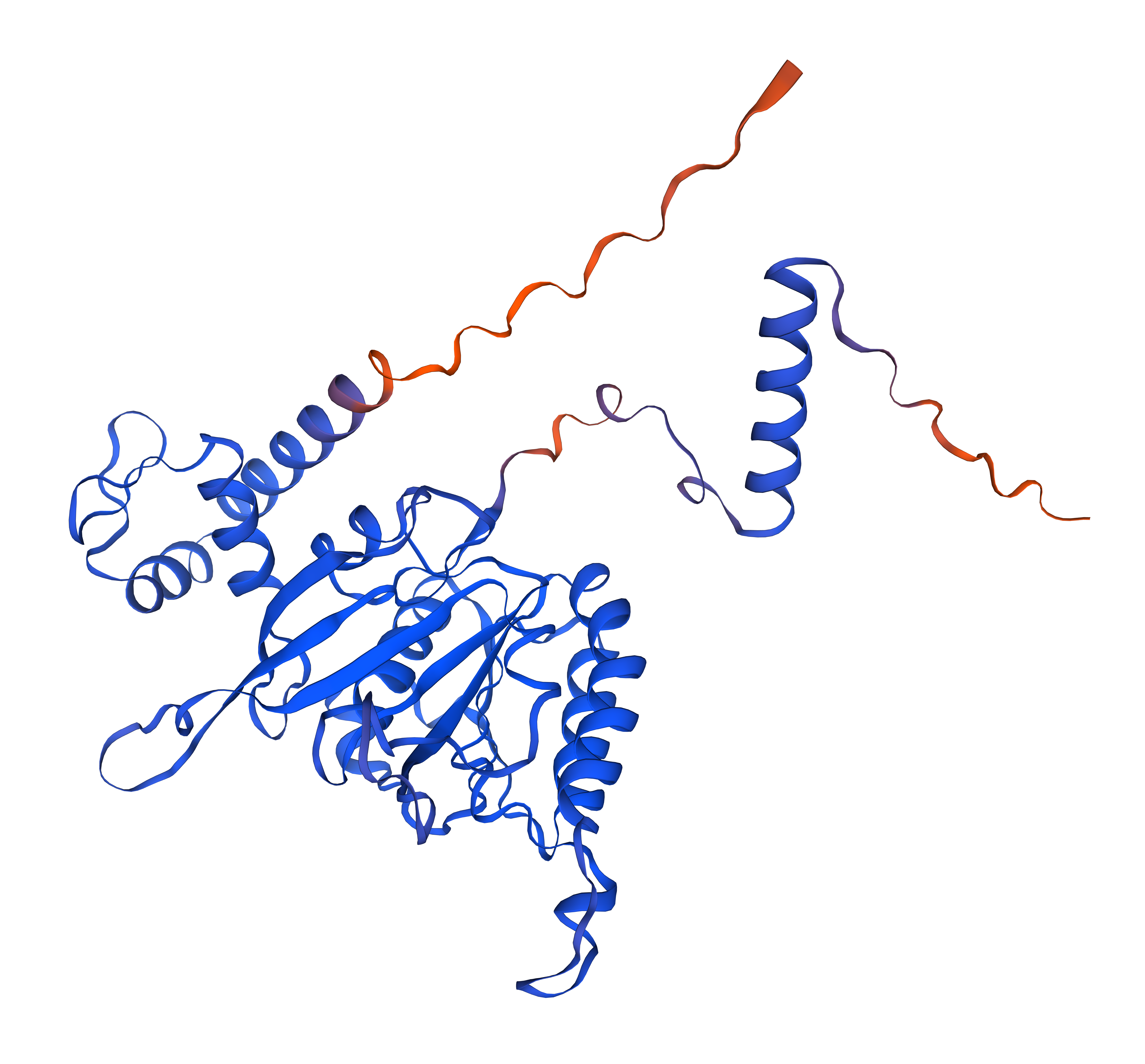
The RecA structure predicted with ESMFold showed a very high confidence score (pLDDT = 94.83) and was compared with previously available structural models, including the SWISS-MODEL homology model based on the crystal structure 2ofo.1 (QMEANDisCo = 0.77 ± 0.05) and the AlphaFold prediction AF-P42443-F1 (average pLDDT = 88.88). All models display the characteristic RecA fold, consisting of a central α/β ATPase domain with a β-sheet core surrounded by multiple α-helices. The arrangement of secondary structural elements, including β-strands, α-helices, and connecting loops, is largely conserved between the three structures. Minor differences are mainly observed in flexible regions such as loops and the C-terminal tail, which are known to exhibit conformational variability. Overall, the high pLDDT value obtained with ESMFold indicates that the predicted structure is highly reliable and consistent with experimentally derived and AI-predicted RecA models.
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
- Input this sequence into ESMFold and compare the predicted structure to your original.
The “New Sequence” generated by ProteinMPNN is not merely a mutated version of the original; it’s a de novo design tailored for the provided 3D protein structure (PDB 1XP8). ProteinMPNN operates on the principle of inverse protein folding, where instead of predicting the 3D structure from a given amino acid sequence, it takes a fixed 3D backbone and designs an amino acid sequence that is predicted to be compatible with and stable within that structure.
| 
|
|---|---|
| 
|
How the new sequence is generated
1. Structural Input: The model uses the atomic coordinates of the protein backbone (from PDB 1XP8) as its primary input.
2. Per-Position Probability: For each amino acid position along the chain, ProteinMPNN evaluates the local structural environment (neighboring residues, local geometry). Based on this context, it predicts a probability distribution over all 20 standard amino acids (and sometimes ‘X’ for ambiguous or masked regions) for that specific position.
3. Sampling: A new amino acid is then sampled from this probability distribution for each position. The sampling_temp parameter (set to 0.1 in our run) influences this sampling: a lower temperature means the model is more likely to pick the highest-probability amino acid, leading to more conservative designs, while a higher temperature introduces more diversity.
4. Novelty and Mutations: The resulting “New Sequence” is thus a sequence that ProteinMPNN predicts will stably adopt the input 3D conformation. Differences between this generated sequence and the “Native Sequence” (from the original PDB) represent designed mutations. These mutations aim to optimize the sequence for the given fold, potentially improving stability or introducing new functions.
Amino acid probability map
Each row on the y-axis represents a different amino acid, and each column on the x-axis corresponds to a specific position in the protein sequence. The color intensity at each cell (intersection of an amino acid and a position) indicates the average probability that ProteinMPNN assigned to that amino acid at that position.
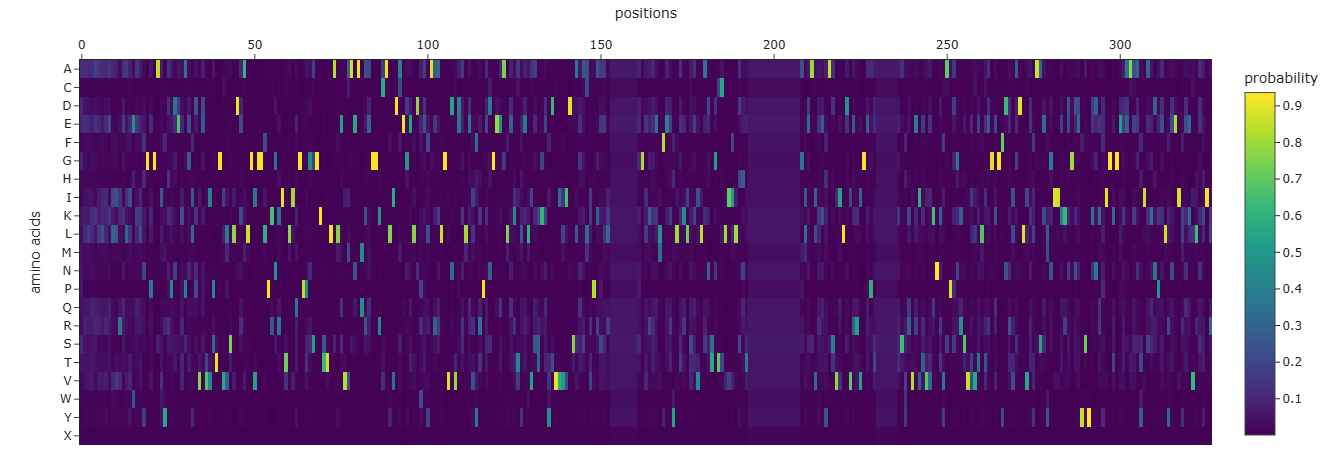
Interpretation
Hotter/Brighter Colors (yellow, white): Indicate a high probability for that specific amino acid at that particular position. This suggests that the model strongly prefers or predicts that amino acid to be structurally compatible at that site. These positions are often critical for the protein’s fold or function, or simply have strong local preferences.
Colder/Darker Colors (blue, purple): Indicate a low probability for that amino acid at that position. The model considers these amino acids unlikely or incompatible with the structural context of that site.
Vertical Stripes of Hot Colors: If a column (a specific position) shows a very bright stripe concentrated on one or a few amino acids, it means that position is highly constrained or conserved. The model has a very strong preference for only a few types of amino acids there.
Horizontal Stripes/Scattered Hot Colors: If a position has several amino acids with moderately high probabilities, it suggests more variability or plasticity at that site. The structure can tolerate different amino acids there.
The “New Sequence” is derived from these probability distributions. The amino acid selected for each position in the new sequence would typically be one of the high-probability amino acids shown in the heatmap for that specific position, especially with a low sampling_temp.
| 
|
|---|
GENERATED SEQUENCE
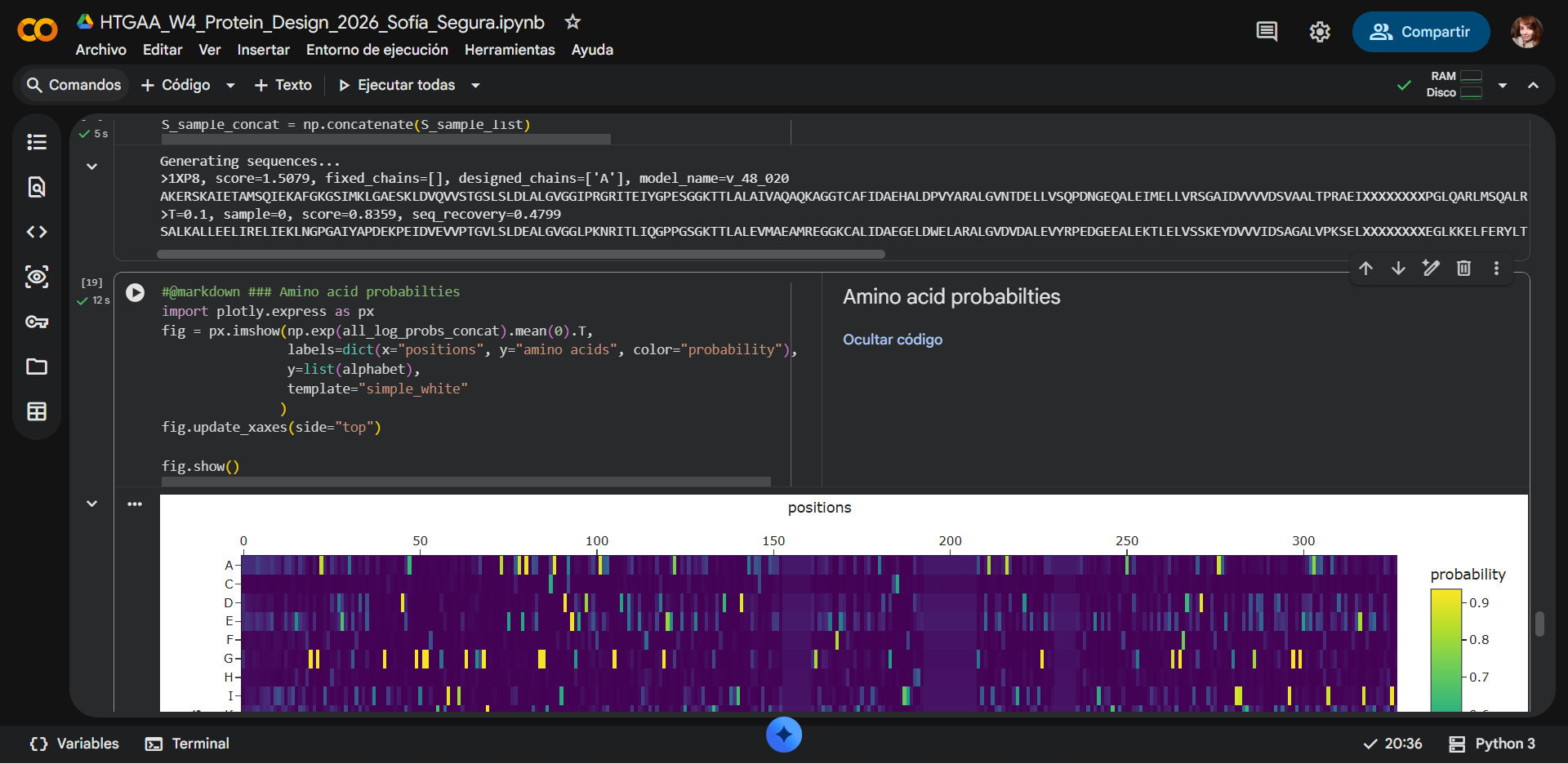
Generating sequences…
1XP8, score=1.5369, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020
AKERSKAIETAMSQIEKAFGKGSIMKLGAESKLDVQVVSTGSLSLDLALGVGGIPRGRITEIYGPESGGKTTLALAIVAQAQKAGGTCAFIDAEHALDPVYARALGVNTDELLVSQPDNGEQALEIMELLVRSGAIDVVVVDSVAALTPRAEIXXXXXXXXPGLQARLMSQALRKLTAILSKTGTAAIFINQVXXXXXXXXXXXXXXXGGRALKFYASVRLDVRKIGQPTXXXXXXVANTVKIKTVKNKVAAPFKEVELALVYGKGFDQLSDLVGLAADMDIIKKAGSFYSYGDERIGQGKEKTIAYIAERPEMEQEIRDRVMAAIR
T=0.1, sample=0, score=0.7948, seq_recovery=0.4497
GELAALLERLARALLWAANGPGAAYAPDERAGLPVEVVPTGVVSLDRALGVGGLPKGRITLIQGPPGSGKTTLALQVVAEAQRAGGKAALIDMEGELDEARAAALGVDVAALERYRPENGEEALEKTLELVSSGEYDVVVIDSLQAAVPKAELXXXXXXXXAGLRAELFARYLARLKEVLAGTGTCLIILHHTXXXXXXXXXXXXXXXGIEAVREAASVILDVRRVGEPEXXXXXXRSWRVEIRVVKNTVAPAGRSVVVTLTEGEGFDRIADLVEEAARLGIIERDGNKYSYKNKFIGEGEKAAAATIAKDPALEEEIRREVLERIR
New Sequence:GELAALLERLARALLWAANGPGAAYAPDERAGLPVEVVPTGVVSLDRALGVGGLPKGRITLIQGPPGSGKTTLALQVVAEAQRAGGKAALIDMEGELDEARAAALGVDVAALERYRPENGEEALEKTLELVSSGEYDVVVIDSLQAAVPKAELXXXXXXXXAGLRAELFARYLARLKEVLAGTGTCLIILHHTXXXXXXXXXXXXXXXGIEAVREAASVILDVRRVGEPEXXXXXXRSWRVEIRVVKNTVAPAGRSVVVTLTEGEGFDRIADLVEEAARLGIIERDGNKYSYKNKFIGEGEKAAAATIAKDPALEEEIRREVLERIR
Part D. Group Brainstorm on Bacteriophage Engineering
Assignees for the following sections
| MIT/Harvard students | Optional |
| Committed Listeners | Required |
- Find a group of ~3–4 students
- Read through the Phage Reading material listed under “Reading & Resources” below.
- Review the Bacteriophage Final Project Goals for engineering the L Protein:
- Increased stability (easiest)
- Higher titers (medium)
- Higher toxicity of lysis protein (hard)
- Brainstorm Session
- Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
- Write a 1-page proposal (bullet points or short paragraphs) describing:
- Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
- Why do you think those tools might help solve your chosen sub-problem?
- Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
- Include a schematic of your pipeline.
- This resource may be useful: HTGAA Protein Engineering Tools
- Each individually put your plan on your HTGAA website
- Include your group’s short plan for engineering a bacteriophage
Resources
- USDA FoodData Central. Beef, raw — nutritional composition. https://fdc.nal.usda.gov/
- IUPAC Gold Book. Avogadro constant. https://goldbook.iupac.org/
- Nelson, D. L., & Cox, M. M. (2021). Lehninger Principles of Biochemistry (8th ed.). W.H. Freeman.
- Hall, J. E. (2020). Guyton and Hall Textbook of Medical Physiology (14th ed.). Elsevier.
- Alberts, B. et al. (2022). Molecular Biology of the Cell (7th ed.). Garland Science.
- Crick, F. H. C. (1968). The origin of the genetic code. Journal of Molecular Biology, 38(3), 367–379.
- Liu, C. C., & Schultz, P. G. (2010). Adding new chemistries to the genetic code. Annual Review of Biochemistry, 79, 413–444.
- Lobanov, A. V., et al. (2009). Selenocysteine: The 21st amino acid. Journal of Biological Chemistry, 284(44), 28532–28536.
- Weber, A.L., Miller, S.L. Reasons for the occurrence of the twenty coded protein amino acids. J Mol Evol 17, 273–284 (1981). https://doi.org/10.1007/BF01795749
- Doig, A.J. (2017), Frozen, but no accident – why the 20 standard amino acids were selected. FEBS J, 284: 1296-1305. https://doi.org/10.1111/febs.13982
- Young, Travis S. & Schultz, P. G. schultz@scripps.edu. (April, 2010). Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon. Journal of Biological Chemistry, Volume 285, Issue 15, 11039 - 11044. https://doi.org/10.1074/jbc.R109.091306
- Atkins, J., Gesteland, R. The twenty-first amino acid. Nature 407, 463–464 (2000). https://doi.org/10.1038/35035189
- Miles, S. A., Nillama, J. A., & Hunter, L. (2023). Tinker, Tailor, Soldier, Spy: The Diverse Roles That Fluorine Can Play within Amino Acid Side Chains. Molecules, 28(17), 6192. https://doi.org/10.3390/molecules28176192
- Lee, Hyang-Yeol; Lee, Kyung-Hoon; Al-Hashimi, Hashim M.; Marsh, E. Neil G. . (2006). Modulating Protein Structure with Fluorous Amino Acids: Increased Stability and Native-like Structure Conferred on a 4-Helix Bundle Protein by Hexafluoroleucine. Journal of the American Chemical Society, 128(1), 337–343. doi:10.1021/ja0563410
- Buer, B.C. and Marsh, E.N.G. (2012), Fluorine: A new element in protein design. Protein Science, 21: 453-462. https://doi.org/10.1002/pro.2030
- Benjamin C. Buer; E. Neil G. Marsh. (2012). Fluorine: A new element in protein design. , 21(4), 453–462. doi:10.1002/pro.2030
- Buer, B.C., Meagher, J.L., Stuckey, J.A. & Marsh, E.N.G. (2012). Structural basis for the enhanced stability of highly fluorinated proteins, Proc. Natl. Acad. Sci. U.S.A. 109 (13) 4810-4815. https://doi.org/10.1073/pnas.1120112109
- Costantino, A., Pham, L.B.T., Barbieri, L., Calderone, V., Ben-Nissan, G., Sharon, M., et al. Controlling the incorporation of fluorinated amino acids in human cells and its structural impact. Protein Science. 2024; 33(3):e4910. https://doi.org/10.1002/pro.4910
- Zhang, Huimin; Song, Yanling; Zou, Yuan; Ge, Yun; An, Yuan; Ma, Yanli; Zhu, Zhi; Yang, Chaoyong James . (2014). A diazirine-based photoaffinity probe for facile and efficient aptamer–protein covalent conjugation. Chemical Communications, 50(38), 4891–. doi:10.1039/c4cc01528b
- S. Ravindra, C. P. Irfana Jesin, A. Shabashini, G. C. Nandi. (2021). Recent Advances in the Preparations and Synthetic Applications of Oxaziridines and Diaziridines. Catal. 363, 1756. https://doi.org/10.1002/adsc.202001372
- Jian Fan, Qingyao Shu, Yi-Ming Li b, Jing Shi. (2022). Efficient synthesis of terminal-diazirine-based histone peptide probes. Tetrahedron Letters Volume 102, 153878. https://doi.org/10.1016/j.tetlet.2022.153878
- Famiano, M.A., Boyd, R.N., Kajino, T. et al. Amino Acid Chiral Selection Via Weak Interactions in Stellar Environments: Implications for the Origin of Life. Sci Rep 8, 8833 (2018). https://doi.org/10.1038/s41598-018-27110-z
- Ronald Breslow, The origin of homochirality in amino acids and sugars on prebiotic earth. Tetrahedron Letters, Volume 52, Issue 32, 2011, Pages 4228-4232, ISSN 0040-4039. https://doi.org/10.1016/j.tetlet.2011.06.002
- Engel, M. H.; Macko, S. A. . (1997). . Nature, 389(6648), 265–268. https://doi.org/10.1038/38460
- M.H Engel; S.A Macko. (2001). The stereochemistry of amino acids in the Murchison meteorite. , 106(1-2), 0–45. https://doi.org/10.1016/s0301-9268(00)00123-6
- Glavin, D.P., Elsila, J.E., McLain, H.L., Aponte, J.C., Parker, E.T., Dworkin, J.P., Hill, D.H., Connolly, H.C., Jr. and Lauretta, D.S. (2021), Extraterrestrial amino acids and L-enantiomeric excesses in the CM2 carbonaceous chondrites Aguas Zarcas and Murchison. Meteorit Planet Sci, 56: 148-173. https://doi.org/10.1111/maps.13451
- OpenAI. (2026). ChatGPT (GPT-5.2) [Large language model]. https://chat.openai.com/
- GeminiAI. (2026). Gemini (Gemini 2.5 Flash) [Large language model].
Tools
- HTGAA Protein Engineering Tools spreadsheet
- NGLViewer: NGL Viewer is a collection of tools for web-based molecular graphics. WebGL is employed to display molecules like proteins and DNA/RNA with a variety of representations.
- PyMOL(https://pymol.org/edu/?q=educational): PyMOL is a user-sponsored molecular visualization system on an open-source foundation, maintained and distributed by Schrödinger.
- Practical PyMOL for Beginners
- Video Tutorials: Video 1 Video2 (and tons more… just search “PyMOL tutorial” in youtube).
- Cheat Sheet
- Advanced Cheat Sheet
- Chimera: A highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles.
- Chimera Tutorials
- Video Tutorials: Video 1 Video 2 (and tons more… just search “Chimera tutorial” in youtube).
- VMD: A molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting
- VMD Tutorials
- Video Tutorials: Video 1 Video 2 (and tons more… you know the drill)
- https://search.foldseek.com/search
Phage Reading
- Identification MS2 lysis protein dependency on DnaJ
- Mutational analysis of the MS2 lysis protein L
- Characterization of the MS2 lysis protein properties
- Phage therapy: From biological mechanisms to future directions
- Phage Therapy: Past, Present and Future
- Generative design of novel bacteriophages with genome language models