HTGAA - Week 5: Protein Design Part II
My Homework
WEEK 5 - BIOINFORMATICS PART II
This week we learn how cutting-edge AI and protein language models are used to design functional proteins and peptides “in silico”.
Lecture (Tues, Mar 3)
Protein Design Part II
Gabriele Corso ▶️Recording
Pranam Chatterjee ▶️Recording
Recitation (Wed, Mar 4)
Phage Therapy
(▶️Recording | 💻Slides)
Suvin Sundararajan, Dominika Wawrzyniak
Part 1: SOD1 Binder Peptide Design (From Pranam)
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Challenge:
- Design short peptides that bind mutant SOD1.
- Then decide which ones are worth advancing toward therapy.
Available models:
- PepMLM: target sequence-conditioned peptide generation via masked language modeling
- PeptiVerse: therapeutic property prediction
- moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
A: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
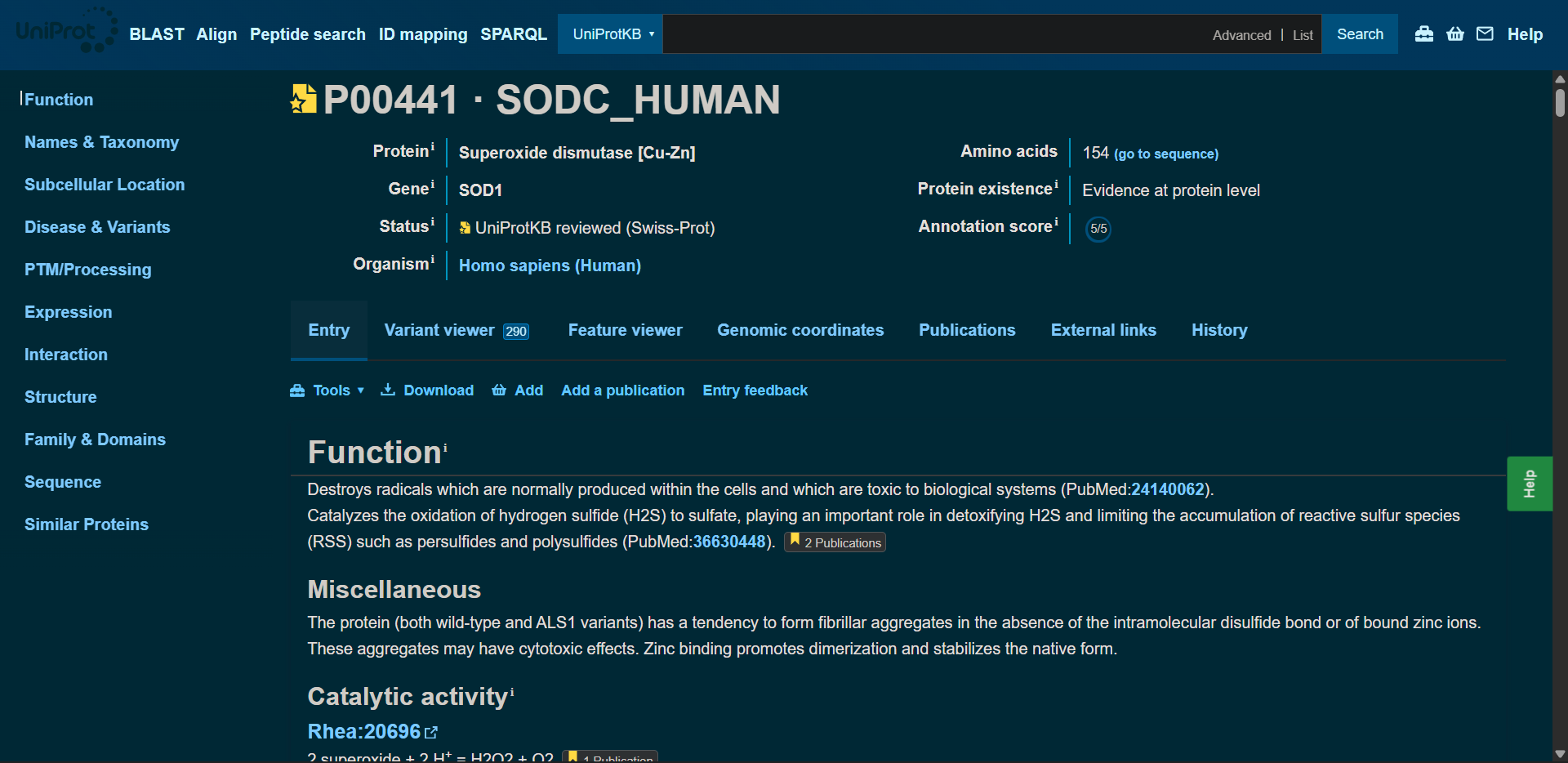
UniProt - SOD1 (P00441) sequence
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutation A → V
Mutated sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
- To the generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
PepMLM Colab used:
| 
|
|---|---|
| 
|
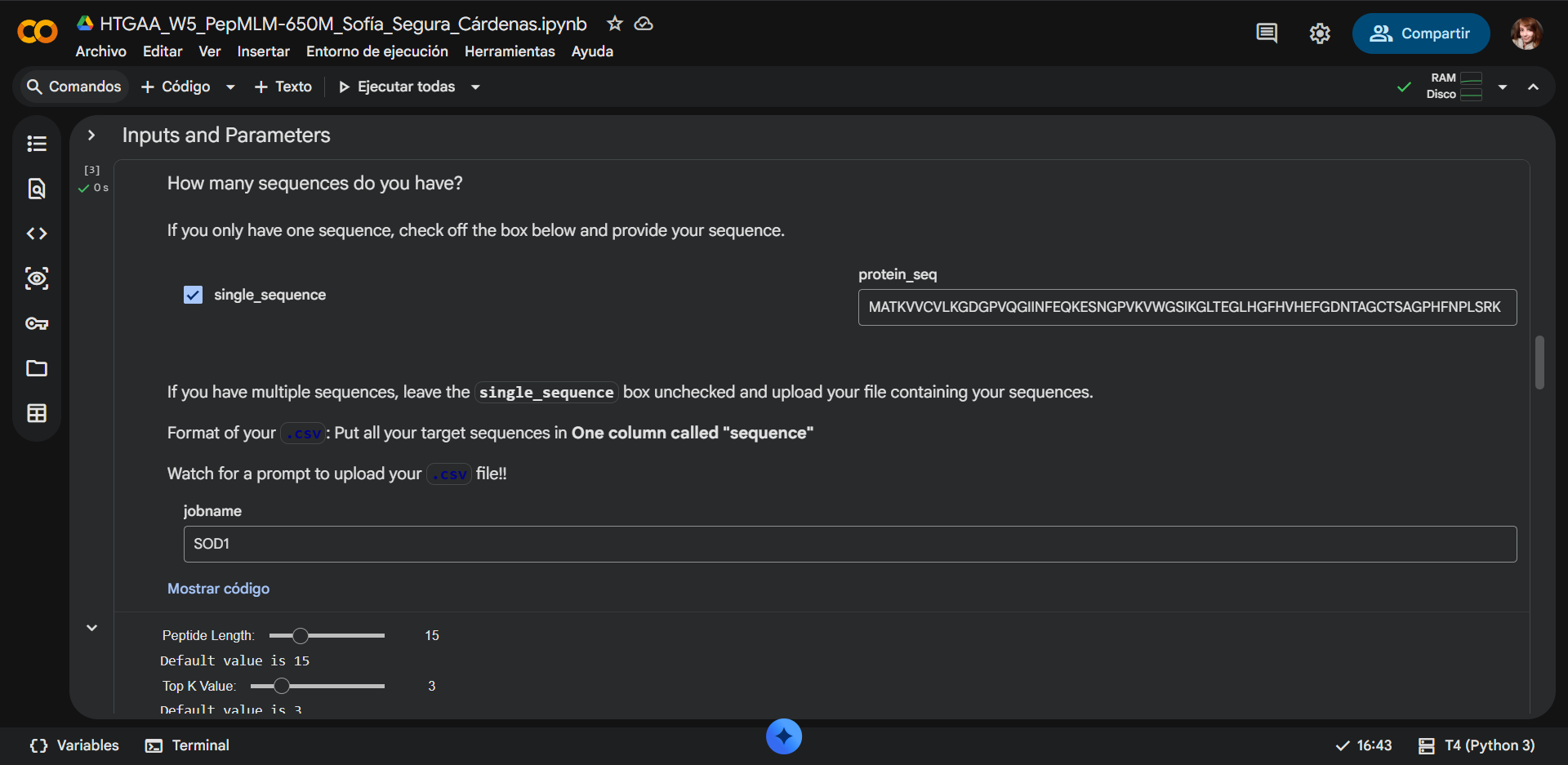
The default values where changed as follows:
- Updated Peptide Length: 12
- Updated Top K Value: 3
- Updated num_binders: 16
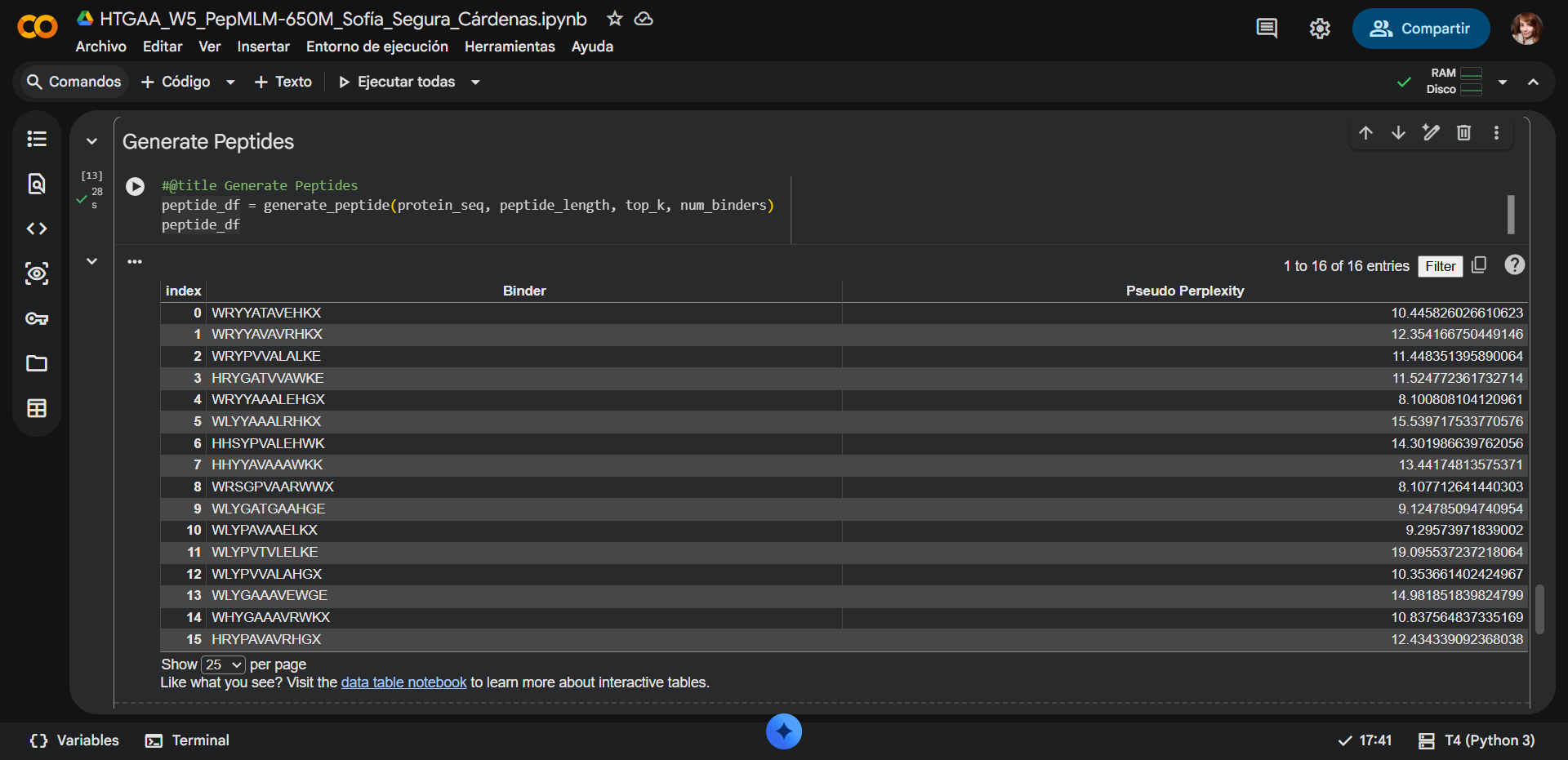
The list of the 16 generated peptides and the 17 one added for control:
PepMLM assigns a pseudo-perplexity score to each generated peptide, reflecting the model’s confidence in the sequence given the target protein context. Lower pseudo-perplexity values indicate higher model confidence and a better fit to the learned sequence distribution of potential binders.
| Index | Binder | Pseudo Perplexity | X |
|---|---|---|---|
| 0 | WRYYATAVEHKX | 10.445826 | Yes |
| 1 | WRYYAVAVRHKX | 12.354167 | Yes |
| 2 | WRYPVVALALKE | 11.448351 | No |
| 3 | HRYGATVVAWKE | 11.524772 | No |
| 4 | WRYYAAALEHGX | 8.100808 | Yes |
| 5 | WLYYAAALRHKX | 15.539718 | Yes |
| 6 | HHSYPVALEHWK | 14.301987 | No |
| 7 | HHYYAVAAAWKK | 13.441748 | No |
| 8 | WRSGPVAARWWX | 8.107713 | Yes |
| 9 | WLYGATGAAHGE | 9.124785 | No |
| 10 | WLYPAVAAELKX | 9.295740 | Yes |
| 11 | WLYPVTVLELKE | 19.095537 | No |
| 12 | WLYPVVALAHGX | 10.353661 | Yes |
| 13 | WLYGAAAVEWGE | 14.981852 | No |
| 14 | WHYGAAAVRWKX | 10.837565 | Yes |
| 15 | HRYPAVAVRHGX | 12.434339 | Yes |
| 16 | FLYRWLPSRRGG | N/A | No |
Several generated peptides contain the residue X, which represents an ambiguous or unknown amino acid in protein sequence notation. In peptide design workflows, X typically appears when the model has uncertainty about the most probable residue at that position. Because X cannot be synthesized or interpreted structurally, these peptides are generally considered lower-confidence candidates for downstream therapeutic design and may be deprioritized in later filtering steps.
Observed sequence pattern
Many of the generated peptides begin with W, H, or the motif WR. Examples include sequences such as WRYY…, WLY…, and HRY…. This pattern suggests that PepMLM may have identified an aromatic and positively charged motif favorable for interaction with SOD1.
A possible explanation is related to the chemical properties of these residues:
- W (Tryptophan) can participate in hydrophobic and aromatic interactions, which often stabilize protein–peptide binding interfaces.
- R, H, and K (Arginine, Histidine, Lysine) are positively charged residues that can contribute to electrostatic interactions with negatively charged regions on the protein surface.
Together, these features may help promote stable binding between the designed peptides and the mutant SOD1 protein.
Selection of the four best candidate peptides
To select candidates for further evaluation, peptides were prioritized based on:
- Low pseudo-perplexity scores (higher model confidence)
- Absence of ambiguous residues (X)
- Reasonable sequence composition for peptide stability
| Peptide | Pseudo Perplexity | Justification |
|---|---|---|
| WLYGATGAAHGE | 9.1248 | Lowest perplexity among sequences without ambiguous residues; strong model confidence. |
| WRYPVVALALKE | 11.4484 | Moderate perplexity and no ambiguous residues; hydrophobic core may favor binding. |
| HRYGATVVAWKE | 11.5248 | Balanced composition with aromatic and hydrophobic residues that may stabilize interactions. |
| HHSYPVALEHWK | 14.3020 | Slightly higher perplexity but still valid; contains aromatic and charged residues that could support binding. |
| FLYRWLPSRRGG | N/A | Known SOD1-binding peptide used as a benchmark |
B: Evaluate Binders with AlphaFold3
Scoring peptides
- Navigate to the AlphaFold Server: alphafoldserver.com
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
- Describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
All AlphaFold predictions were run using a fixed random seed [100] to ensure reproducibility across peptide–protein complex predictions.
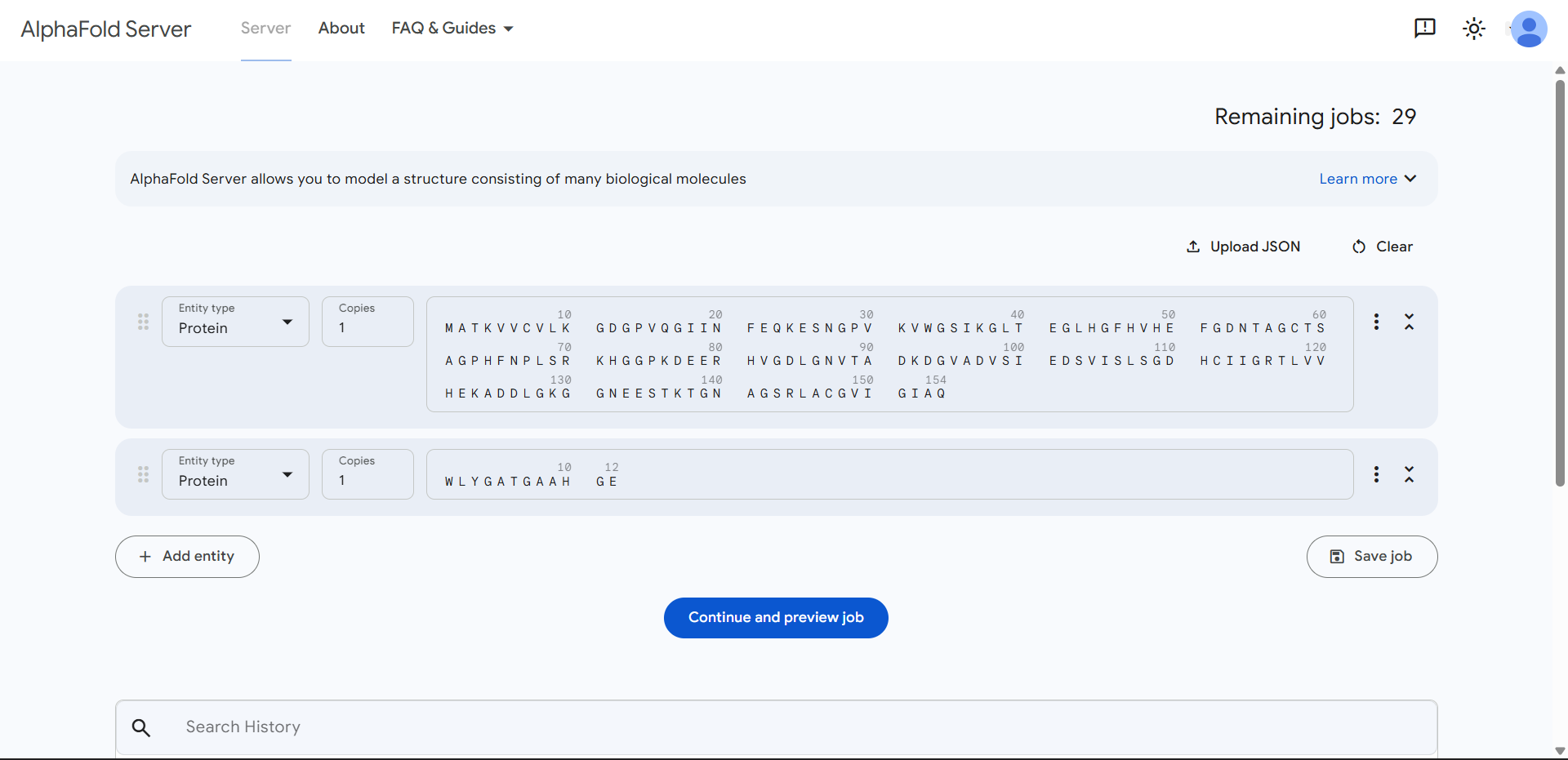
| Run 1: WLYGATGAAHGE | Parameters |
|---|---|
| 
|
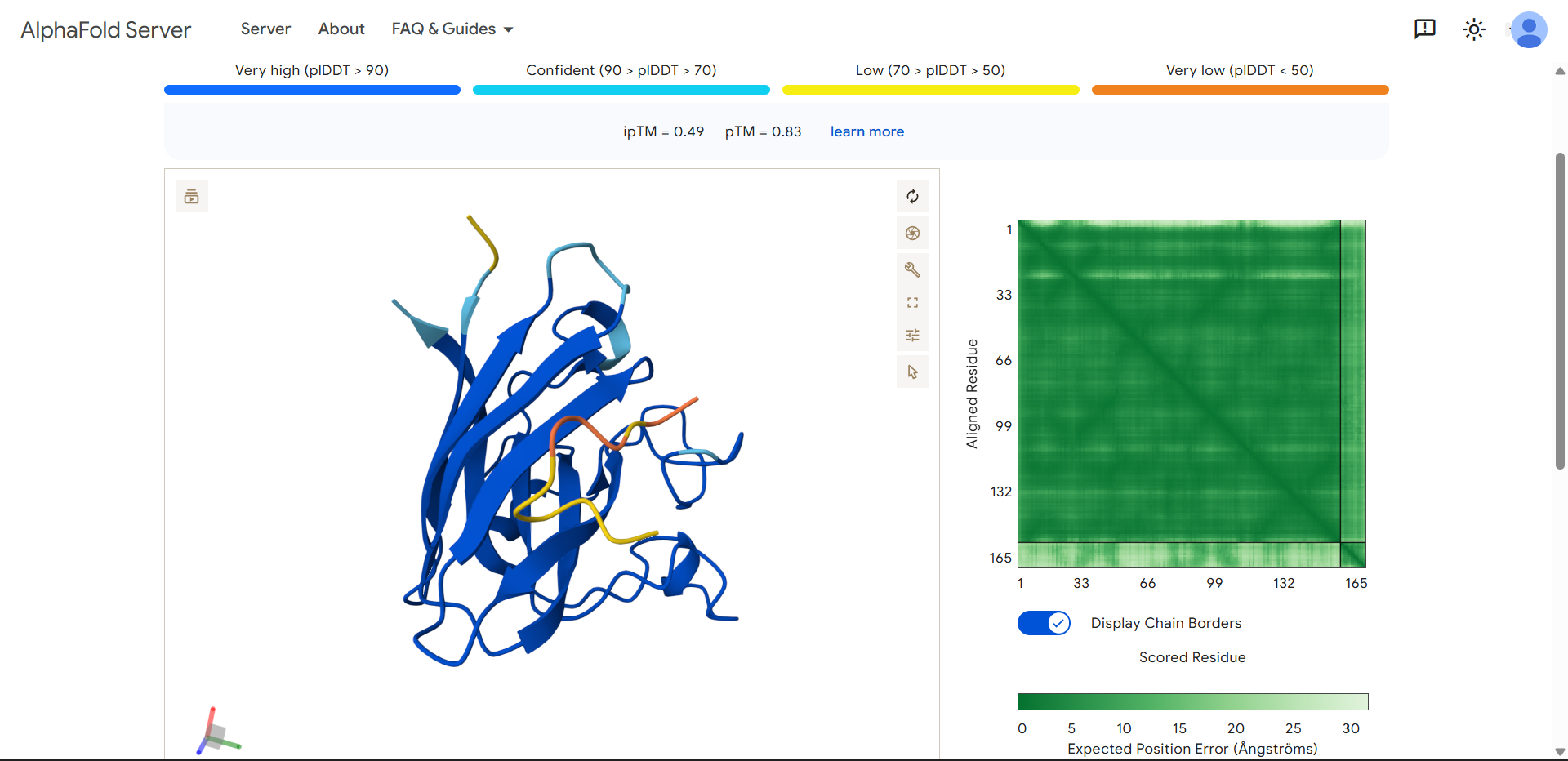
| Run 1 | (ipTM=0.49, pTM=0.83) |
|---|---|
| 
|
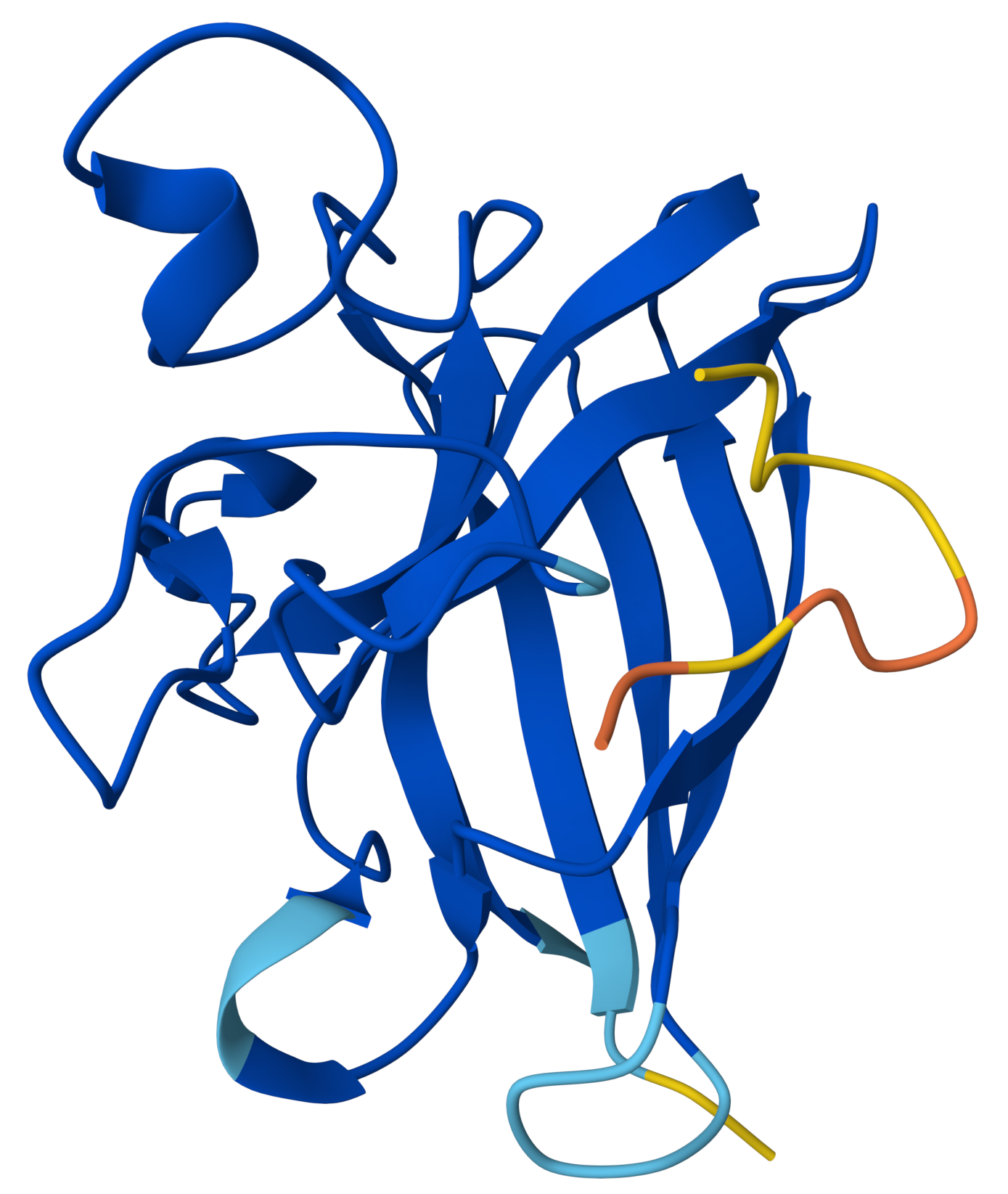
In the structure:
- SOD1 appears dark blue, meaning the protein structure is predicted with very high confidence.
- The peptide is yellow/orange, meaning low confidence in its position and structure.
This usually indicates that AlphaFold is uncertain about the peptide’s binding pose, which is consistent with your ipTM = 0.49.
The peptide
WLYGATGAAHGEproduced an ipTM score of 0.49, indicating very low confidence in the predicted protein–peptide interaction. The overall structure of SOD1 was predicted with high confidence (dark blue pLDDT values), while the peptide displayed lower confidence scores (yellow/orange). Structural inspection shows the peptide positioned along the surface of the SOD1 β-barrel, rather than binding near the N-terminal region where the A4V mutation is located. The low pLDDT values suggest that the peptide adopts a flexible or weakly defined binding conformation, consistent with a surface-associated interaction rather than a tightly bound interface.
Does it localize near the N-terminus where A4V sits?
No. The peptide does not appear to bind near the N-terminal region where the A4V mutation is located. Instead, it is positioned further along the side of the protein.
Does it engage the β-barrel region or approach the dimer interface?
Yes. The peptide is located along the surface of the SOD1 β-barrel, which is the central structural feature of the protein composed of several β-strands (the arrow-shaped ribbons in the structure). This suggests a surface interaction with the β-barrel region.
No. The model shows only a single SOD1 monomer, so the dimer interface is not present in this prediction. Therefore, the peptide cannot be interacting with the dimer interface in this model.
Does it appear surface-bound or partially buried?
It appears surface-associated but weakly constrained. The peptide is positioned near the surface of the protein, but the yellow/orange coloring indicates low structural confidence, meaning AlphaFold is not strongly confident about the exact binding pose. This suggests that the peptide may transiently interact with the protein surface rather than forming a stable, well-defined interface.
| Run 2: WRYPVVALALKE | Parameters |
|---|---|
| 
|
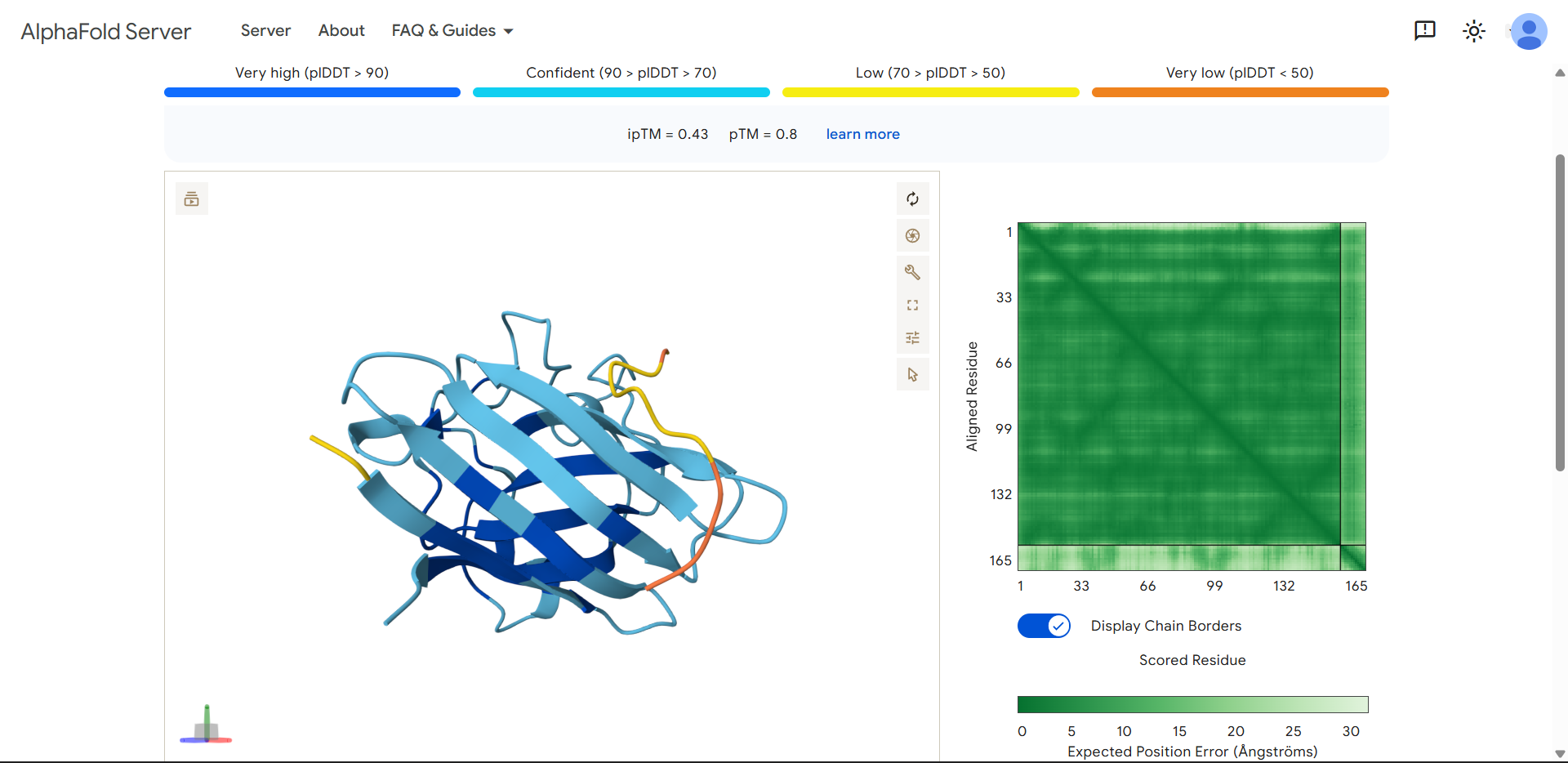
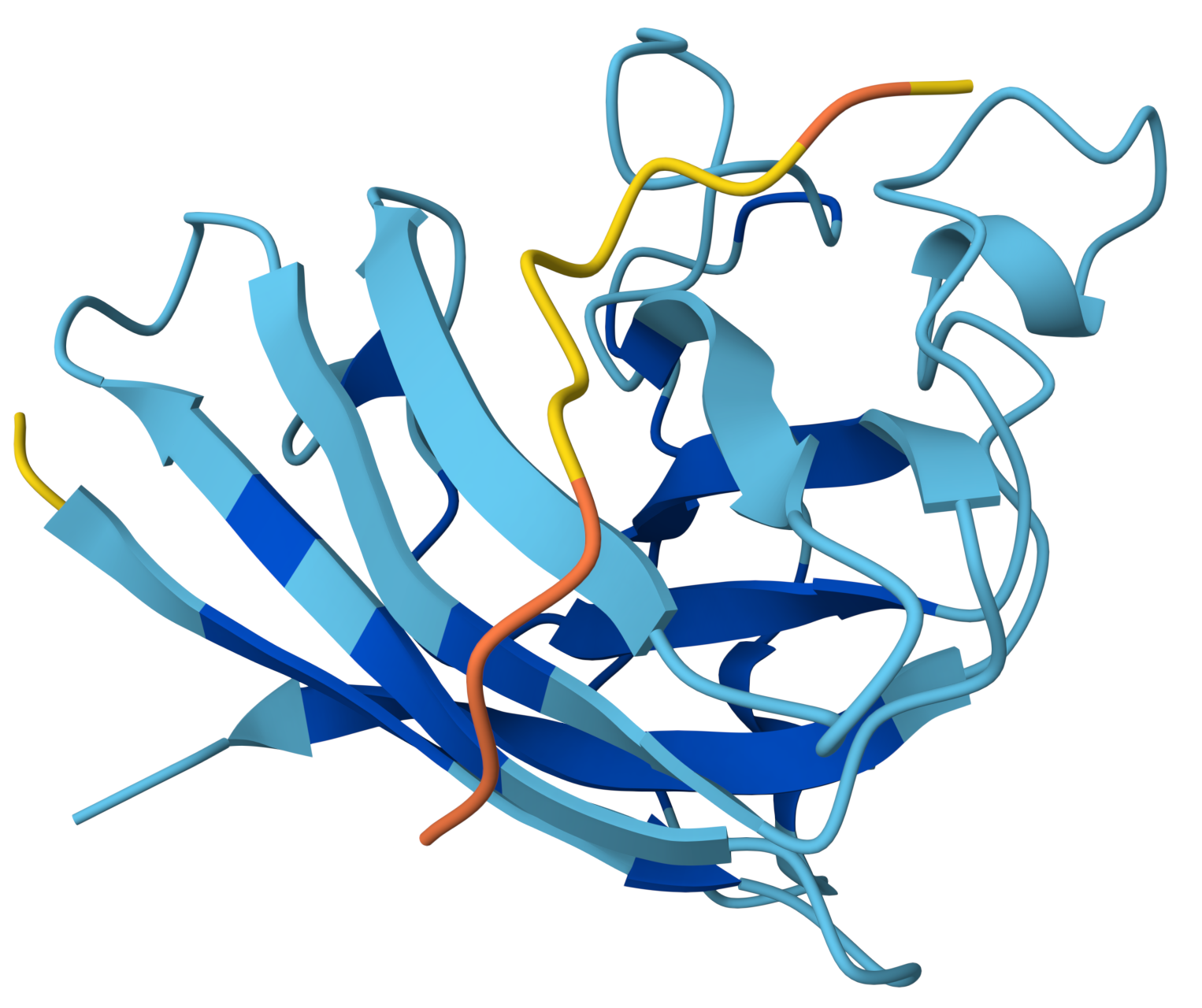
| Run 2 | (ipTM=0.43, pTM=0.80) |
|---|---|
| 
|
Compared to the first peptide:
- ipTM decreased slightly (0.49 → 0.43) → weaker predicted interaction
- pTM also decreased (0.83 → 0.80) → more structural perturbation in SOD1
Important observation: protein color changes
The protein is no longer uniformly dark blue. This suggests:
- Local decreases in pLDDT
- Possible structural perturbations induced by the peptide
The peptide may be destabilizing local regions of SOD1 or AlphaFold is uncertain about the interface, propagating uncertainty into nearby residues. A peptide can appear to interact more broadly but still produce lower confidence, indicating a less stable or more disruptive interaction.
The peptide
WRYPVVALALKEproduced an ipTM score of 0.43, indicating moderate but still low confidence in the predicted protein–peptide interface. The peptide appears to align along the surface of the β-barrel, forming broader contact with the protein compared to the first design. However, it does not localize near the N-terminal region where the A4V mutation resides, and no interaction with the dimer interface can be assessed. The peptide shows partial structural definition, with a central region of moderate confidence and flexible termini. Notably, the SOD1 structure exhibits localized decreases in confidence, suggesting possible structural perturbation or uncertainty induced by the peptide. Overall, the interaction appears surface-bound and weakly defined, without a clear binding pocket or stable interface.
Does it localize near the N-terminus where A4V sits?
No, not clearly. The peptide is positioned along the side of the β-barrel, not near the top region where the N-terminus (and A4V mutation) is located. Therefore, it does not appear to target the mutation site directly.
Does it engage the β-barrel region or approach the dimer interface?
Yes, more convincingly than Run 1. The peptide runs along the surface of the β-sheets, appearing to align with the β-barrel architecture. This suggests a surface-guided interaction, possibly stabilized by:
- hydrophobic residues (V, L, A)
- aromatic residue (W, Y)
However, it still does not insert into a defined binding pocket.
No. Again, only a monomer is modeled, so the dimer interface is absent. No conclusions can be drawn about dimer stabilization.
Does it appear surface-bound or partially buried?
Partially surface-bound, partially flexible. The central region of the peptide (yellow) suggests moderate confidence (~70 pLDDT). The ends (orange) remain highly flexible/unresolved. This indicates:
- Some transient or weak interaction with the protein surface
- No stable, well-defined binding conformation
| Run 3: HRYGATVVAWKE | Parameters |
|---|---|
| 
|
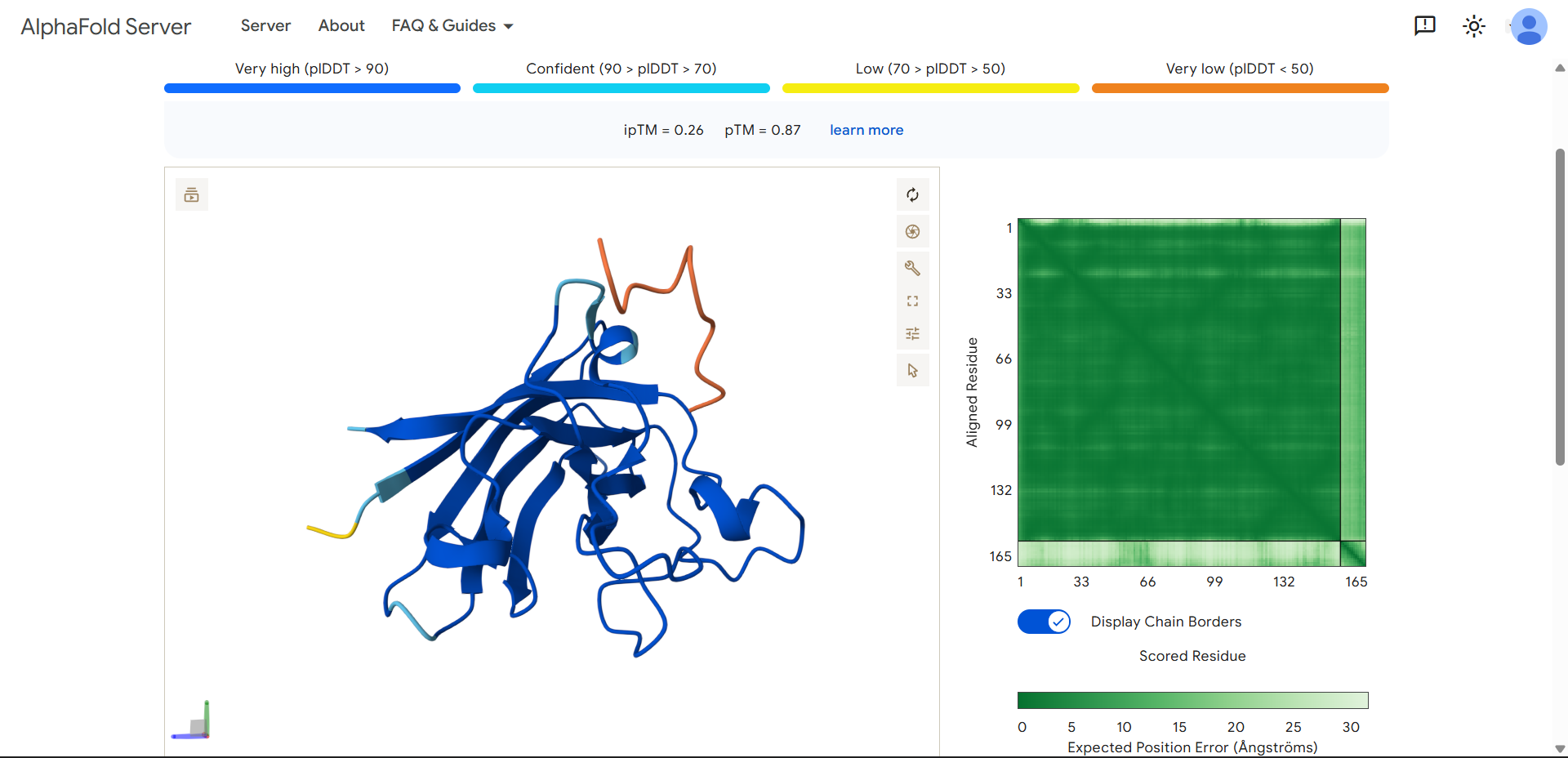
| Run 3 | (ipTM=0.26, pTM=0.87) |
|---|---|
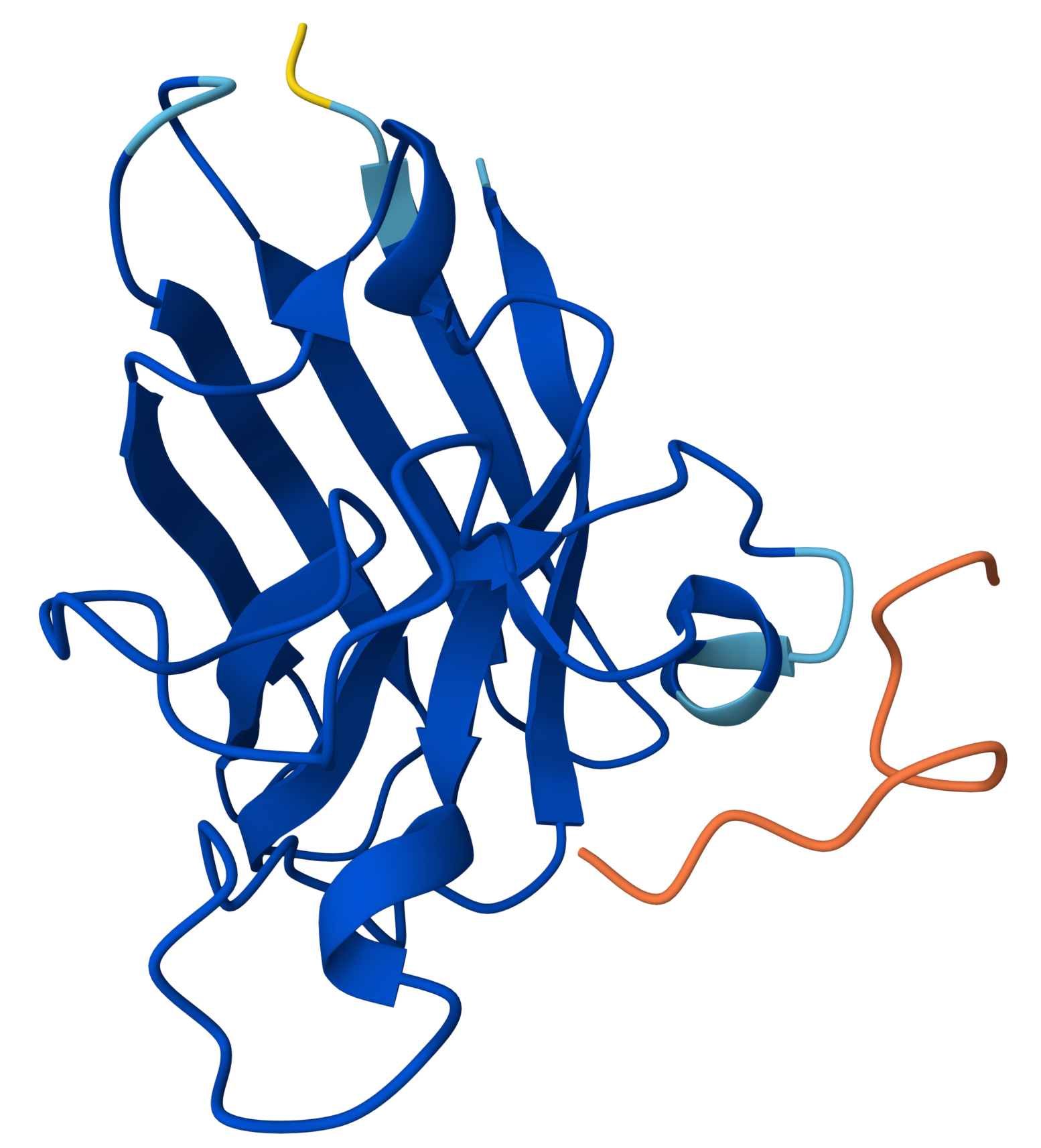
| 
|
- The protein is predicted extremely well
- The peptide is not interacting meaningfully at all
The peptide
HRYGATVVAWKEproduced an ipTM score of 0.26, indicating very low confidence in the predicted protein–peptide interaction, while the overall SOD1 structure was predicted with high confidence (pTM = 0.87). The peptide appears completely detached from the protein, with no visible interaction with the β-barrel or any defined binding region. It does not localize near the N-terminal region where the A4V mutation is located, and no interaction with the dimer interface can be assessed. The peptide exhibits very low confidence (orange coloring) across most of its length, suggesting high flexibility and lack of a stable conformation. Overall, this model indicates no meaningful binding interaction, representing the weakest candidate among the peptides tested.
Does it localize near the N-terminus where A4V sits?
No. The peptide is located far from the N-terminal region of SOD1. It does not approach the top portion of the structure where the A4V mutation resides.
Does it engage the β-barrel region or approach the dimer interface?
- No. Unlike Run 2, this peptide does not even align along the β-barrel surface. It is clearly spatially separated from the structured core of the protein.
- No. Again, only a monomer is modeled, so the dimer interface is not present.
Does it appear surface-bound or partially buried?
Completely detached, his is actually the cleanest negative result so far! This are the key observation:
- The peptide is far away from the protein
- It is colored mostly orange, indicating very low confidence and high flexibility
- There is no visible interaction interface
- This is essentially a non-binding prediction.
Why this happens
Even though the sequence contains:
- H (charged)
- W/Y (aromatic)
- hydrophobic residues (V, A)
The arrangement and context of residues matters more than composition. This peptide likely does not form a compatible interface geometry, remains too flexible to stabilize binding or is treated by AlphaFold as an independent chain.
| Run 4: HHSYPVALEHWK | Parameters |
|---|---|
| 
|
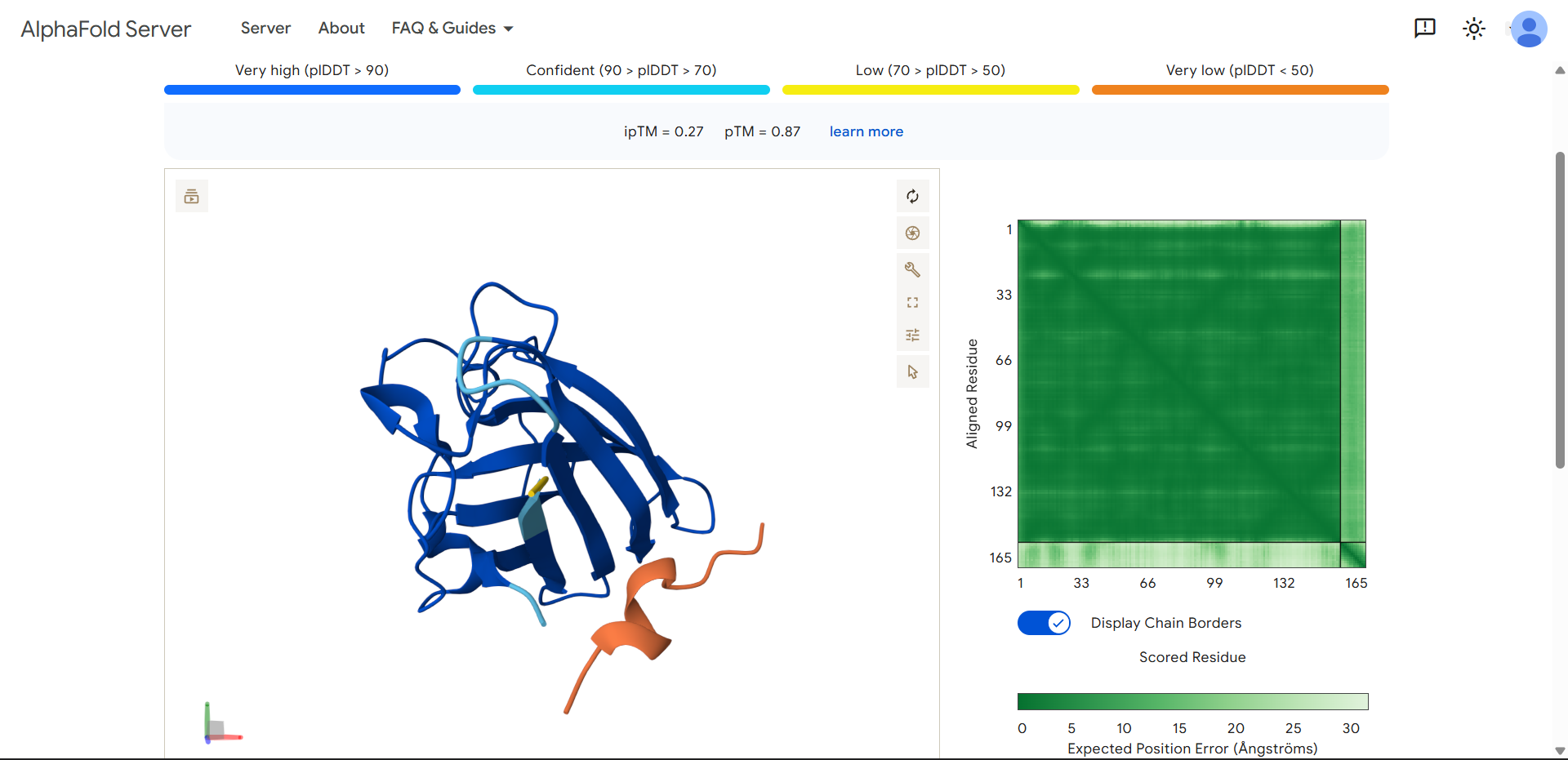
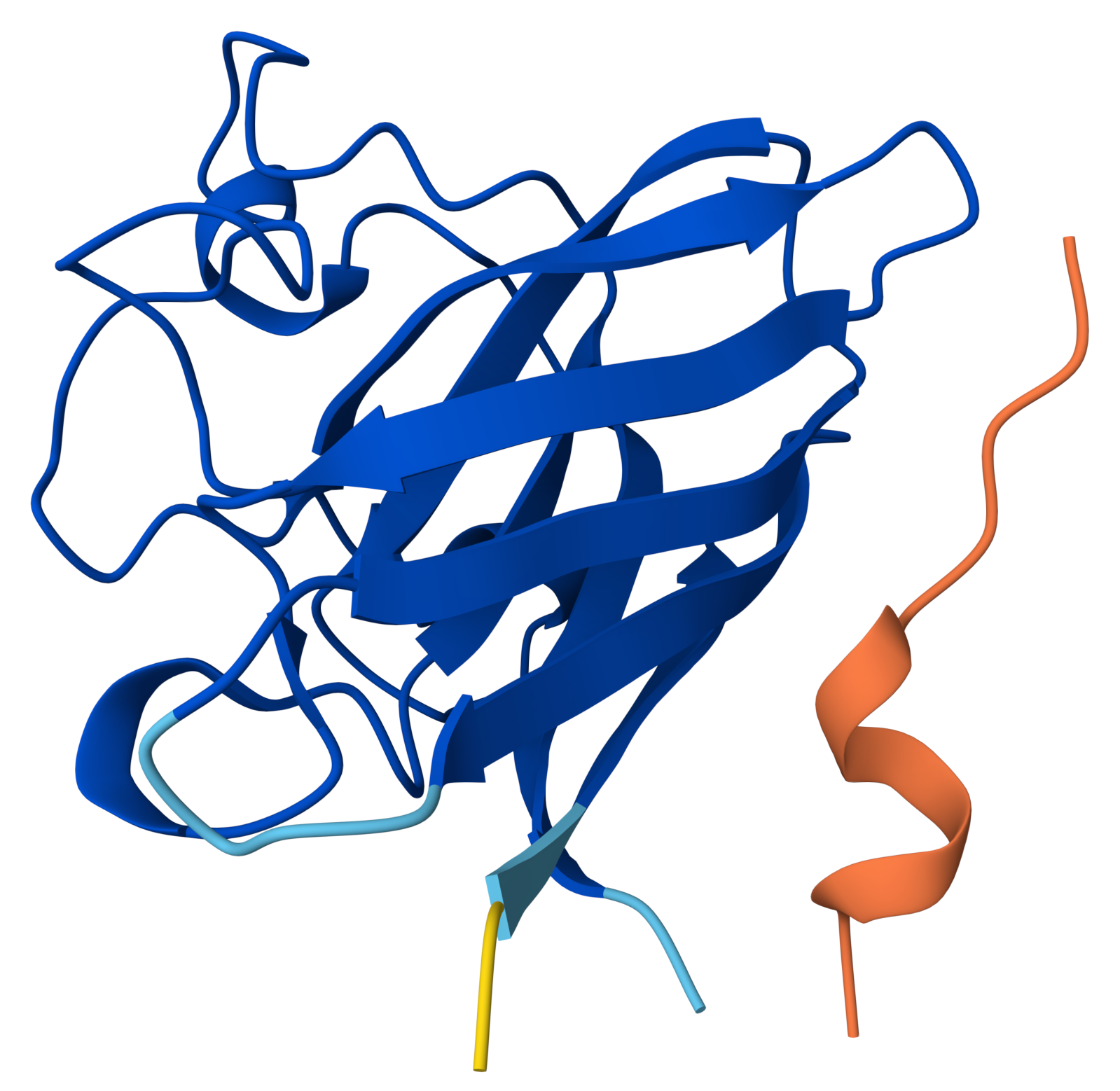
| Run 4 | (ipTM=0.27, pTM=0.87) |
|---|---|
| 
|
Same pattern as Run 3:
- Protein is very well predicted
- Interaction is essentially absent
Important observation: peptide secondary structure
The peptide looks “thicker” and more structured (helix-like or sheet-like), it may be forming a transient secondary structure (likely α-helix). However, internal folding ≠ binding. This means, the peptide can stabilize itself but still fails to interact with SOD1.
This suggests: Binding requires complementarity, not just structure.
Even with:
- aromatic residues (Y, W)
- charged residues (H)
The peptide does not match the geometry or chemistry of the binding surface.
The peptide
HHSYPVALEHWKproduced an ipTM score of 0.27, indicating very low confidence in the predicted protein–peptide interaction, while the SOD1 structure was predicted with high confidence (pTM = 0.87). The peptide appears fully detached from the protein, with no observable interaction with the β-barrel or the N-terminal region containing the A4V mutation. Interestingly, unlike other non-binding peptides, this sequence adopts a more compact and partially structured conformation, suggesting the formation of internal secondary structure. Despite this, the peptide does not form a stable interface with SOD1, indicating that self-folding alone is insufficient for binding. Overall, this model represents a non-binding case with increased peptide structural definition.
Does it localize near the N-terminus where A4V sits?
No. The peptide is clearly distant from the N-terminal region and does not approach the area where the A4V mutation is located.
Does it engage the β-barrel region or approach the dimer interface?
- No. There is no contact with the β-barrel surface. The peptide is positioned away from the structured core of the protein.
- No. As in all previous runs, only a monomer is modeled, so the dimer interface is not represented.
Does it appear surface-bound or partially buried?
Detached, but structurally more defined than previous cases. This is the key difference:
- The peptide is still far from the protein (no interaction)
- But unlike Run 3, it is not just a random flexible chain
- It appears to form a more compact, partially folded structure
| Run 5: FLYRWLPSRRGG | Parameters |
|---|---|
| 
|
| Run 5 | (ipTM=0.30, pTM=0.78) |
|---|---|
| 
|
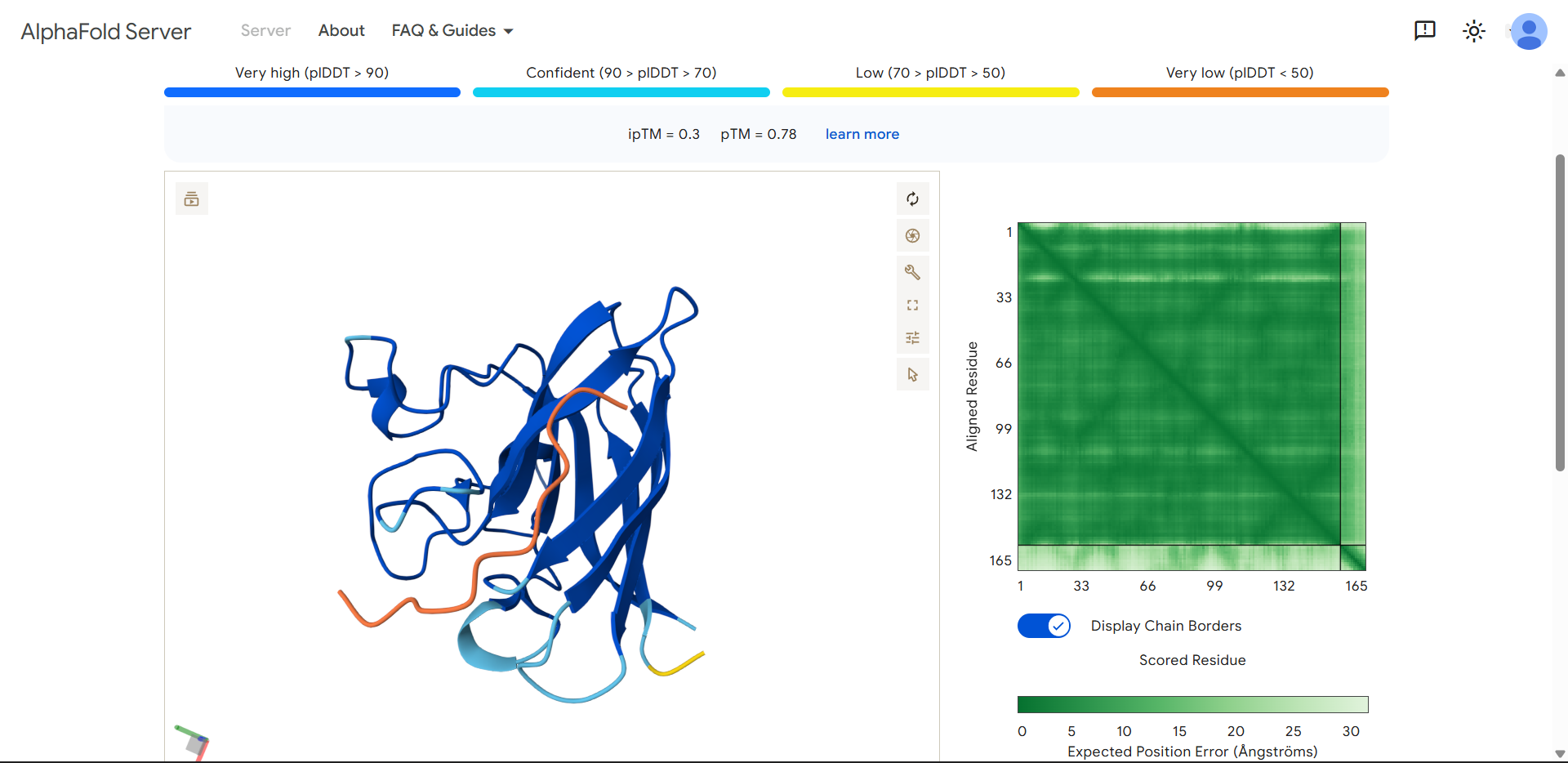
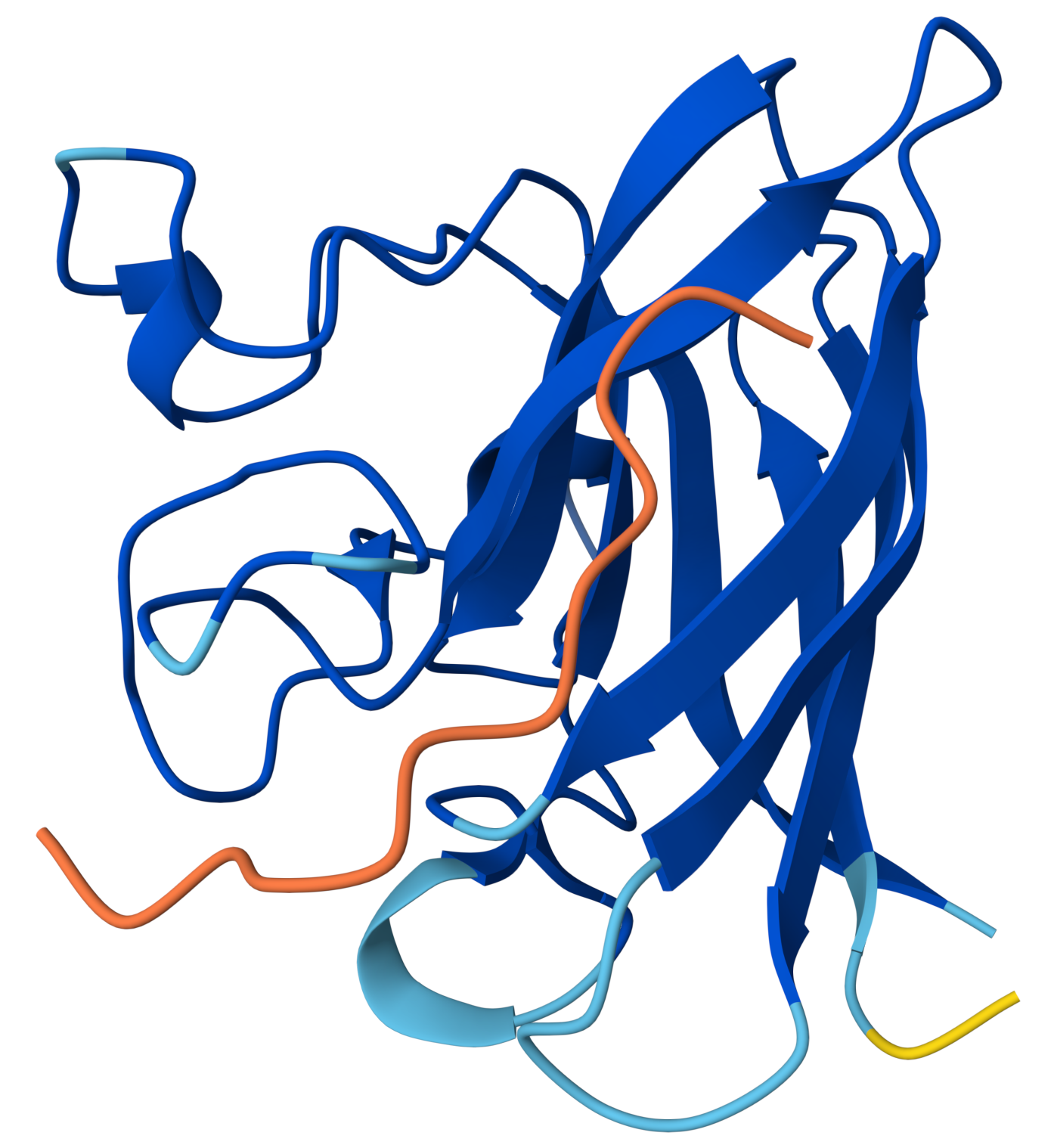
The protein structure is still predicted well, but the interaction between the peptide and SOD1 is predicted very poorly!
The control peptide
FLYRWLPSRRGGproduced an ipTM score of 0.30, indicating very low confidence in the predicted protein–peptide interface. While the overall fold of SOD1 was predicted with reasonable confidence (pTM = 0.78), the peptide displayed very low pLDDT values across its entire length, suggesting high structural uncertainty. Visual inspection shows that the peptide lies loosely along the surface of the β-barrel, but it does not form a well-defined binding interface and does not localize near the N-terminal region where the A4V mutation occurs. Instead, the peptide appears highly flexible and partially detached from the protein surface.
Does it localize near the N-terminus where A4V sits?
No. The peptide does not appear to bind near the N-terminal region of SOD1. The N-terminus is located in the upper portion of the structure, while the peptide is positioned toward the lower region of the protein. Therefore, the peptide does not interact with the region where the A4V mutation occurs in this prediction.
Does it engage the β-barrel region or approach the dimer interface?
Partially, but only loosely. The peptide lies along the outer surface of the β-barrel, but it does not form a clear or well-defined binding interface. It appears to pass across the surface rather than docking into a specific pocket.
No. The model again contains only a single SOD1 monomer, so the dimer interface is not present in this prediction. Therefore, the peptide cannot be interacting with the dimer interface.
Does it appear surface-bound or partially buried?
It appears largely unbound and highly flexible. The peptide is colored orange across nearly its entire length, indicating very low pLDDT (<50). This means AlphaFold has very little confidence in the peptide’s structure or position. This suggests that the peptide does not form a stable interaction with the protein in the predicted model and may be essentially floating near the protein surface.
Final results
| Run | Peptide | Seed | ipTM | pTM | Protein confidence |
|---|---|---|---|---|---|
| 1 | WLYGATGAAHGE | 100 | 0.49 | 0.83 | stable |
| 2 | WRYPVVALALKE | 100 | 0.43 | 0.80 | slightly perturbed |
| 3 | HRYGATVVAWKE | 100 | 0.26 | 0.87 | stable |
| 4 | HHSYPVALEHWK | 100 | 0.27 | 0.87 | stable |
| 5 | FLYRWLPSRRGG | 100 | 0.30 | 0.78 | stable |
Across all predictions, the PepMLM-generated peptides exhibited a range of interaction behaviors with Superoxide dismutase 1, but none achieved high-confidence binding according to AlphaFold ipTM scores. The best-performing designs (WLYGATGAAHGE and WRYPVVALALKE) showed moderate interface confidence (ipTM ≈ 0.43–0.49) and appeared to interact weakly along the β-barrel surface, although without forming well-defined binding pockets or localizing near the N-terminal region containing the A4V mutation. In contrast, other peptides (HRYGATVVAWKE and HHSYPVALEHWK) displayed little to no interaction, remaining largely detached from the protein despite in some cases adopting partial secondary structure. Surprisingly, the known binder (FLYRWLPSRRGG) also yielded a low ipTM score (0.30) and showed no clear binding interface in the predicted model. Overall, none of the PepMLM-generated peptides clearly matched or exceeded the known binder in terms of predicted binding confidence; however, several designs performed comparably or slightly better in silico. These results highlight important limitations of structure-based prediction for short, flexible peptides, suggesting that low-confidence AlphaFold outputs do not necessarily rule out experimental binding, and that additional validation methods would be required to accurately assess peptide affinity.
C: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of the peptides!
For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
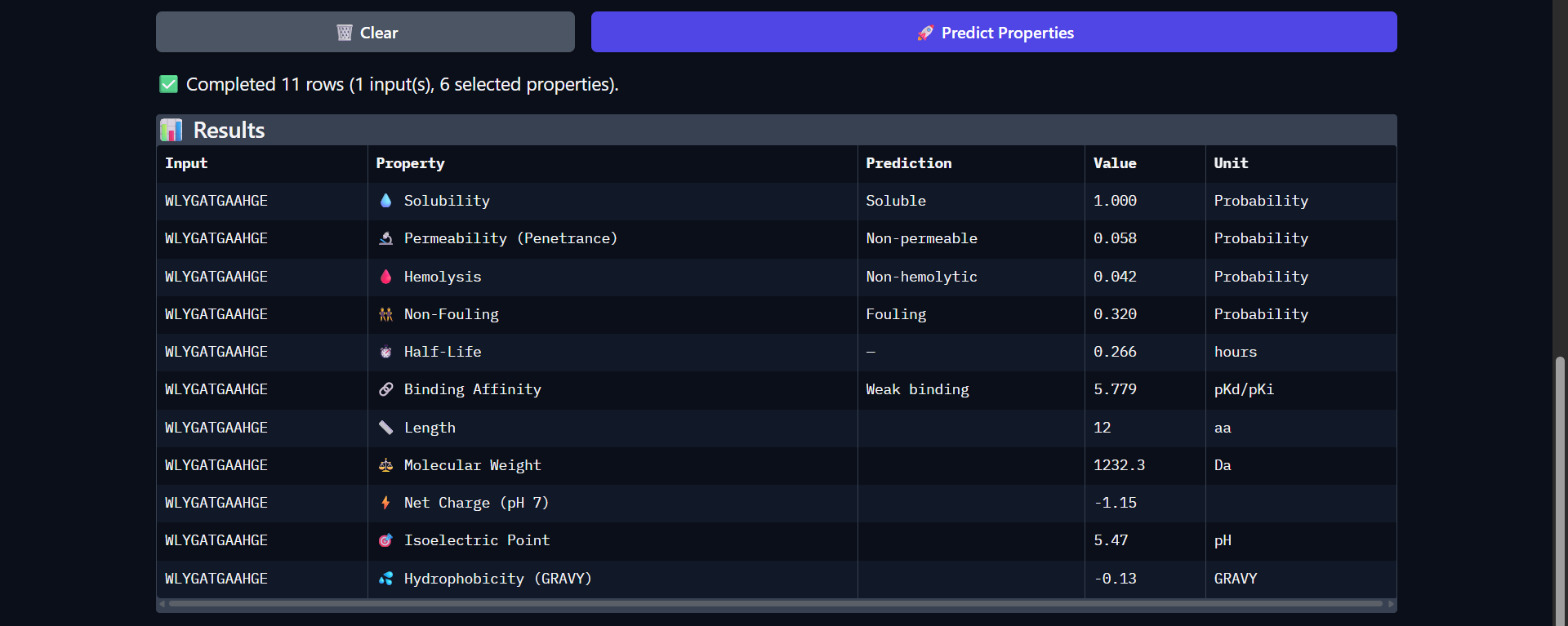
| Run 1: WLYGATGAAHGE |
|---|
|
|
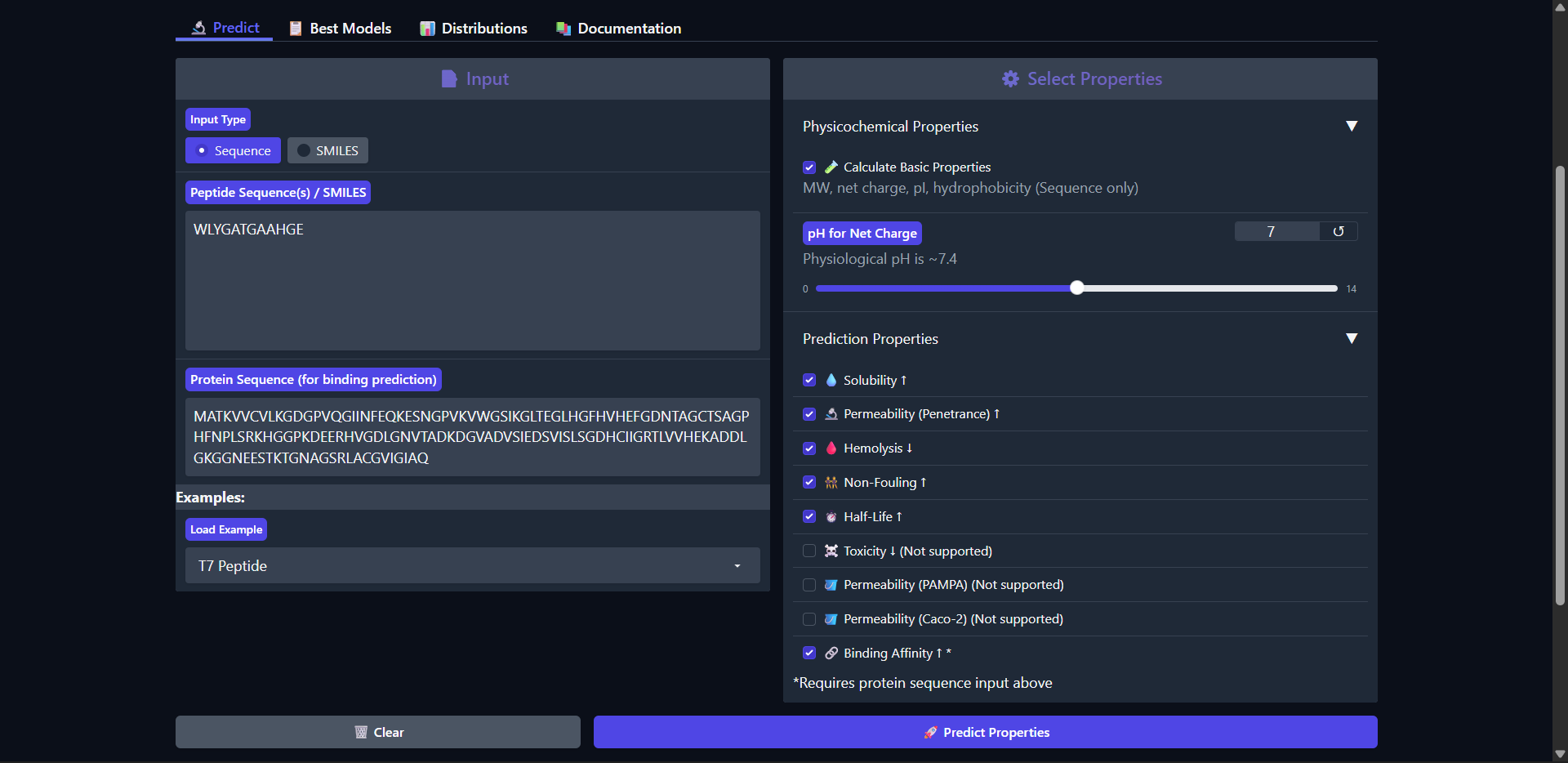
Good drug-like properties, but weak efficacy
The peptide WLYGATGAAHGE shows a favorable therapeutic profile despite only moderate structural interaction with Superoxide dismutase 1 predicted by AlphaFold (ipTM ≈ 0.49). It is predicted to be highly soluble (1.000) and non-hemolytic (0.042), which are desirable properties for therapeutic development. However, the peptide is classified as non-permeable (0.058) and has a relatively short predicted half-life (0.266 hours), which may limit its bioavailability. The predicted binding affinity is weak (pKd/pKi = 5.779), consistent with the moderate and surface-level interaction observed structurally. The peptide carries a slight negative charge at physiological pH (-1.15) and exhibits near-neutral hydrophobicity (GRAVY = -0.13), suggesting a balanced but not strongly interacting physicochemical profile. Overall, while structural predictions suggest limited binding strength, the peptide demonstrates good safety and solubility characteristics, making it a reasonable candidate for further optimization rather than immediate therapeutic application.
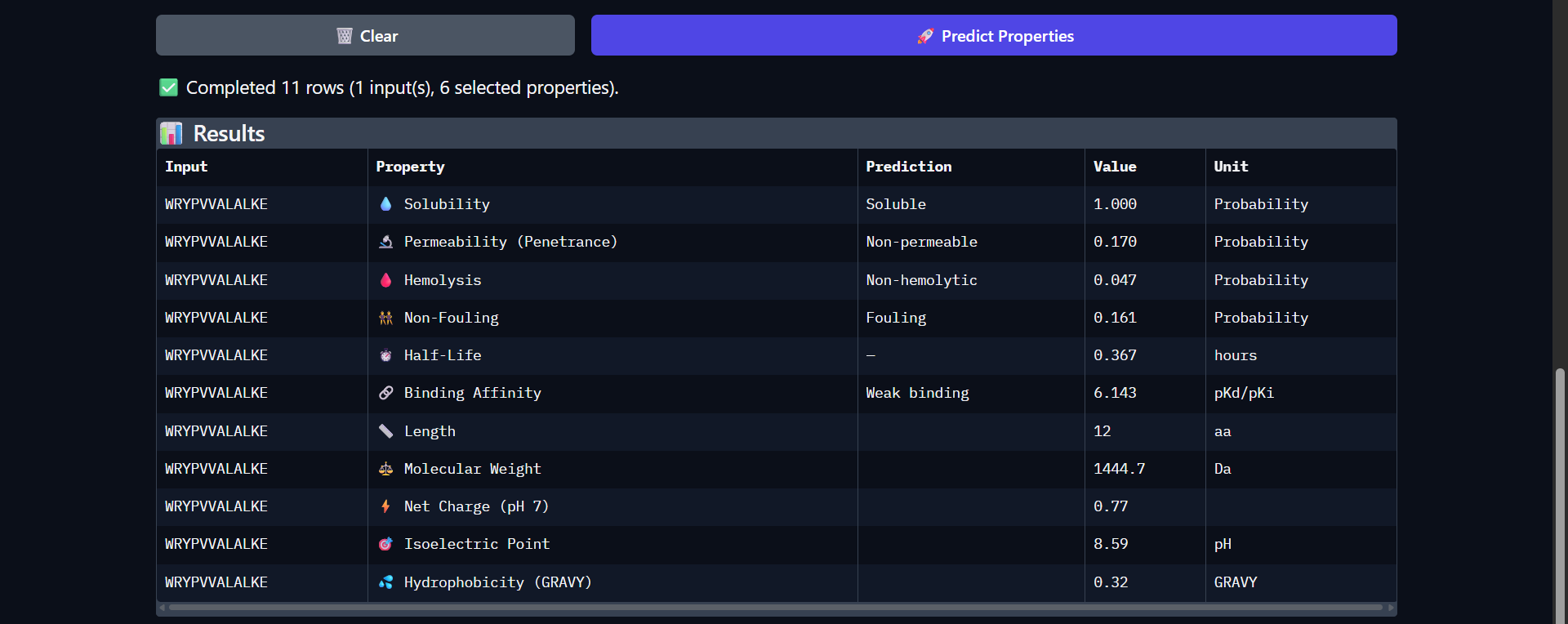
| Run 2: WRYPVVALALKE |
|---|
|
|
The peptide WRYPVVALALKE shows a slightly improved predicted binding affinity (pKd/pKi = 6.143) compared to WLYGATGAAHGE, which is consistent with its somewhat more extensive surface interaction observed in AlphaFold (ipTM ≈ 0.43). Like the previous peptide, it is predicted to be highly soluble (1.000) and non-hemolytic (0.047), indicating a favorable safety profile. However, it remains non-permeable (0.170) and exhibits only a modest increase in predicted half-life (0.367 hours). Notably, this peptide is more hydrophobic (GRAVY = 0.32) and carries a slightly positive charge at physiological pH (0.77), which may contribute to its somewhat improved binding affinity through enhanced surface interactions. Despite these improvements, the peptide is still classified as a weak binder, and the interaction observed structurally remains surface-level and not well-defined. Overall, this peptide demonstrates a better balance between binding potential and physicochemical properties compared to Run 1, although significant limitations remain for therapeutic application.
| Run 3: HRYGATVVAWKE |
|---|
|
|
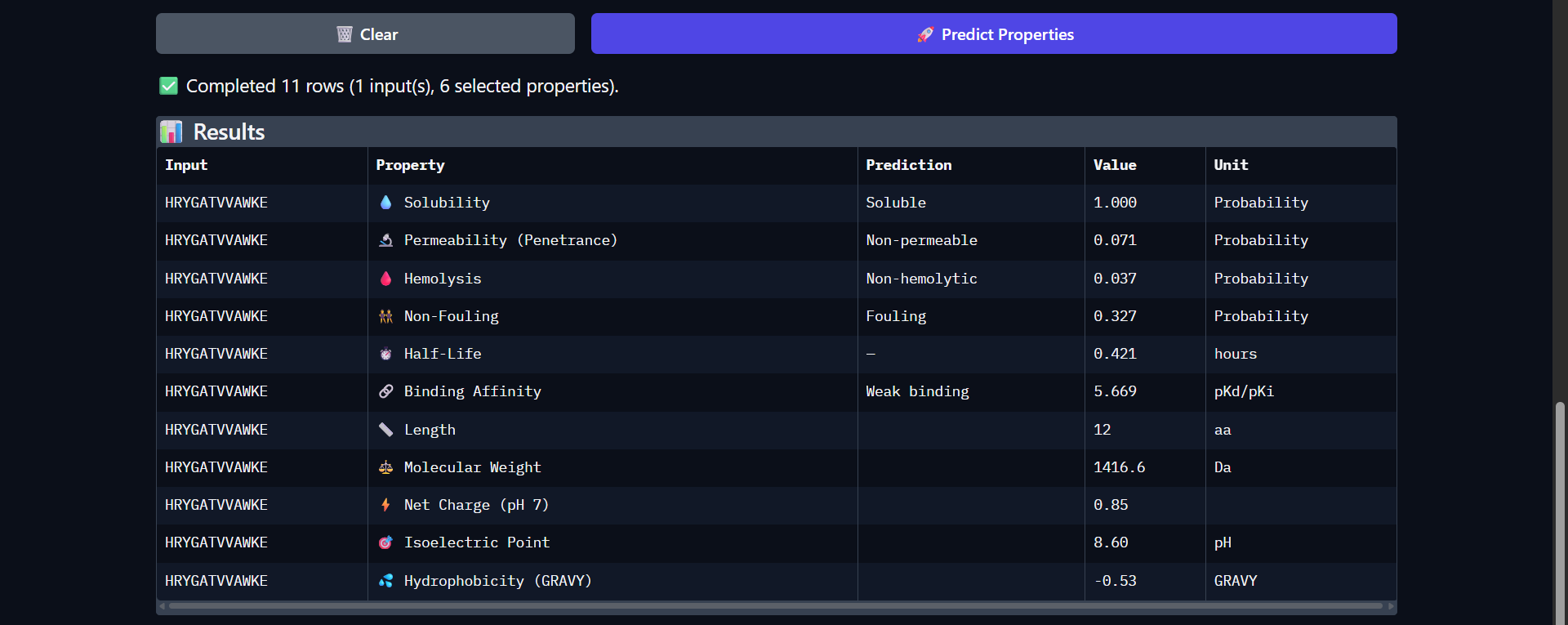
The peptide HRYGATVVAWKE shows a weaker predicted binding affinity (pKd/pKi = 5.669) compared to the previous candidates, which is consistent with the very low interaction confidence observed in AlphaFold (ipTM ≈ 0.26). Structurally, this peptide appeared fully detached from Superoxide dismutase 1, indicating no meaningful binding interaction. Despite this, the peptide retains favorable therapeutic properties, including high solubility (1.000) and low hemolysis probability (0.037). It also exhibits one of the longest predicted half-life so far (0.421 hours) among the tested peptides. However, it remains non-permeable (0.071) and shows relatively high fouling potential (0.327). The peptide carries a positive net charge (0.85) but is overall more hydrophilic (GRAVY = -0.53), which may reduce its ability to form stable hydrophobic interactions with the protein surface. Overall, both structural and physicochemical predictions consistently indicate that this peptide is a poor binder, despite having acceptable safety and solubility characteristics.
| Run 4: HHSYPVALEHWK |
|---|
|
|
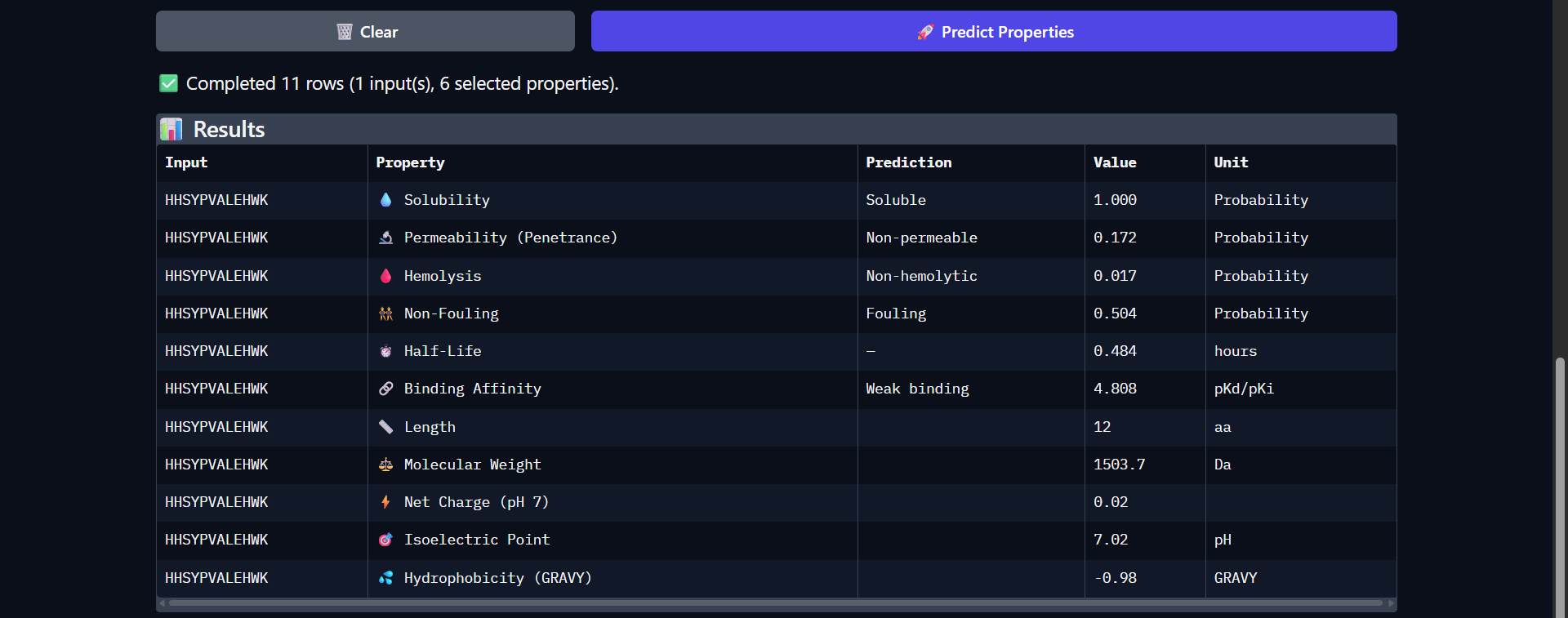
The peptide HHSYPVALEHWK shows the weakest predicted binding affinity among all candidates (pKd/pKi = 4.808), which is consistent with the very low interaction confidence observed in AlphaFold (ipTM ≈ 0.27). Structurally, the peptide appeared fully detached from Superoxide dismutase 1, indicating no meaningful interaction. Despite this, it exhibits several favorable therapeutic properties, including high solubility (1.000) and the lowest hemolysis probability (0.017) among all peptides. It also shows the longest predicted half-life (0.484 hours), suggesting improved stability relative to other candidates. However, it presents the highest fouling propensity (0.504) and remains non-permeable (0.172). The peptide is nearly neutral at physiological pH (net charge ≈ 0.02) and highly hydrophilic (GRAVY = -0.98), which may limit its ability to form stable hydrophobic interactions with the protein surface. Overall, both structural and physicochemical analyses indicate that this peptide is not a viable binder, despite its favorable safety and stability profile.
| Run 5 - Control peptide: FLYRWLPSRRGG |
|---|
|
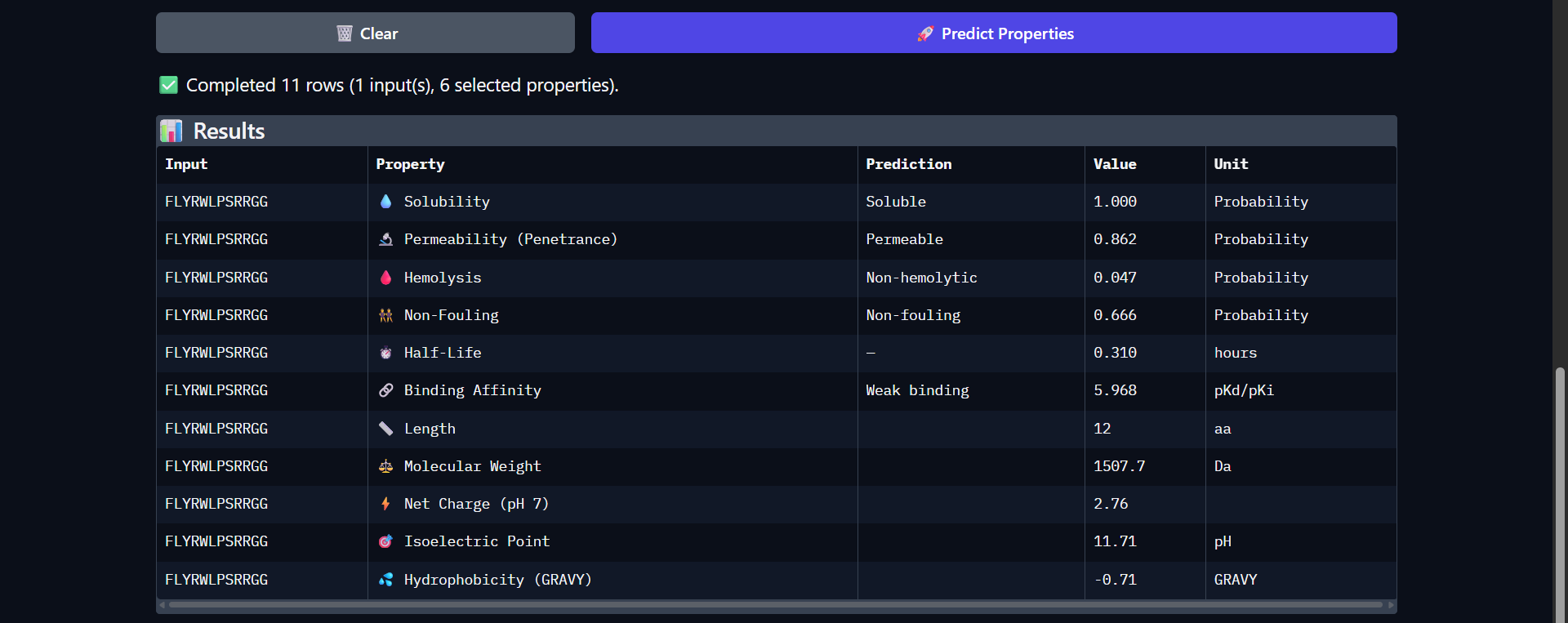
|
The control peptide FLYRWLPSRRGG exhibits a distinct physicochemical profile compared to the PepMLM-generated candidates. While its predicted binding affinity remains in the weak range (pKd/pKi = 5.968), consistent with the low interaction confidence observed in AlphaFold (ipTM ≈ 0.30), it demonstrates several advantageous therapeutic properties. Notably, it is predicted to be highly permeable (0.862), in contrast to all generated peptides, which were non-permeable. Additionally, it is classified as non-fouling (0.666) and non-hemolytic (0.047), indicating favorable biocompatibility. The peptide carries a strong positive charge (2.76) and a high isoelectric point (11.71), which may facilitate interactions with negatively charged cellular membranes and contribute to its permeability. Despite these advantages, its binding affinity and structural predictions do not indicate a strong or well-defined interaction with Superoxide dismutase 1. Overall, the control peptide highlights a trade-off between cellular delivery properties and binding specificity, suggesting that effective therapeutic peptides must balance both aspects.
Final insights
| Peptide | ipTM | Affinity | Permeability | Key takeaway |
|---|---|---|---|---|
| WLYGATGAAHGE | 0.49 | 5.78 | ❌ | best structure |
| WRYPVVALALKE | 0.43 | 6.14 | ❌ | best affinity |
| HRYGATVVAWKE | 0.26 | 5.67 | ❌ | no binding |
| HHSYPVALEHWK | 0.27 | 4.81 | ❌ | worst binder |
| FLYRWLPSRRGG | 0.30 | 5.97 | ✅ | best delivery properties |
Winner peptide! 😀
Among the evaluated candidates, WRYPVVALALKE represents the best balance between predicted binding and therapeutic properties. This peptide exhibited the highest predicted binding affinity (pKd/pKi = 6.143) and showed moderate interaction with SOD1 in AlphaFold predictions, suggesting some potential for target engagement. While it remains non-permeable and displays only moderate stability, it is highly soluble and non-hemolytic, indicating a favorable safety profile. In comparison, other peptides either showed weaker binding or no interaction, while the control peptide demonstrated superior permeability but no improved binding. Therefore, WRYPVVALALKE would be the most suitable candidate to advance, as it provides the best compromise between binding potential and acceptable physicochemical properties, and could be further optimized to improve delivery and stability.
D: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
How to:
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook:
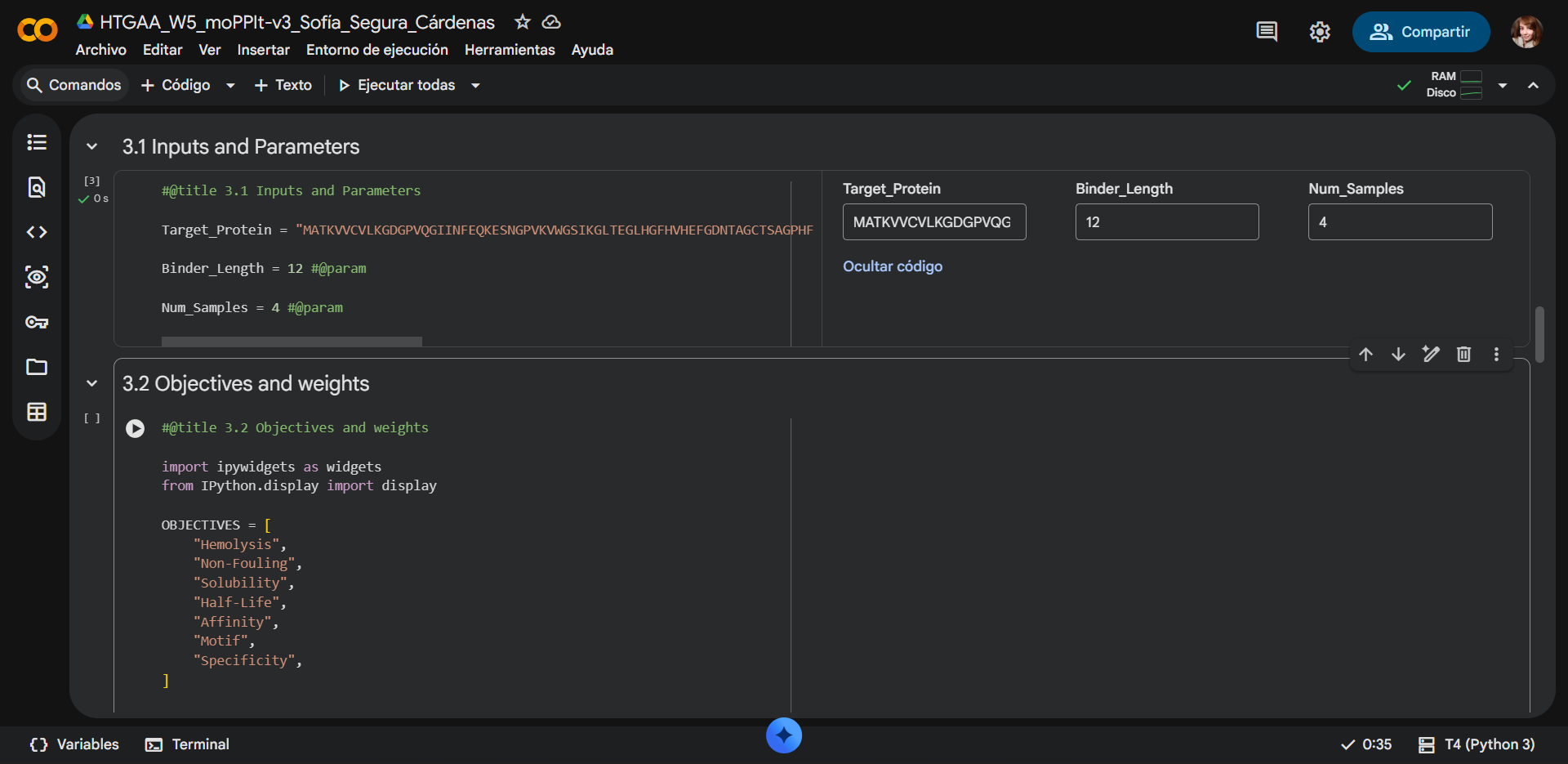
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
moPPIt Colab used:
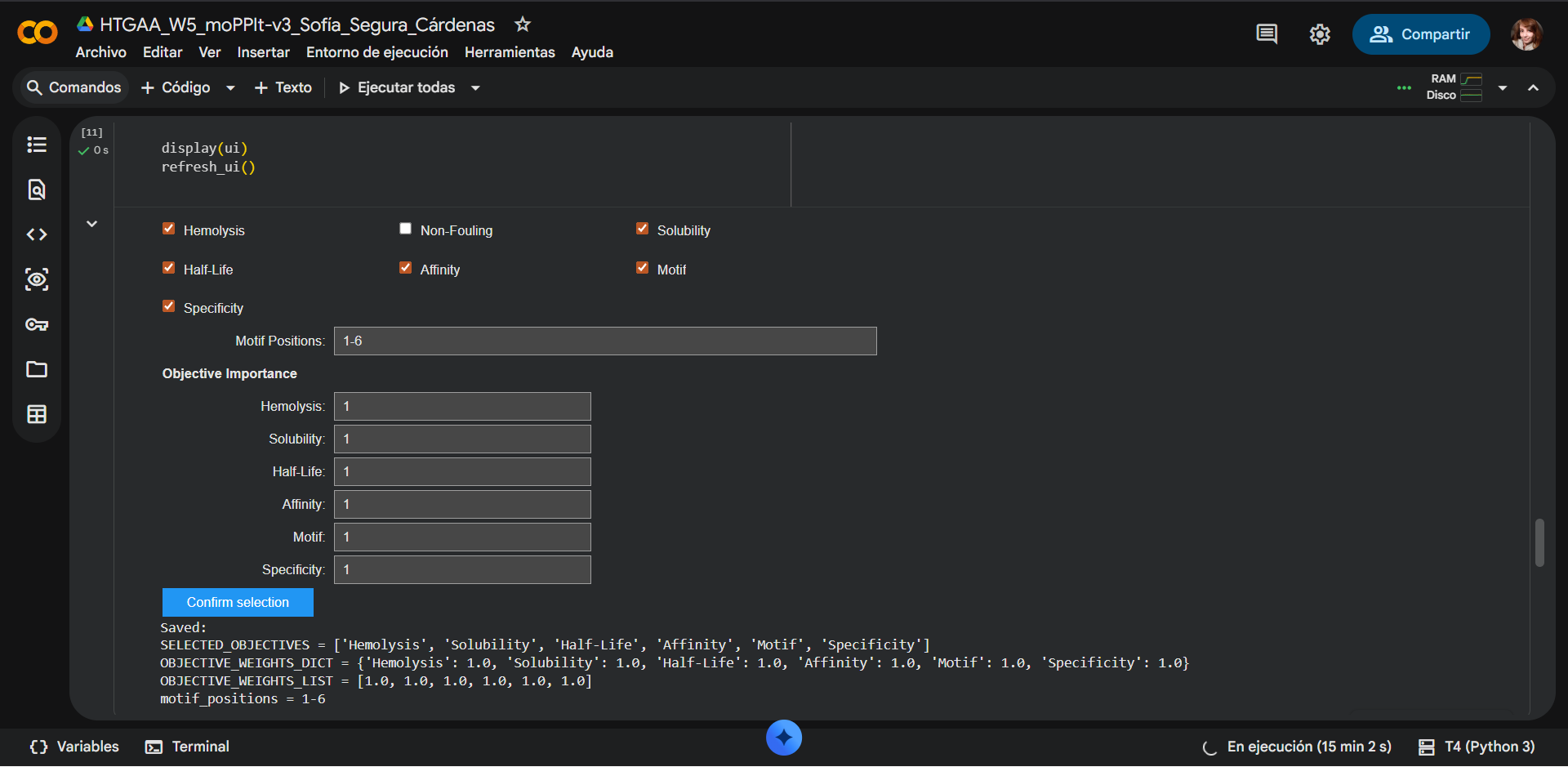
The motif positions were constrained to residues 1–6 of the peptide to bias binding toward the N-terminal region of SOD1, where the A4V mutation is located (residues 1–10). By restricting the motif to the N-terminal portion of the peptide, the design encourages early contact formation between the peptide and the target region of interest. This does not enforce a one-to-one positional interaction but instead promotes favorable orientation and interaction propensity near the mutation site. Additionally, limiting motif positions to 1–5 reduces the search space, improving computational efficiency while maintaining biologically relevant targeting.
| 
|
|---|---|
| 
|
| 
|
Due to computational limitations in the Colab environment, particularly related to GPU memory, it was not feasible to optimize all properties simultaneously in moPPIt while generating multiple peptide candidates. Including all objectives significantly increases the complexity of the multi-objective optimization process, leading to higher memory usage and instability during execution. As a result, it was necessary to reduce the number of properties selected to successfully generate peptide sequences.
Selected properties
| Property | Objective importance | Selection |
|---|---|---|
| Hemolysis | 1 | Yes |
| Non-Fouling | 0 | No |
| Solubility | 1 | Yes |
| Half-Life | 1 | Yes |
| Affinity | 1 | Yes |
| Motif | 1 | Yes |
| Specificity | 1 | Yes |
The Non-fouling property was sacrificied as an optimization objective. When designing a therapeutic peptide targeting mutant SOD1, the most reasonable property to relax in a multi-objective optimization framework such as moPPIt would be Non-fouling. While properties such as solubility and non-hemolytic behavior are essential for safety and delivery, and binding affinity is the primary objective, some degree of nonspecific interaction may be tolerated during early-stage design to enhance binding strength. Specificity can often be improved in later optimization steps, whereas insufficient binding cannot be easily rescued. Therefore, allowing partial fouling enables exploration of sequences with stronger interaction potential, which can subsequently be refined for selectivity.
Generated binders
| Run | Binder | Hemolysis | Solubility | Half-Life | Affinity | Motif | Specificity |
|---|---|---|---|---|---|---|---|
| 6 | WILIKKLGGSTA | 0.912 | 0.50 | 5.063 | 5.824 | 0.030 | 0.853 |
| 7 | KTEEEWKALFAD | 0.915 | 0.58 | 12.482 | 6.501 | 0.011 | 0.712 |
| 8 | ETPTEIAQKLKE | 0.923 | 0.67 | 4.499 | 5.145 | 0.612 | 0.724 |
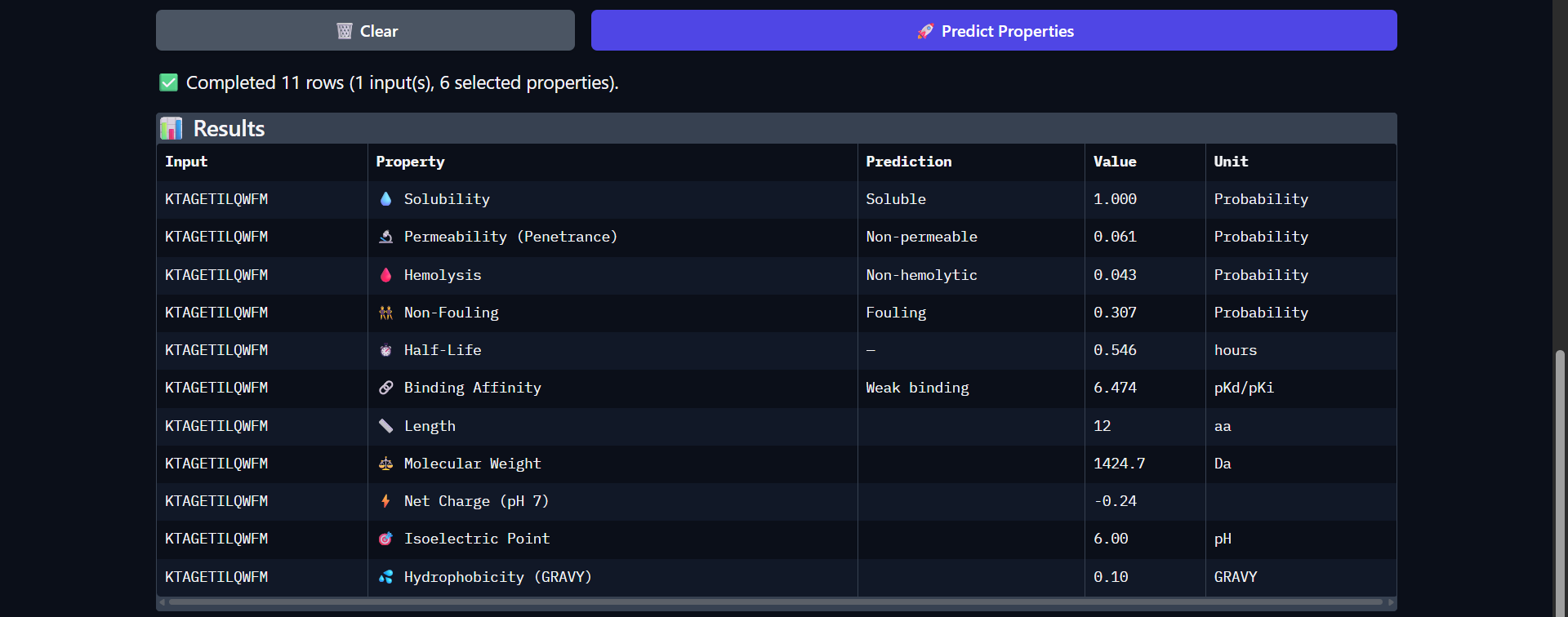
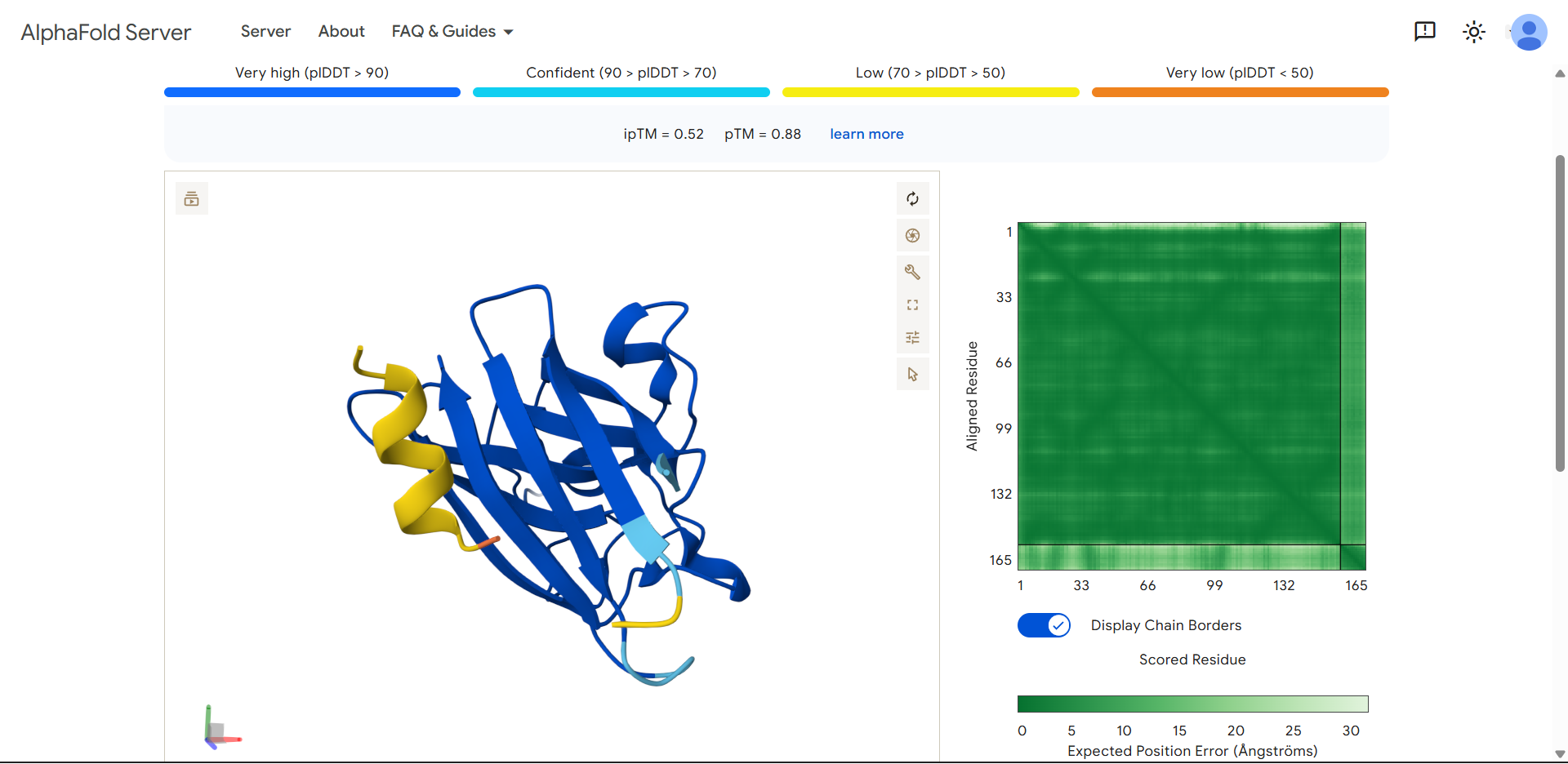
| 9 | KTAGETILQWFM | 0.939 | 0.50 | 7.405 | 6.474 | 0.599 | 0.609 |
All moPPIt-generated peptides are strongly predicted to be hemolytic
Many high-affinity peptides resemble antimicrobial peptides, which are inherently hemolytic due to their ability to disrupt lipid membranes.
All moPPIt-generated peptides exhibited very high hemolysis probabilities (>0.9), indicating a strong tendency to disrupt cellular membranes. This is likely a consequence of the optimization strategy, where specificity (non-fouling) was excluded and binding affinity was prioritized. As a result, the model favored sequences with physicochemical properties similar to membrane-active peptides, such as high charge and amphipathicity, which are known to correlate with hemolytic activity. This highlights an important trade-off in peptide design: improving binding and target interaction can inadvertently increase toxicity. Therefore, although these peptides may have promising binding characteristics toward Superoxide dismutase 1, their high hemolytic potential makes them unsuitable for direct therapeutic application without further optimization.
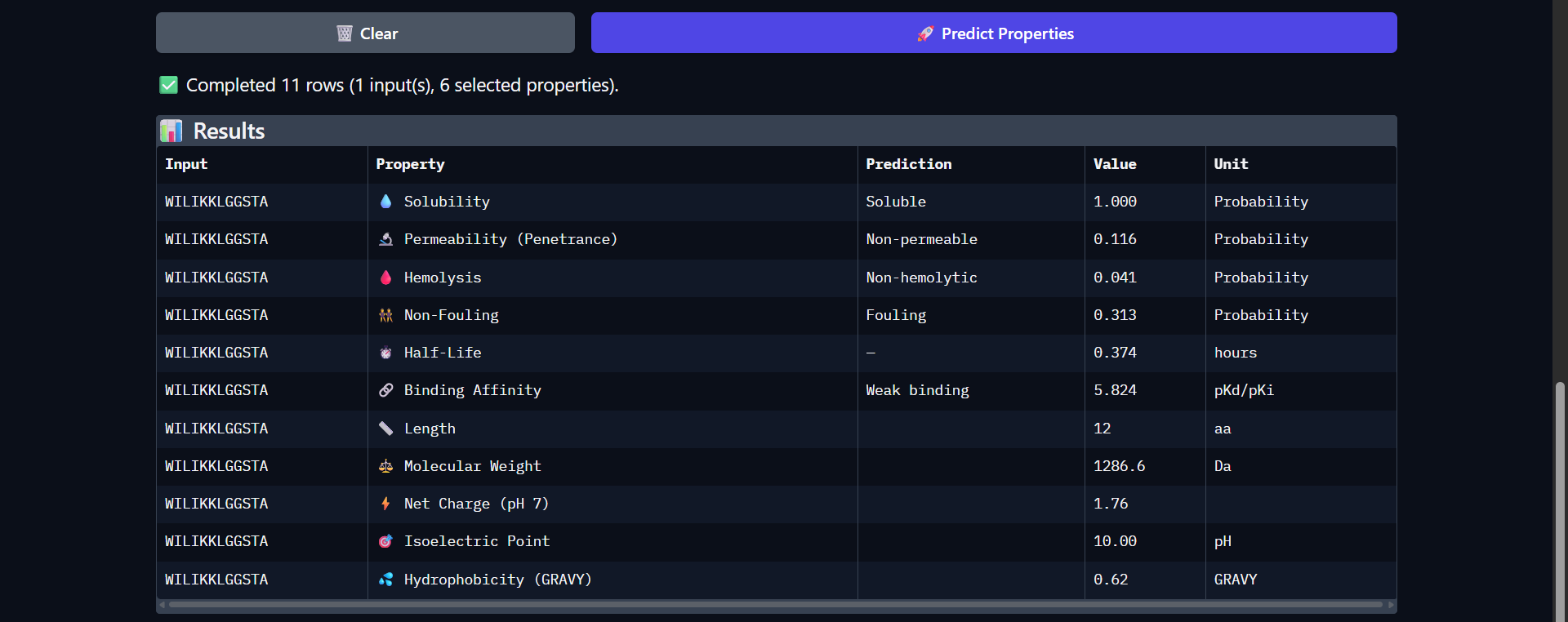
| Run 6: WILIKKLGGSTA - ipTM = 0.4, pTM = 0.83 |
|---|
|
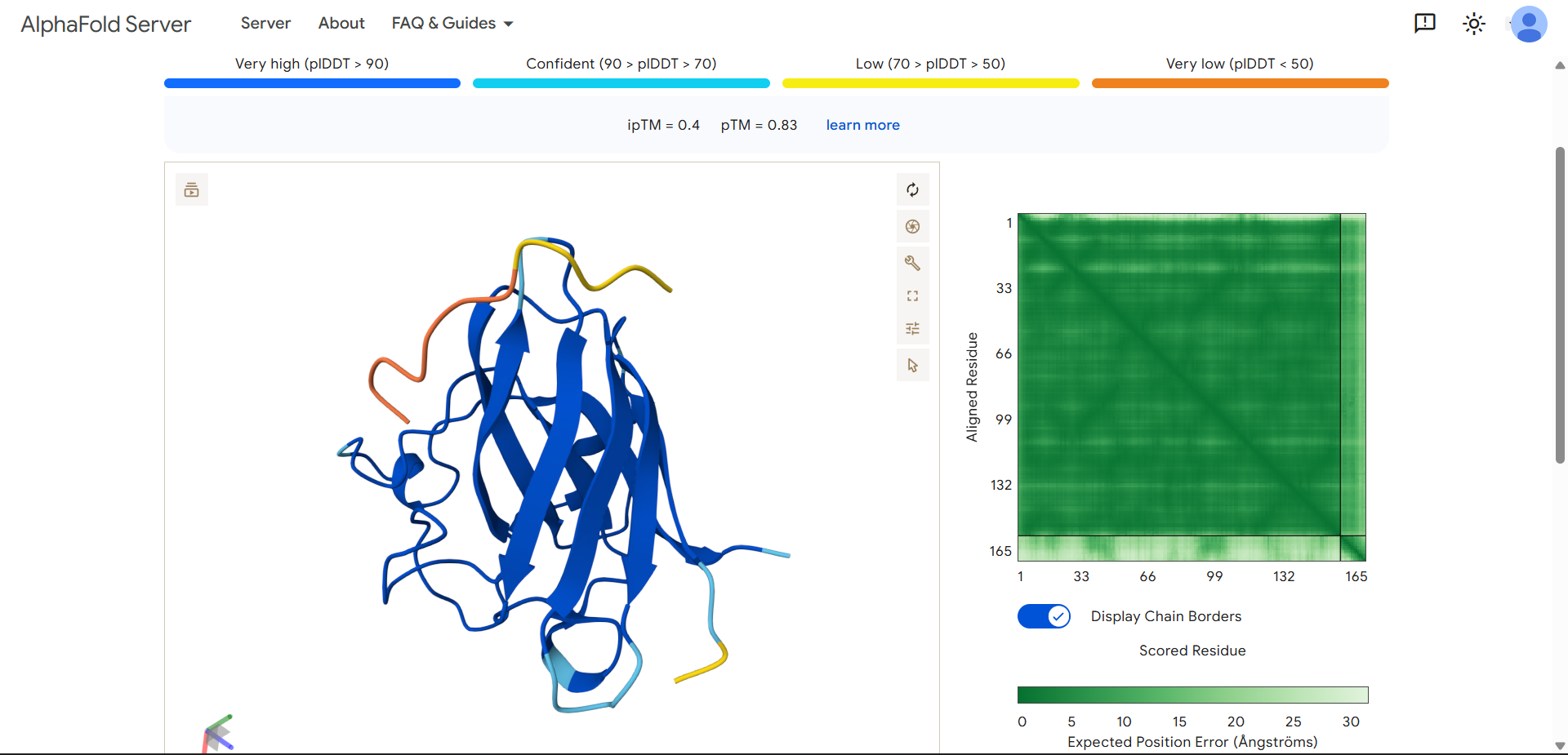
|
| Run 7: KTEEEWKALFAD - ipTM = 0.35, pTM = 0.87 |
|---|
|
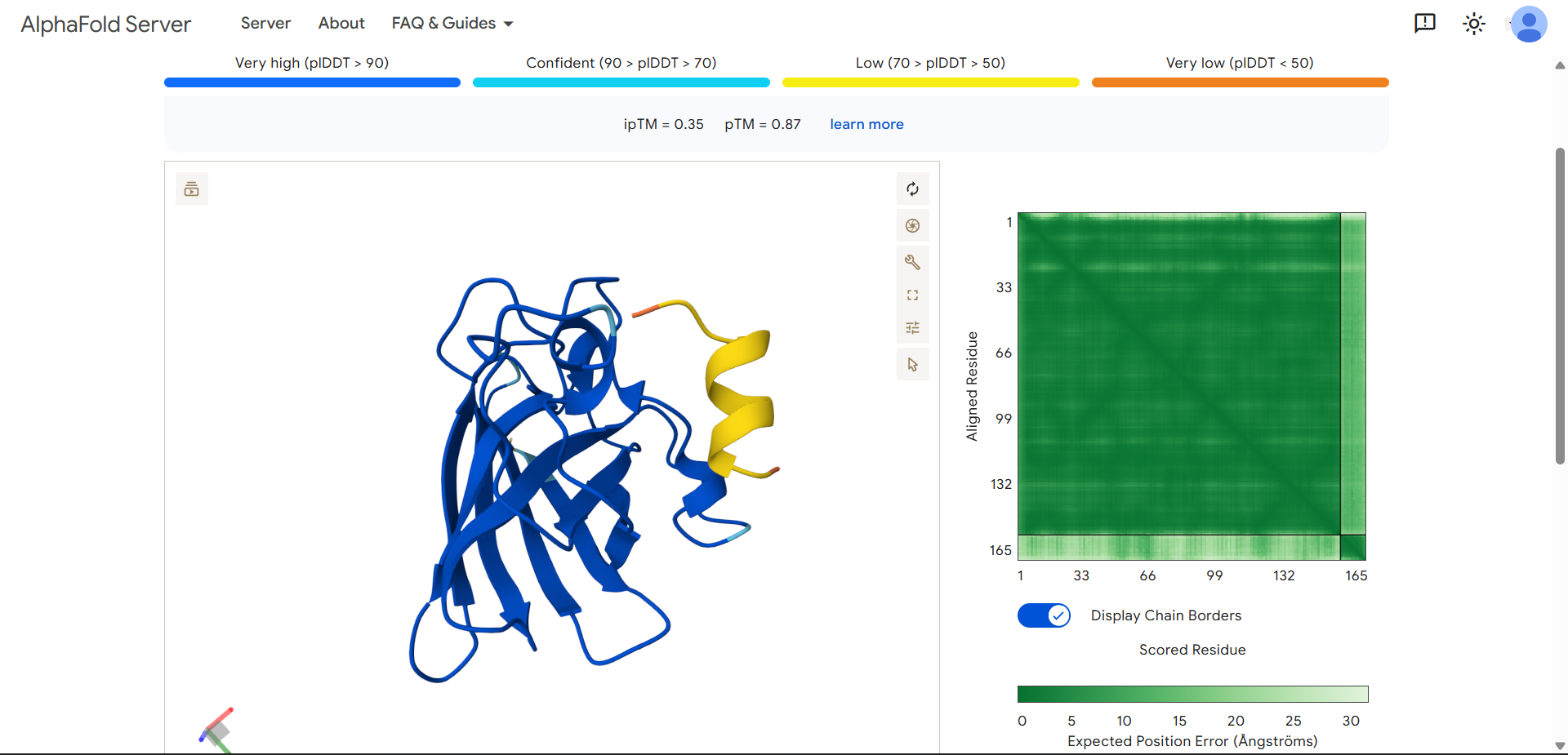
|
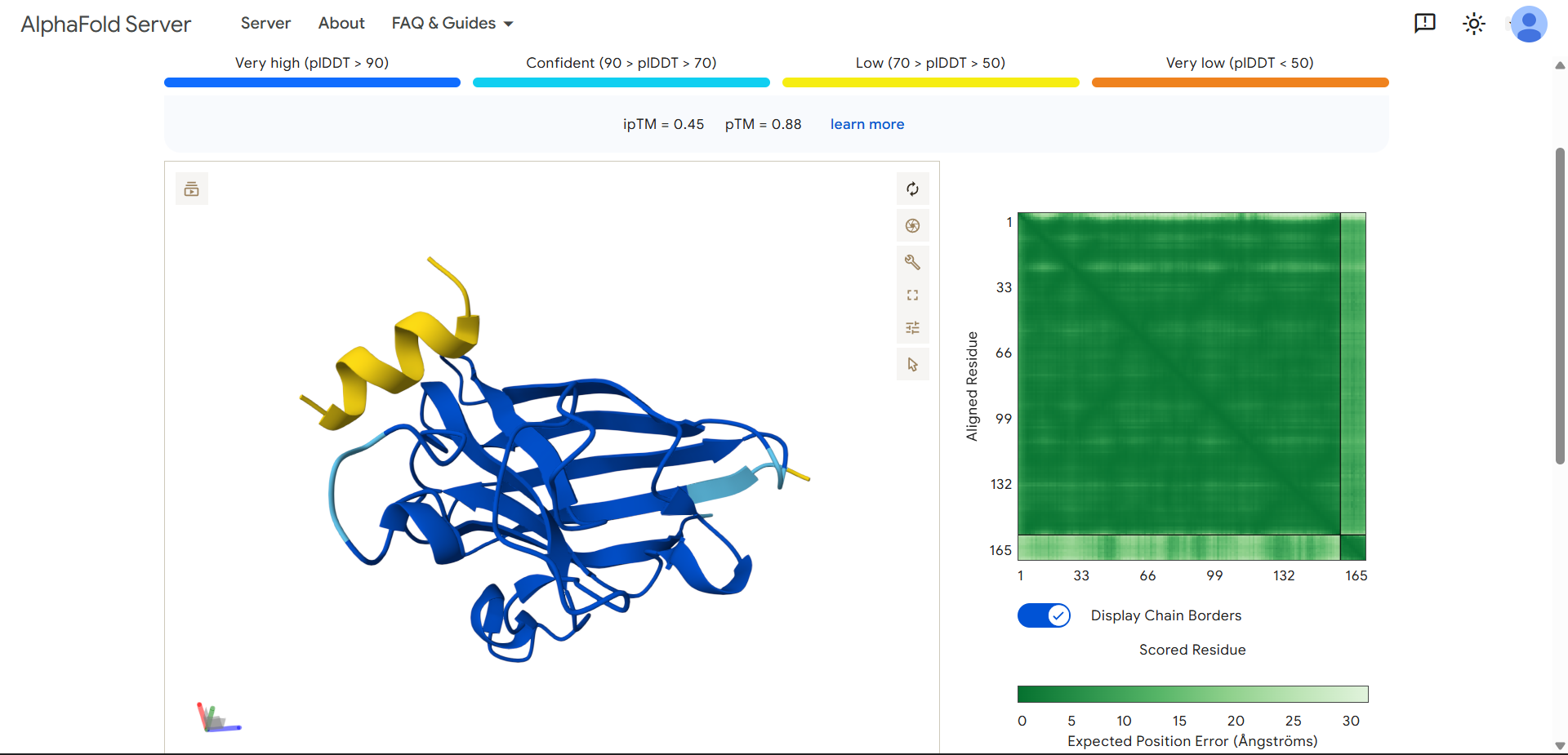
| Run 8: ETPTEIAQKLKE - ipTM = 0.45, pTM = 0.88 |
|---|
|
|
| Run 9: KTAGETILQWFM - ipTM = 0.52, pTM = 0.88 |
|---|
|
|
Although the generated peptides (Runs 6–9) exhibit favorable physicochemical properties (such as high solubility, low predicted hemolysis, and acceptable structural stability) the structural predictions obtained from AlphaFold and PeptiVerse indicate that they do not achieve the intended functional objective of binding to the N-terminal region of the mutated protein.
Specifically:
- The ipTM values (0.35–0.52) suggest low confidence in protein–peptide interactions, indicating that binding is likely weak or non-specific.
- In contrast, the pTM values (~0.83–0.88) are relatively high, reflecting accurate prediction of the overall protein structure, but this does not imply successful peptide binding.
- Visual inspection in AlphaFold shows that:
- The peptides do not localize to the N-terminal region (residues 1–4), which was the intended binding site.
- Instead, they remain dispersed near the β-barrel, without forming stable or consistent interactions.
- The peptides appear in yellow coloration, particularly in Runs 7–9, corresponding to moderate confidence scores (pLDDT ~50–70), which suggests structural flexibility or lack of a well-defined binding conformation.
- The mutated protein remains in dark blue, indicating that its structural integrity is preserved, but without evidence of functional interaction with the peptides.
Resources
- HTGAA Protein Engineering Tools spreadsheet
- AlphaFold Server. https://alphafoldserver.com/
- PeptiVerse. ChatterjeeLab. https://huggingface.co/spaces/ChatterjeeLab/PeptiVerse
- Chen, L.T., Quinn, Z., Dumas, M. et al. (2025). Target sequence-conditioned design of peptide binders using masked language modeling. Nat Biotechnol. https://doi.org/10.1038/s41587-025-02761-2
- Chen, T., Dumas, M., Watson, R., et al. (2023). PepMLM: Target Sequence-Conditioned Generation of Therapeutic Peptide Binders via Span Masked Language Modeling. arXiv. https://doi.org/10.48550/arXiv.2310.03842
- Chen, T., Quinn, Z., Mishra, K., et al. (2026). moPPIt: De Novo Generation of Motif-Specific and Functionally Active Peptide Binders via Discrete Flow Matching. https://doi.org/10.1101/2024.07.31.606098
- OpenAI. (2026). ChatGPT (GPT-5.2) [Large language model]. https://chat.openai.com/