Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Based on Protocol for Phusion™ High-Fidelity PCR Master Mix with GC Buffer (https://www.neb.com/en-gb/protocols/protocol-for-phusion-high-fidelity-pcr-master-mix-with-gc-buffer-m0532)
Phusion DNA Polymerase - a high-fidelity enzyme that synthesises new DNA strands with 3’→5’ exonuclease activity for proofreading dNTPs - building blocks for DNA synthesis (200 µM each at 1X concentration) MgCl₂ - Cofactor required for polymerase activity (1.5 mM at 1X concentration) Possibly optional as the question doesn’t mention it (GC Buffer - optimised buffer for amplifying difficult templates with high GC content or secondary structure) DMSO (optional additive) - helps denature secondary structures in GC-rich or difficult templates (recommended at 3% final concentration)
What are some factors that determine primer annealing temperature during PCR?
Primer Tm as in melting temperature, primer length, GC content, primer pair Tm similarity
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR vs. Restriction Enzyme Digests
Comparison Table
| Aspect | PCR | Restriction Enzyme Digests |
|---|---|---|
| Mechanics | Amplifies specific DNA regions using primers | Cuts DNA at specific recognition sequences |
| Flexibility | More flexible; doesn’t require specific cut sites | Limited by existing restriction sites in sequence |
| Mutations | Can introduce mutations via primer design | Cannot introduce mutations |
| End Products | Variable ends based on primer design | Creates predictable, precise ends |
| Time needed | ~90 minutes with thermal cycling | Faster (~1-2 hours incubation) |
| Template amount | Can work with small amounts of template | Requires sufficient DNA quantity |
| Sticky ends | Not applicable | Can create compatible sticky ends |
| When to use | When introducing mutations into sequences, no suitable restriction sites exist in the DNA, from small amounts of template DNA | When restriction sites are conveniently located in the sequence, Creating compatible sticky ends for cloning, Cutting plasmids for traditional cloning applications, Precise, predictable cuts are required |
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Ensure primers have 20-22 bp overhangs that are complementary between fragments
After PCR and DpnI digest, purify the DNA using the Zymo DNA Clean & Concentrator kit and measure concentration using Nanodrop/Qubit. The concentration should be above ~30 ng/µL.
Run the PCR products on an agarose gel to verify correct band sizes. Mix 3 µL of sample with 3.3 µL of 6x Loading Dye, run at ~100 mV for 15 min, and compare against a DNA ladder. Calculate your predicted digest on Benchling to verify the correct band size matches what you see on the gel
Ensure only unmethylated PCR products (not the original methylated template plasmid) are present through DpnI Treatment, reducing background colonies from unmutated template.
How does the plasmid DNA enter the E. coli cells during transformation?
By diffusion when the cells are shocked
Describe another assembly method in detail (such as Golden Gate Assembly) Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate assembly is a molecular cloning technique used to join DNA fragments in a single reaction. Methods for golden gate include MoClo, GoldenBraid 2.0, Mobius assembly and EMMA. It’s an efficient and cost-effective means to generate construct variants. Type IIS restriction enzymes are used to assemble multiple fragments in a single reaction. It’s also important to make sure that the Type IIS included in the process is not present in the vector or insert sequences
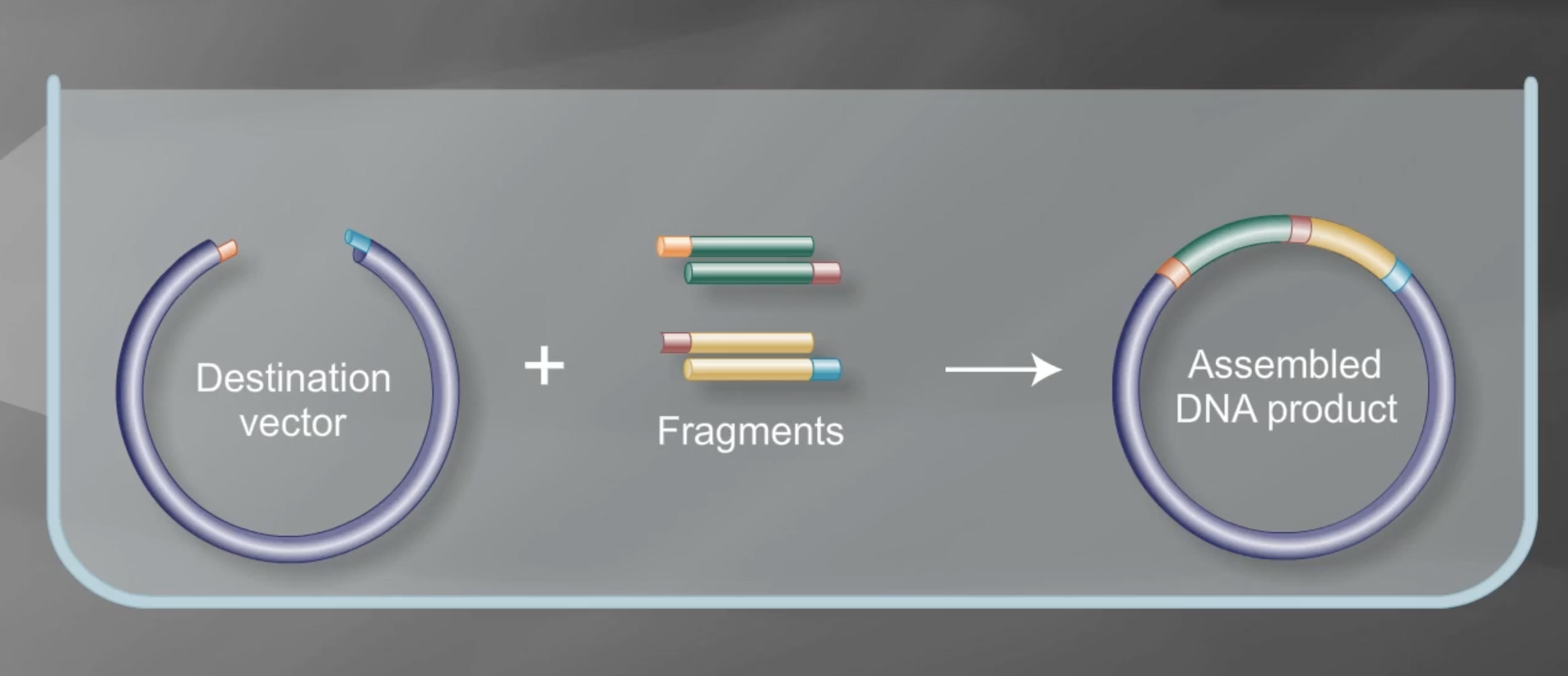 Screenshot taken from https://www.youtube.com/watch?v=JuiJASyG9gc
Screenshot taken from https://www.youtube.com/watch?v=JuiJASyG9gc
Model this assembly method with Benchling or Asimov Kernel!
As we don’t have access to Asimove I’ll be modelling this with Benchling
First, I added the DNA sequense of the well-known green fluorescent protein (GFP), which is native to the jellyfish Aequorea victoria
Changed the GGTCTC to GGTCTT to be able to work with the Golden Gate Assembly. Then I’ve added the plasmid to the project. I used the same one described in the lab. Then I checked for the presence of GGTCTC to use as the typeII enzyme.. there were 2 of those. Checked the other ones 2 of those again. Basically all three Type IIS enzymes (BsaI, BbsI, and Esp3I) have recognition sites in the mUAV plasmid
I ended up editing the GGTCTC ones as they were all in the base parts of the plasmid so they didn’t affect much. ( GGTCTC → GGTCTT )
Then I figured out that amilCP gene starts at 2114 and ends at 2779 Complement: A↔T, G↔C
I then loooked for stop codons and found the TAA one. For the primer design i also needed the 20 protein sequence of the GFP which was atgtctaaaggtgaagaatt. Basically my primer components ended up being BsaI site: GGTCTC, Overhang A: AATG (includes the ATG start codon) and the binding region which is those first 20 bp of GFP = atgtctaaaggtgaagaatt. Then for the reverse binding region, the positions of 666 to 685 (20 before the TAA) are TGGTCTTGTTAGAATTTGTT therefore reverse complement ones are AACAAATTCTAACAAGACCA
End primers are GFP Forward 5’-GGTCTCAATGTCTAAAGGTGAAGAATT-3’ and GFP Reverse 5’-GAGACCAAGCAACAAATTCTAACAAGACCA-3'
Then as for the backbone forward primer 5’-GGTCTCGCTTAAGCTTCAAATAAAACGAA-3’ as it is (taagcttcaaataaaacgaa) where GGTCTC is a BsaI recognition site, G a spacer base, and CTTAAGCTTCAAATAAAACGAA a binding region.
Backbone Reverse ended up being 5’-GAGACCCATTTTAGTATTTCTCCTCTTTCT-3'
Then knowing all this it’s time to do the Golden Gate Assembly
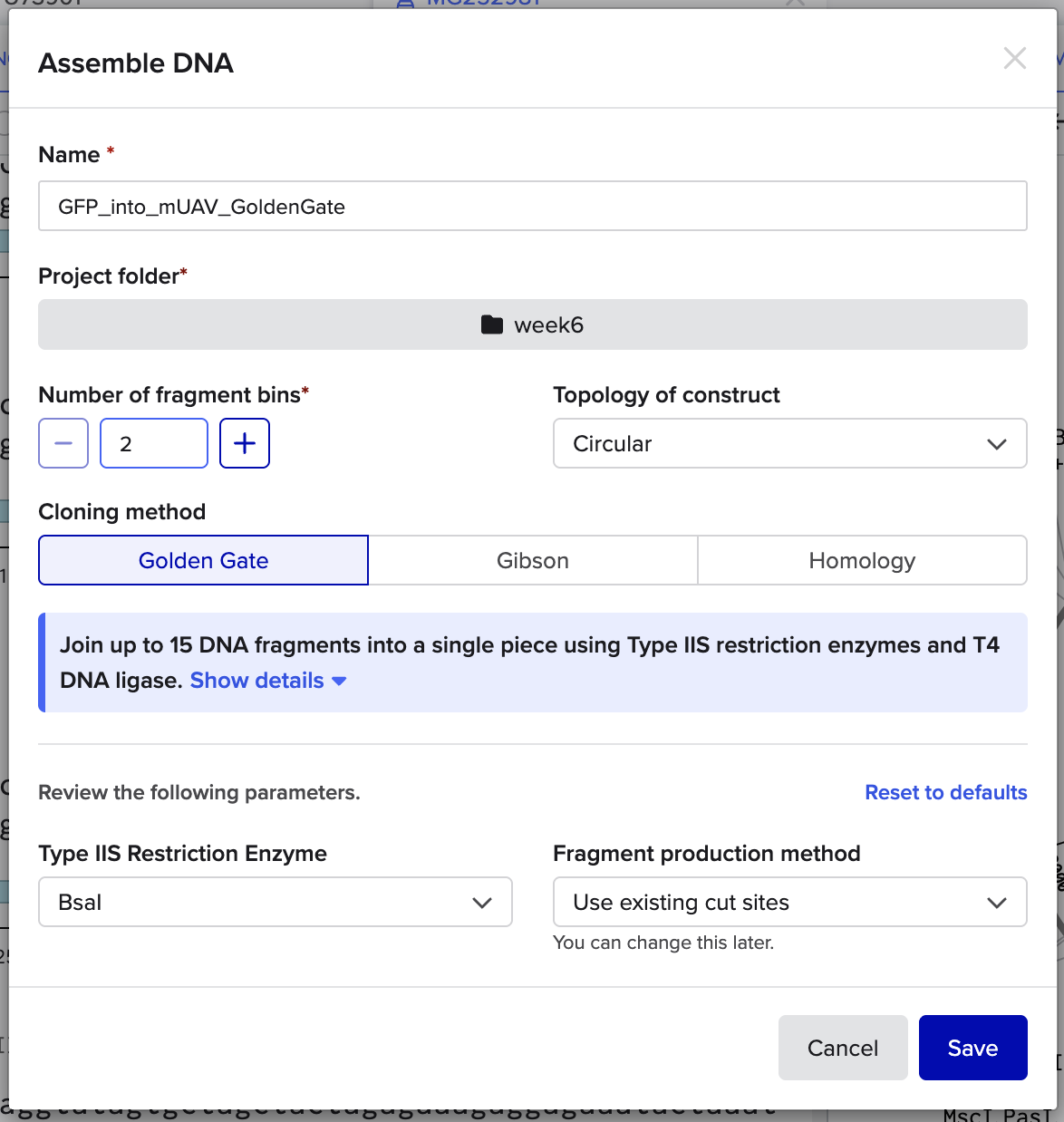
Needed to change the setting from use cut lines otherwise i was getting an error
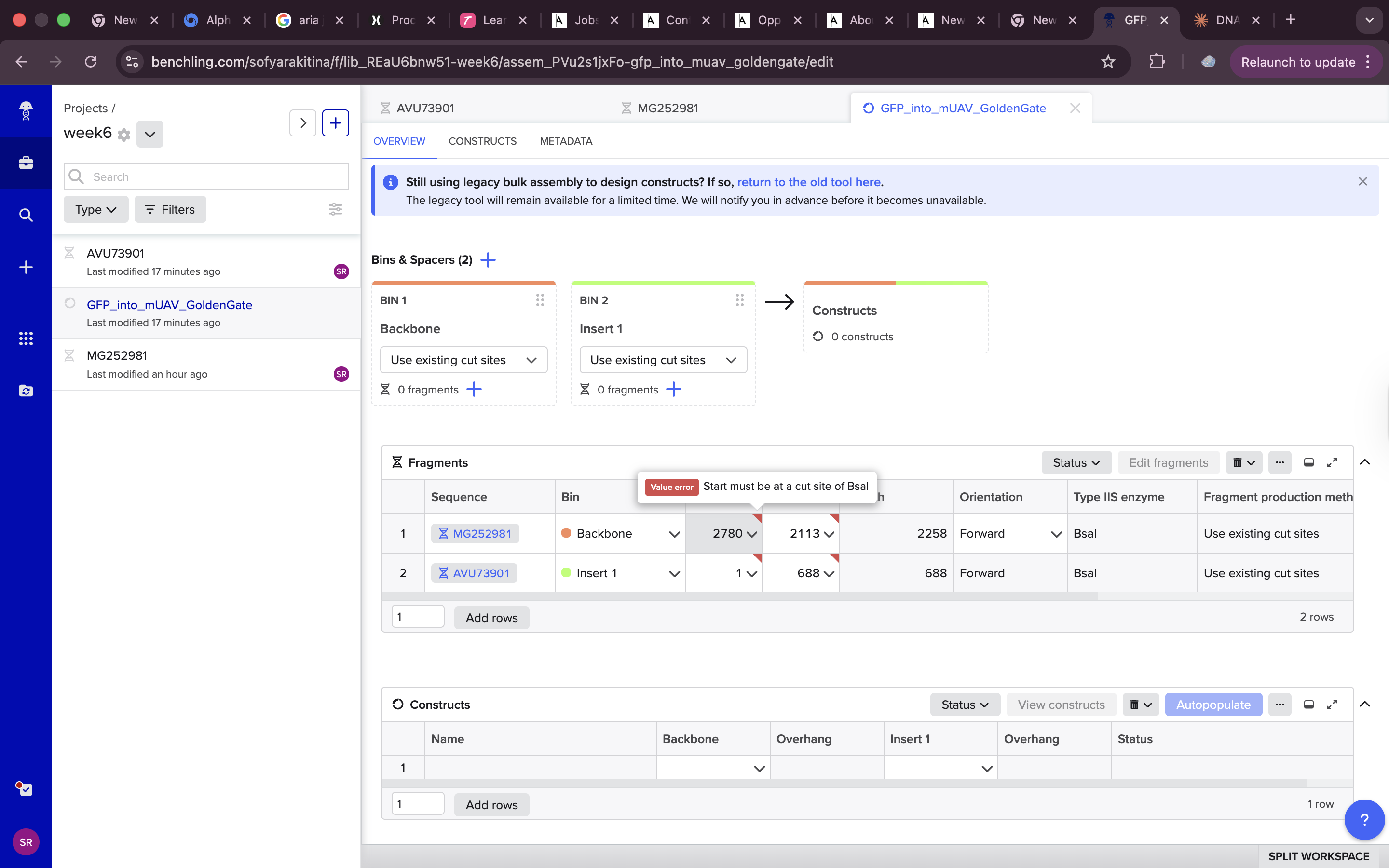 but other than that it was a pretty straightforward process
but other than that it was a pretty straightforward process
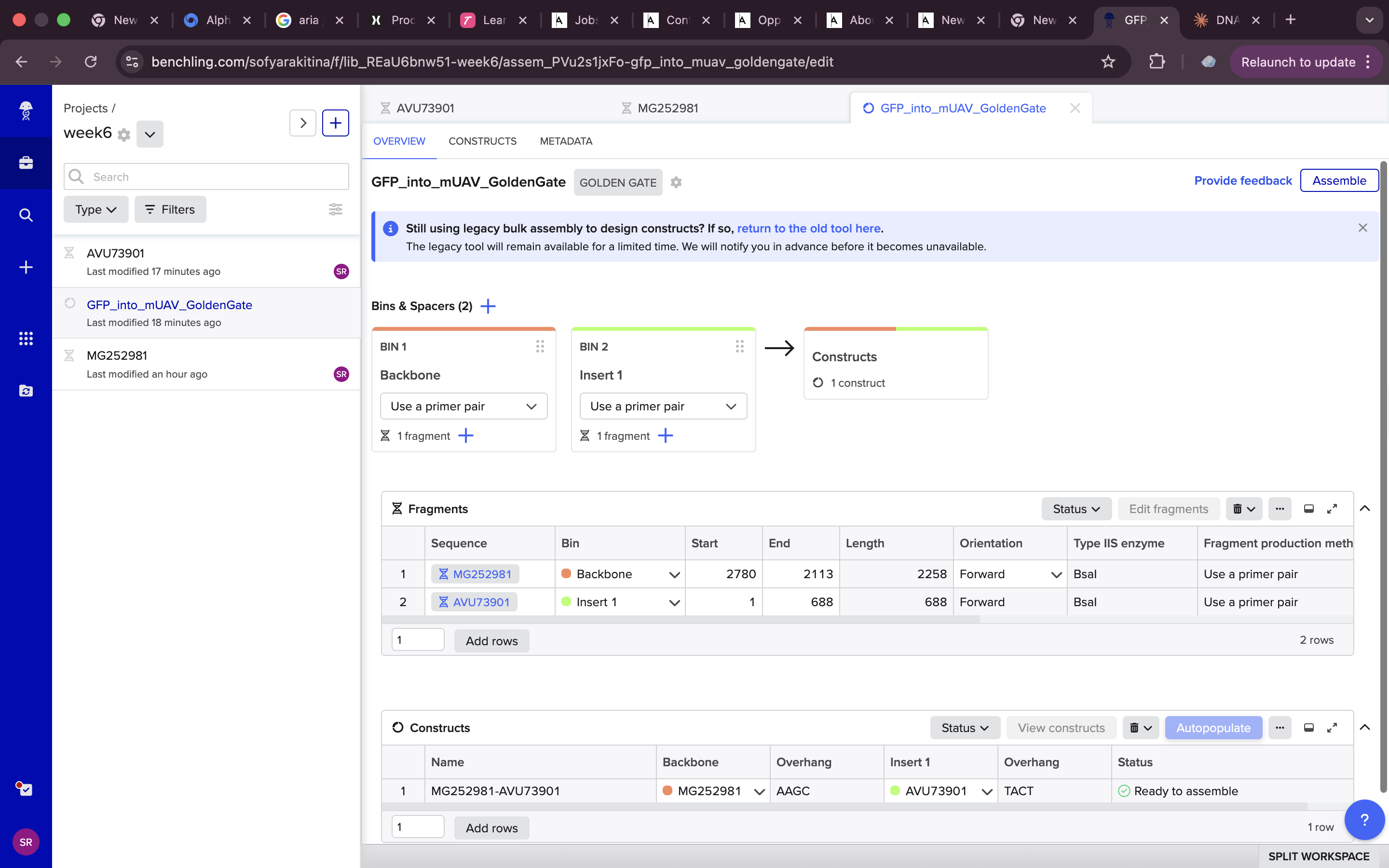
and here are some final setting that I used
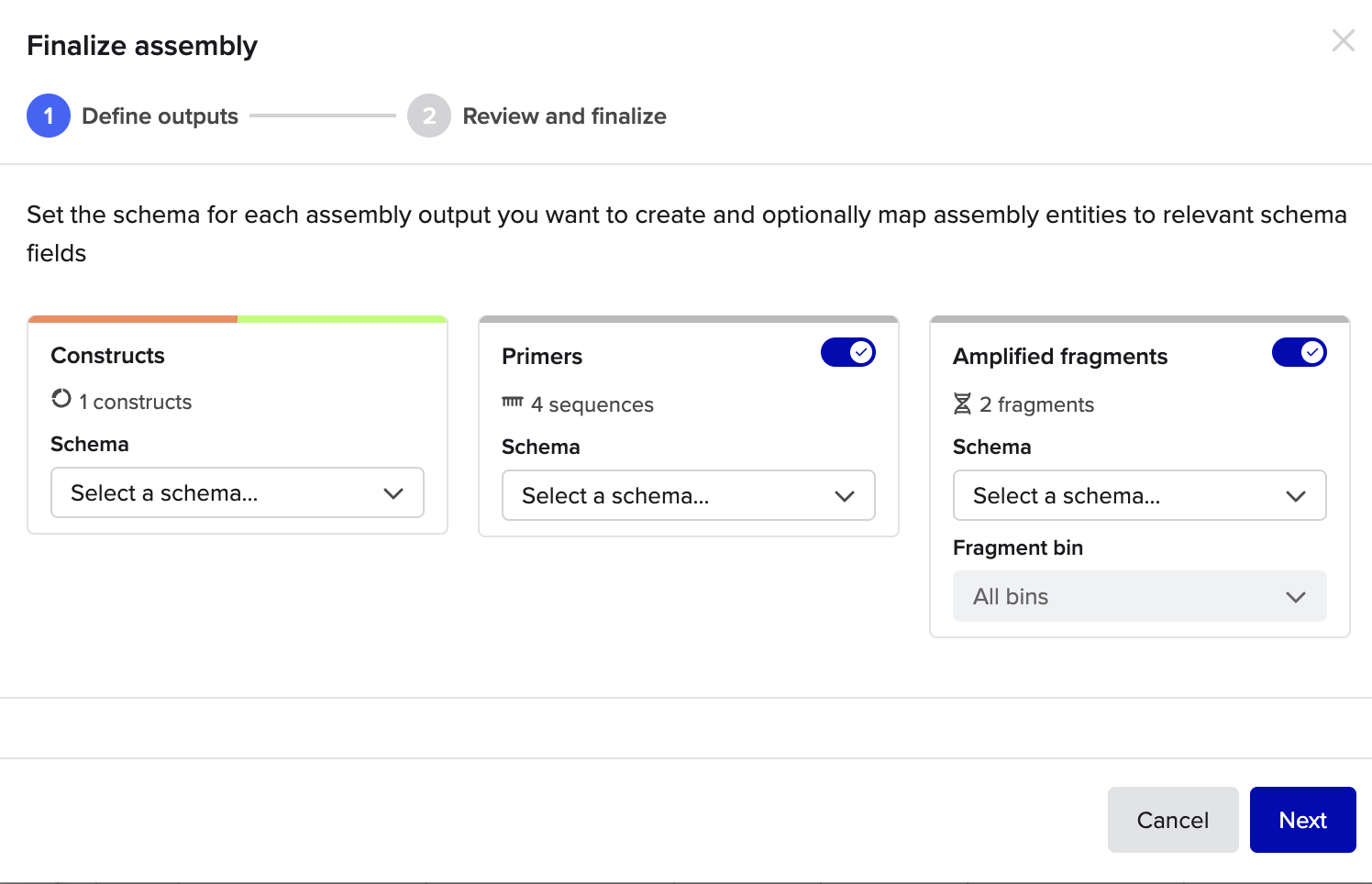
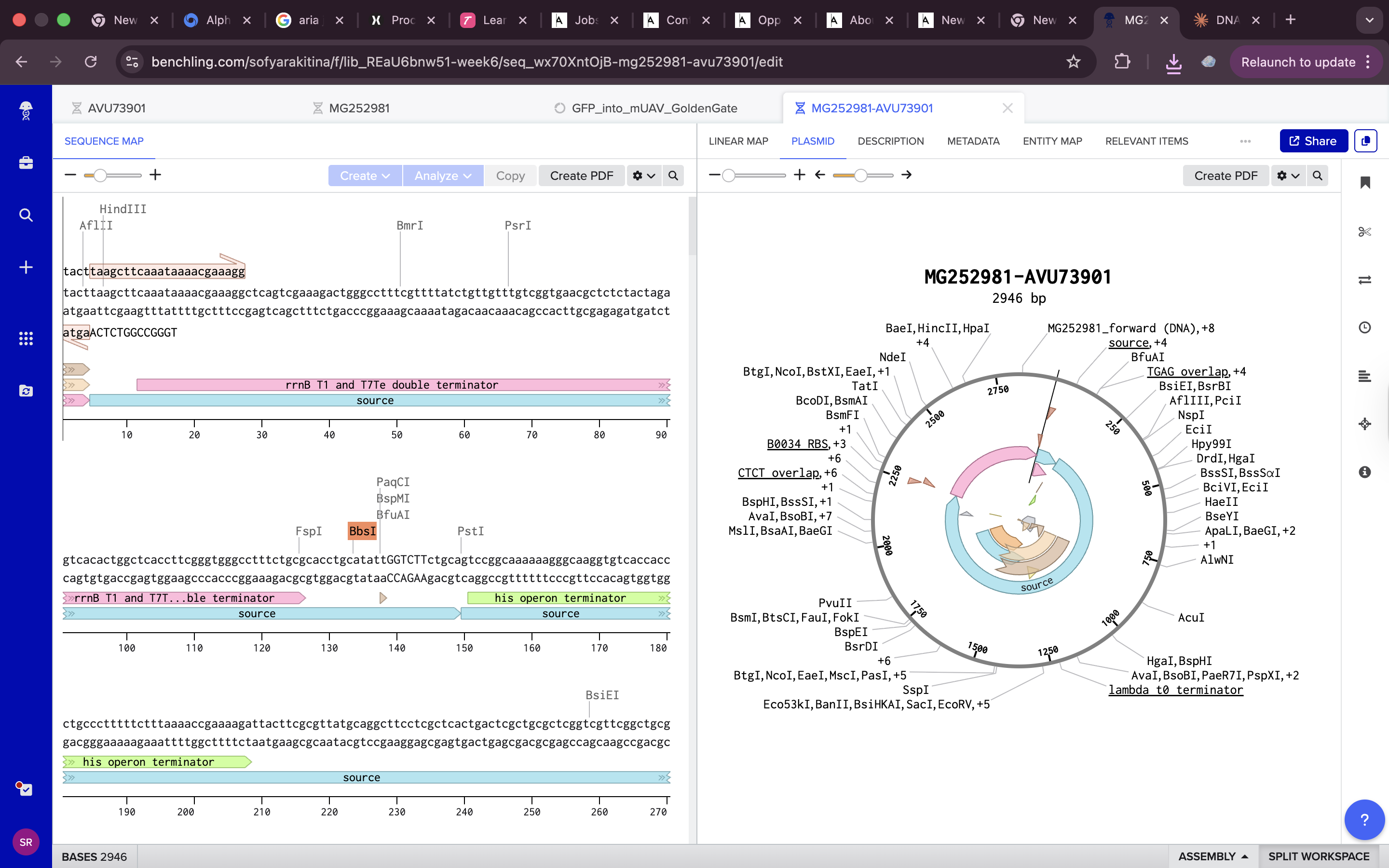
all done !!! https://benchling.com/s/seq-7kPMc9tGfTBvUaQHkniq?m=slm-VIKLDWpNc1o4GKeuM4Ii
Assignment: Asimov Kernel
(didn’t get access)