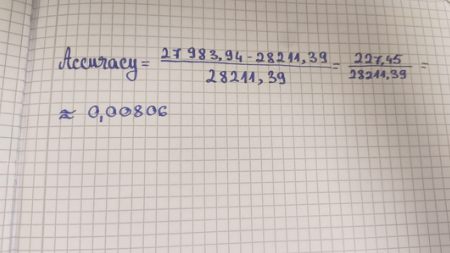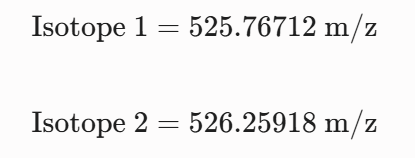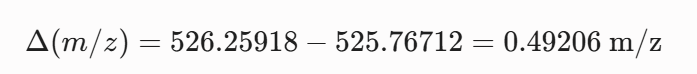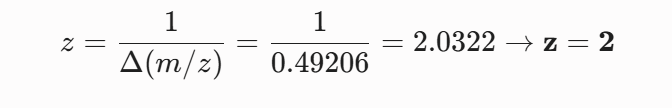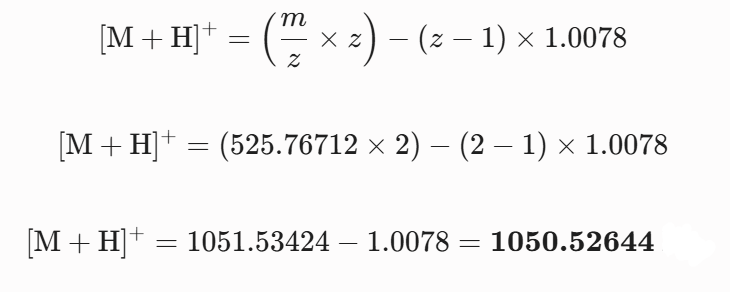First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. My idea: • The use of biotechnology to make pills or capsules , that reduce the glucose spike after a meal rich in carbohydrates . • My idea came from some podcasts which have as principal subject the book “Glucose revolution” by Jessie Inchauspe and also from her videos . • -I think that if the pills contains mulberry leaf extract , acetic acid , eriocitrin, the result be long term and will came faster. • The pills should be useful for everyone , diabetics , nondiabetics, prediabetics • The addition of mulberry leaf extract to sucrose resulted in a significantly lower glycemic response and insulinemic response compared to a matched placebo (sucrose alone). The change in blood glucose measurements were significantly lower at 15 min (p < 0.001), 30 min (p < 0.001), 45 min (p = 0.008), and 120 min (p < 0.001) and plasma insulin measurements were significantly lower at 15 min (p < 0.001), 30 min (p < 0.001), 45 min (p < 0.001), 60 min (p = 0.001) and 120 min (p < 0.001). The glucose iAUC (- 42%, p = 0.001), insulin iAUC (- 40%, p < 0.001), peak glucose (- 40.0%, p < 0.001) and peak insulin (- 41%, p < 0.001) from baseline were significantly lower for white mulberry leaf extract compared with the placebo. White mulberry leaf extract was well tolerated and there were no reported adverse events. • This study evaluated the potential effectiveness of different doses of Eriomin® on hyperglycemia and insulin resistance associated with other metabolic biomarkers in prediabetic individuals. Prediabetes patients (n = 103, 49 ± 10 years) were randomly divided into four parallel groups: (a) Placebo; (b) Eriomin 200 mg; (c) Eriomin 400 mg; and (d) Eriomin 800 mg. Assessment of biochemical, metabolic, inflammatory, hepatic, renal, anthropometric markers, blood pressure, and dietary parameters were performed during 12 weeks of intervention. Treatment with all doses of Eriomin (200, 400, and 800 mg) had similar effects and altered significantly the following variables: blood glucose (-5%), insulin resistance (-7%), glucose intolerance (-7%), glycated hemoglobin (-2%), glucagon (-6.5%), C-peptide (-5%), hsCRP (-12%), interleukin-6 (-13%), TNFα (-11%), lipid peroxidation (-17%), systolic blood pressure (-8%), GLP-1 (+15%), adiponectin (+19%), and antioxidant capacity (+6%). Eriomin or placebo did not influence the anthropometric and dietary variables. Short-term intervention with Eriomin, at doses of 200, 400, or 800 mg/day, benefited glycemic control, reduced systemic inflammation and oxidative stress, and reversed the prediabetic condition in 24% of the evaluated patients. these studies were taken from : https://pubmed.ncbi.nlm.nih.gov/36644880/https://pubmed.ncbi.nlm.nih.gov/33858439/https://pubmed.ncbi.nlm.nih.gov/31183921/ 2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy. Main objective: -Ensuring the responsible and rigorous use of nutrition engineering technologies; 2.1 Patient safety: Testing carefully and prevention of the extracts side effects; 2.2 Equity of access: Preventing inequitable access to the pills , where only small groups of population can use them. the benefits have to be accessible for everyone , both in terms of cost and the information about them, which can convince they to buy them to improve them lifestyle and in order to enjoy carbohydrate -rich meals.
Part 1: Benchling & In-silico Gel Art
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. You might find Ronan’s website a helpful tool for quickly iterating on designs!
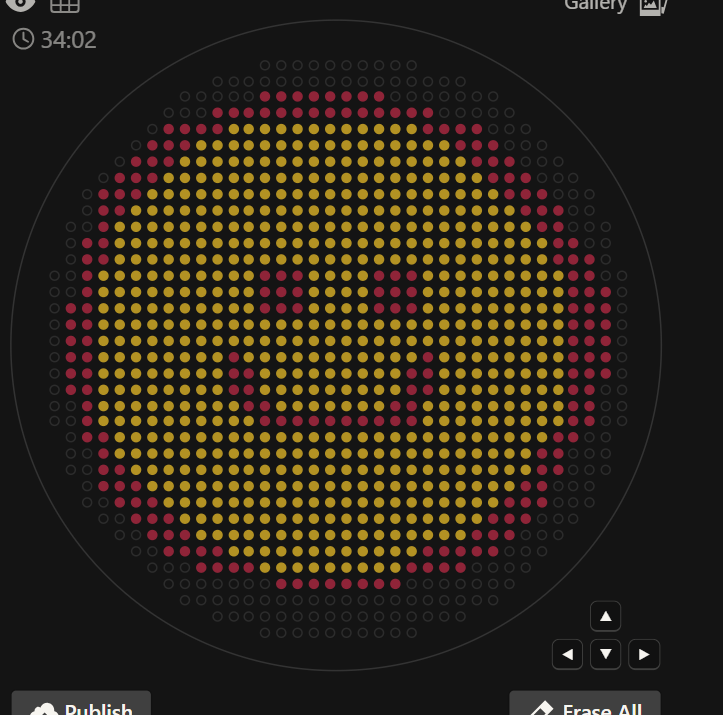
1 Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. I searched some images and when I found what I wanted I made a screenshot of it and I put it in openntrons.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1 What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
General homework questions
1 Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Waters Part I — Molecular Weight
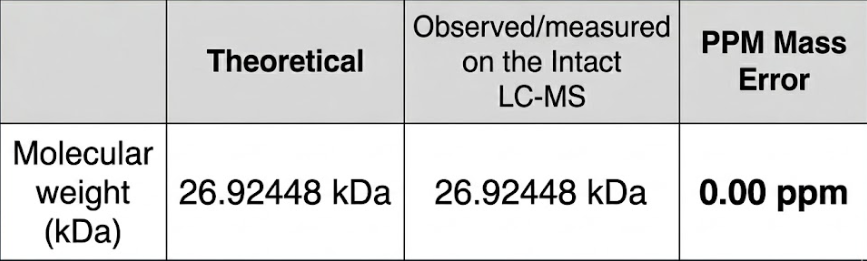
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I did not have a chance to contribute, but I will try to become a TA this fall! 😉
Subsections of Homework
Week 1 HW: Principles and Practices
1. First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
My idea:
• The use of biotechnology to make pills or capsules , that reduce the glucose spike after a meal rich in carbohydrates .
• My idea came from some podcasts which have as principal subject the book “Glucose revolution” by Jessie Inchauspe and also from her videos .
• -I think that if the pills contains mulberry leaf extract , acetic acid , eriocitrin, the result be long term and will came faster.
• The pills should be useful for everyone , diabetics , nondiabetics, prediabetics
• The addition of mulberry leaf extract to sucrose resulted in a significantly lower glycemic response and insulinemic response compared to a matched placebo (sucrose alone). The change in blood glucose measurements were significantly lower at 15 min (p < 0.001), 30 min (p < 0.001), 45 min (p = 0.008), and 120 min (p < 0.001) and plasma insulin measurements were significantly lower at 15 min (p < 0.001), 30 min (p < 0.001), 45 min (p < 0.001), 60 min (p = 0.001) and 120 min (p < 0.001). The glucose iAUC (- 42%, p = 0.001), insulin iAUC (- 40%, p < 0.001), peak glucose (- 40.0%, p < 0.001) and peak insulin (- 41%, p < 0.001) from baseline were significantly lower for white mulberry leaf extract compared with the placebo. White mulberry leaf extract was well tolerated and there were no reported adverse events.
• This study evaluated the potential effectiveness of different doses of Eriomin® on hyperglycemia and insulin resistance associated with other metabolic biomarkers in prediabetic individuals. Prediabetes patients (n = 103, 49 ± 10 years) were randomly divided into four parallel groups: (a) Placebo; (b) Eriomin 200 mg; (c) Eriomin 400 mg; and (d) Eriomin 800 mg. Assessment of biochemical, metabolic, inflammatory, hepatic, renal, anthropometric markers, blood pressure, and dietary parameters were performed during 12 weeks of intervention. Treatment with all doses of Eriomin (200, 400, and 800 mg) had similar effects and altered significantly the following variables: blood glucose (-5%), insulin resistance (-7%), glucose intolerance (-7%), glycated hemoglobin (-2%), glucagon (-6.5%), C-peptide (-5%), hsCRP (-12%), interleukin-6 (-13%), TNFα (-11%), lipid peroxidation (-17%), systolic blood pressure (-8%), GLP-1 (+15%), adiponectin (+19%), and antioxidant capacity (+6%). Eriomin or placebo did not influence the anthropometric and dietary variables. Short-term intervention with Eriomin, at doses of 200, 400, or 800 mg/day, benefited glycemic control, reduced systemic inflammation and oxidative stress, and reversed the prediabetic condition in 24% of the evaluated patients.
these studies were taken from :
https://pubmed.ncbi.nlm.nih.gov/36644880/https://pubmed.ncbi.nlm.nih.gov/33858439/https://pubmed.ncbi.nlm.nih.gov/31183921/2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Main objective:
-Ensuring the responsible and rigorous use of nutrition engineering technologies;
2.1 Patient safety:
Testing carefully and prevention of the extracts side effects;
2.2 Equity of access:
Preventing inequitable access to the pills , where only small groups of population can use them. the benefits have to be accessible for everyone , both in terms of cost and the information about them, which can convince they to buy them to improve them lifestyle and in order to enjoy carbohydrate -rich meals.
3 Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
3.1 Implementing strict regulation
-Purpose :To ensure that the process is correct and the result is not harmful.
-Desing: A set of rules it will be created and will stay as a contract that companies and researchers have to sign .
-Assumptions: There might be resistance from industries or individuals who don’t thinks these rules are necessary for an already existing product;
-Risks of Failure &Success: If the regulations are too strict , it could limit the research or the process for making them;
3.2 Establishing public funding initiatives
-Purpose: To make the pills accessible for many categories of people , without lost profit.
-Design: Government and international organizations would collaborate to fund nutritional research and subsidize treatments for low-income population.
-Assumptions: The companies might not like the idea and the case and they won’t be interested to found such initiatives.
-Risks of Failure &Success: If public funding is not sufficient , the program could fail, but if it works it will be very useful for many people.
3.3 Public education and awareness campaigns
-Purpose: To make people to understand how glucose-lowering capsules work or their benefits and risks.
-Desing: Some necessary things for campaigns are educative materials, like brochures and videos. Also will be making workshops or webinars with healthcare.
Assumptions : People will engage with and trust the educational materials;
Risks of Failure &Success: Low participation or attention from the public and well-informed users may increase demand faster than supply;
Does the option:
Action1:Pre-market testing
Action2:Labeling & transparency
Action3:Public education
Enhance Biosecurity
• By preventing incidents
1
2
3
• By helping respond
2
2
1
Foster Lab Safety
• By preventing incident
1
2
3
• By helping respond
2
2
1
Protect the environment
• By preventing incidents
1
2
3
• By helping respond
2
2
1
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
2
1
1
• Not impede research
3
2
1
• Promote constructive applications
1
2
1
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Based on scoring, I would prioritize Action1. Action2 and 3 are almost as supportive as the first one, but secondary measure. This is a very correct approach, but not very usual. Capsules that are safe for use with the intention of limiting possible incidents in the lab as well as in the nature. The capsules rated best for the prevention of incidents and promoting safe uses. Even if the cost is higher which leads to research struggles the results ale benefical.
Homework Questions from Professor Jacobson: [Lecture 2 slides]
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The error rate of polymerase is 1:106.The human genome contains approximately 3 billion base pairs(3x109).Without correction , this would result in aprox. 3000 errors every time a cell divides. Biologicaly after replication, the secondary protein systems scans the DNA to fix remaining discrepancies.Much of the human genome is non-coding (introns) or “dormant,” meaning many mutations occur in areas that don’t affect cell function.For an average human protein (~345 amino acids / 1036 bp), there are roughly 10^157 possible DNA sequences that could code for it, because most amino acids are represented by multiple codons.
Homework Questions from Dr. LeProust: [Lecture 2 slides]
What’s the most commonly used method for oligo synthesis currently?
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Why can’t you make a 2000bp gene via direct oligo synthesis?
The gold standarts is solid-phase phsphoramidite chemical syntesis. Synthesizing strands longer than 200 nucleotides is difficult due to stepwise error accumulation. Even with a 99.5% efficiency rate per step, the mathematical probability of a perfect sequence drops exponentially as length increases.By 200 bases, the yield of the “perfect” product is very low. Additionally, side reactions and physical crowding on the synthesis support hinder the process.Why 2000bp is Impossible via Direct SynthesisA 2000bp gene (4000 total nucleotides) cannot be made in one go because the cumulative error rate would result in zero functional product. Instead, scientists synthesize small “oligos” and then stitch them together using biological assembly methods.
**Homework Question from George Church: [Lecture 2 slides]
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
[(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:**
The 10 Essential Amino AcidsThe ten amino acids that most animals (including humans) cannot produce on their own are:Arginine (R)*, Histidine (H), Isoleucine (I), Leucine (L), Lysine (K), Methionine (M), Phenylalanine (F), Threonine (T), Tryptophan (W), and Valine (V).*Note: Arginine is often considered “semi-essential,” especially for growth.View of the “Lysine Contingency"The “Lysine Contingency” (from Jurassic Park) is a flawed scientific concept for two main reasons:Vertebrate Biology: No vertebrate can synthesize lysine naturally. Therefore, “knocking out” the ability for dinosaurs to make it doesn’t change anything—they were already dependent on their diet for it.Ecological Reality: In the wild, lysine is abundant in plants and other animals. If the dinosaurs escaped, they would simply eat lysine-rich food (like soy or meat), rendering the laboratory “failsafe” completely useless.
Week 2-DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.You might find Ronan’s website a helpful tool for quickly iterating on designs!
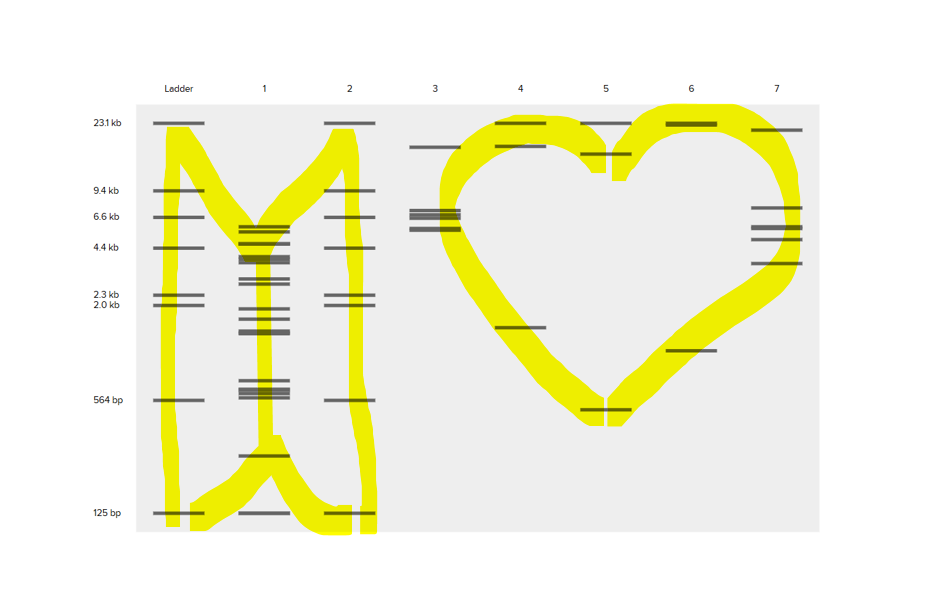
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
I didn’t have acces to a lab ,so I studied the protocol.
Part 3: DNA Design Challenge
3.1. Choose your protein.
I choose Dsup-Damage suppressor protein from Ramazzottius varieornatus-tardigrade, because Dsup is a fascinating protein, unique to extremophile tardigrades, which binds to nucleosomes and protects DNA from damage caused by radiation , desiccation and reactive oxigen species.This makes it highly relevant for applications in biotechnology, such as enhancing radiation resistance in cells for space exploration, cancer therapy, or environmental remediation. It’s also an intrinsically disordered protein, offering insights into novel DNA-protection mechanisms beyond traditional repair pathways.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I used uniprot to find Dusp protein amino acid sequence. uniprot-P0DOW4
This is the protein sequence:MASTHQSSTEPSSTGKSEETKKDASQGSGQDSKNVTVTKGTGSS
ATSAAIVKTGGSQGKDSSTTAGSSSTQGQKFSTTPTDPKTFSSDQKEKSKSPAKEVPS
GGDSKSQGDTKSQSDAKSSGQSQGQSKDSGKSSSDSSKSHSVIGAVKDVVAGAKDVAG
KAVEDAPSIMHTAVDAVKNAATTVKDVASSAASTVAEKVVDAYHSVVGDKTDDKKEGE
HSGDKKDDSKAGSGSGQGGDNKKSEGETSGQAESSSGNEGAAPAKGRGRGRPPAAAKG
VAKGAAKGAAASKGAKSGAESSKGGEQSSGDIEMADASSKGGSDQRDSAATVGEGGAS
GSEGGAKKGRGRGAGKKADAGDTSAEPPRRSSRLTSSGTGAGSAPAAAKGGAKRAASS
SSTPSNAKKQATGGAGKAAATKATAAKSAASKAPQNGAGAKKKGGKAGGRKRK
I reversed translated the amino acid sequnce into nucleotide sequence.
To produce the Dsup protein from this DNA, technologies leveraging the central dogma (DNA → RNA → protein) can be used. I'll describe both cell-dependent and cell-free methods.
Cell-dependent (in vivo): Insert the optimized DNA into an expression vector (e.g., pET-28a with T7 promoter). Transform the plasmid into E. coli (e.g., BL21(DE3) strain) via electroporation or heat shock. Induce expression with IPTG, which activates the T7 RNA polymerase to transcribe the DNA into mRNA. The mRNA is then translated by ribosomes using tRNAs to assemble the protein. Harvest cells, lyse them, and purify Dsup (e.g., via His-tag affinity chromatography if added).
Cell-free (in vitro): Use a cell-free expression system (e.g., PURExpress or wheat germ extract). Transcribe the DNA into mRNA using T7 RNA polymerase, NTPs, and buffers. Then, add the mRNA to a translation mix containing ribosomes, tRNAs, amino acids, energy sources (ATP/GTP), and cofactors. Translation occurs directly, producing the protein without cells. This is faster for screening but lower yield.
Both methods allow scalable production, with cell-dependent being cheaper for large quantities.
3.5.How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
In nature, the Dsup gene in tardigrades is transcribed into mRNA in the nucleus under stress conditions (e.g., radiation). The mRNA is exported to the cytoplasm, where it’s translated into the Dsup protein by ribosomes. Dsup then localizes to the nucleus via its nuclear localization signal, binding nucleosomes to shield DNA.
For alignment (using Benchling-style visualization):
DNA: atggcatccacacaccaatcatcc… (original sequence as in 3.2)
RNA: auggcauccacacaccaaucaucc… (T → U)
Protein: M A S T H Q S S T E P S… (each codon triplet → 1 AA)
This shows the flow: DNA transcribed to RNA (complementary, T→U), RNA translated in triplets (e.g., AUG = M start). Failures like mutations could alter this, but optimization helps overcome expression issues in heterologous systems.
Part 4: Prepare a Twist DNA Synthesis Order
**5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would like to sequence the skin microbiome of individuals with atopic dermatitis to identify the imbalance between beneficial and harmful bacteria, which would help me in creating a personalized treatment.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would chose the third generation sequencing , because it’s essential for distinguishing between related bacterial strands in the microbiome .The essentials steps are that the DNA is passed through a nanopore and as it moves, it creates specific fluctuations in an electrical current, which are then decoded directly into the DNA sequence.The limitations are that it has a higher error rate per base compared to short-read sequencing, though high-coverage analysis successfully mitigates this.
To complete this step, I must first collect and filter my raw input to remove errors. Next, categorize the remaining data into specific groups based on their shared traits. Finally, verify the results against your original requirements to ensure everything is accurate and ready for use.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would synthesize a Synthetic Antimicrobial Peptide (AMP) Genetic Circuit. This system uses a Quorum Sensing “switch”—a promoter that only activates when it detects high concentrations of S. aureus—linked to a gene for an Antimicrobial Peptide (like Omiganan). In patients with atopic dermatitis, the skin’s barrier is overwhelmed by harmful bacteria. By writing a circuit that only triggers in the presence of this specific pathogen, we create a personalized “search-and-destroy” therapeutic that restores microbial balance without the broad-spectrum damage of traditional antibiotics.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
For the synthesis, I would use Silicon-Based DNA Synthesis (Twist Bioscience). This technology uses a silicon chip to “write” thousands of different DNA sequences simultaneously, making it more scalable and cost-effective than old-fashioned plastic plate methods.
To verify that the synthesized DNA perfectly matches my design, I would use Next-Generation Sequencing (NGS). The essential steps include Library Prep , Cluster Generation , Sequencing by Synthesis (identifying bases via fluorescent flashes), and Data Analysis (reassembling the final code). While NGS is highly scalable, its main limitations are its total speed (taking 24–48 hours) and a slight dip in accuracy when encountering homopolymers.
Week 3 HW: Opentrons art
1 Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
I searched some images and when I found what I wanted I made a screenshot of it and I put it in openntrons.
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
I find this article " AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots “, which is prety interesting and I choose to describe it.
The article introduces AssemblyTron, an open-source Python software designed to automate the “Build” step in synthetic biology. It acts as a bond between DNA design software (like j5) and affordable laboratory robots (the Opentrons OT-2). AssemblyTron is a open-source software tool that uses a robot to build DNA automatically, making biological engineering faster, cheaper, and more accurate. It make the work in a shorter time , but with the same quality as a human and give to reaschers mor time to focus on more important things.
Week 4 HW: Protein Design Part I
A
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
A Dalton is equal to 1/12 atom of carbon, so aprox. 1.66x10-27kg.
I will chose beef , because the type isn’t specified . 100g of beef has aporx. 25g protein, so 500g has 125g protein. 1g=6.022x1023 atomic mass units =>125g=7,5275 x 10^25 Dalton
2.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans eat beef but do not become a cow, eat fish but do not become fish, because these foods are decomposed in substances that are used or throw away . They don’t have an impact on our DNA so we don’t become cows or fishes.
3. Why are there only 20 natural amino acids?
There are only 20 natural amino acids because like that they are more stable and it can provide a big variety of proteins.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, it is possible , but I won’t do it.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before enzymes that make them, and before life started amino acids come from abiotic synthesis. Simple inorganic molecules reacted under high-energy conditions.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
While natural L-amino acids form right-handed alpha-helices, a polymer made of D-amino acids would adopt a left-handed conformation.
7. If you make an α-helix using D-amino acids , there will be a left handedness.
Yes, they have been discovered as rare but stable variants. The 3-helix is more tightly wound and elongated, appearing at the termini of regular helices. The pi-helix is wider and shorter, it is found near functional sites in enzymes.
8. Why are most molecular helices right-handed?
Most molecular helices are right-handed because that direction keeps the amino acid side chains from crashing into the backbone.
9. Why do β-sheets tend to aggregate?
• What is the driving force for β-sheet aggregation?
Beta-sheets aggregate because of the hydrophobic effect, where water molecules force non-polar side chains together to hide them from the aqueous environment, making the sheets into stacks. Once they meet, unsatisfied hydrogen bonds along the edges act like a chemical glue, locking the strands into stable fibers called amyloids.
10. Why do many amyloid diseases form β-sheets?
• Can you use amyloid β-sheets as materials?
Sheet aggregation is Mostly governed by strong, repetitive hydrogen bonding that occurs between the backbone NH and C=O groups of neighboring strands. The role of dehydration and burial of the hydrophobic side chains is to assist this process. Besides this, stacking of aromatic rings and dipole interactions contribute by further stabilizing the structure. As a result, aligned -sheets are not only energetically favorable but are also very likely to form larger aggregates.
11. Design a β-sheet motif that forms a well-ordered structure.
For amyloid diseases, misfolded proteins form cross- fibrils due to trapping the exposed aggregation-prone regions in stable -sheet conformations that stack to form very long, persistent fibrils. Besides the pathological implications, the same stability and ability of self-assembly feature of amyloid fibrils can be exploited for generating nanofibers, hydrogels, and scaffolds used in tissue engineering and drug delivery. Forced folding of the -sheet motif in a -hairpin form with the sequence VIVTYGGYTVIV is a good example of this. Here the strands VIVTY and YTVIV are joined through the GG turn. One hydrophobic stacking face and one soluble face are produced by hydrophobic (Val Ile Tyr) and polar (Thr, Tyr) residues alternating along the strand. This sort of arrangement is that of a compact, ordered two-stranded -sheet.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
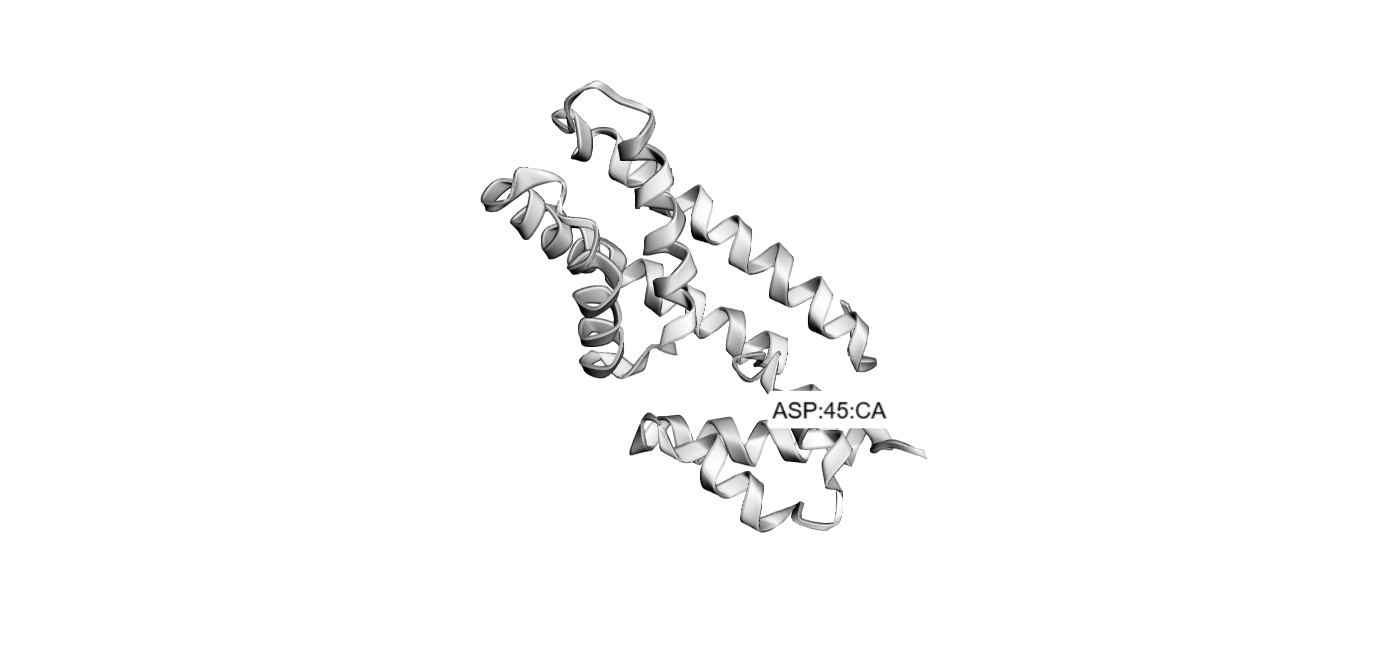
1Briefly describe the protein you selected and why you selected it.
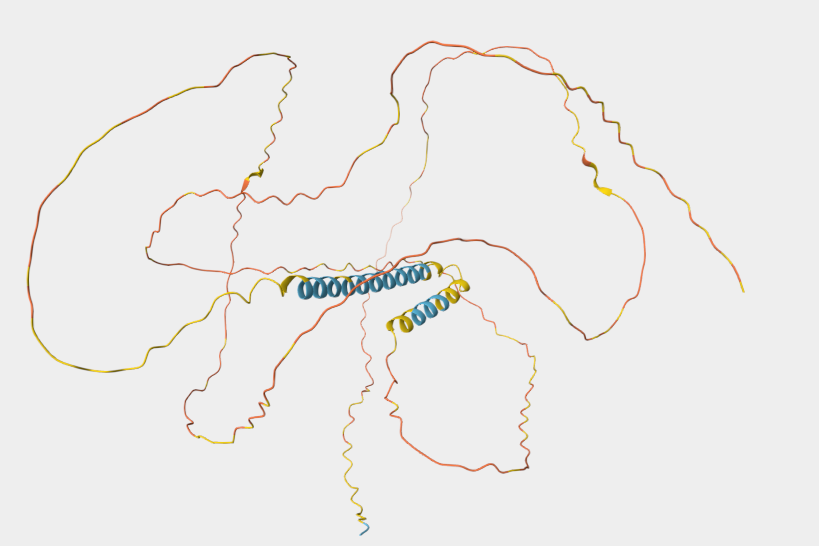
I chose the same protein that I used in homework two, because Dsup is very interseting and I like very uch tardigrades.
2 Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
THe protein ha 445 aminoacids.The most frequent aminoacid is Serine. It apears 84 times.
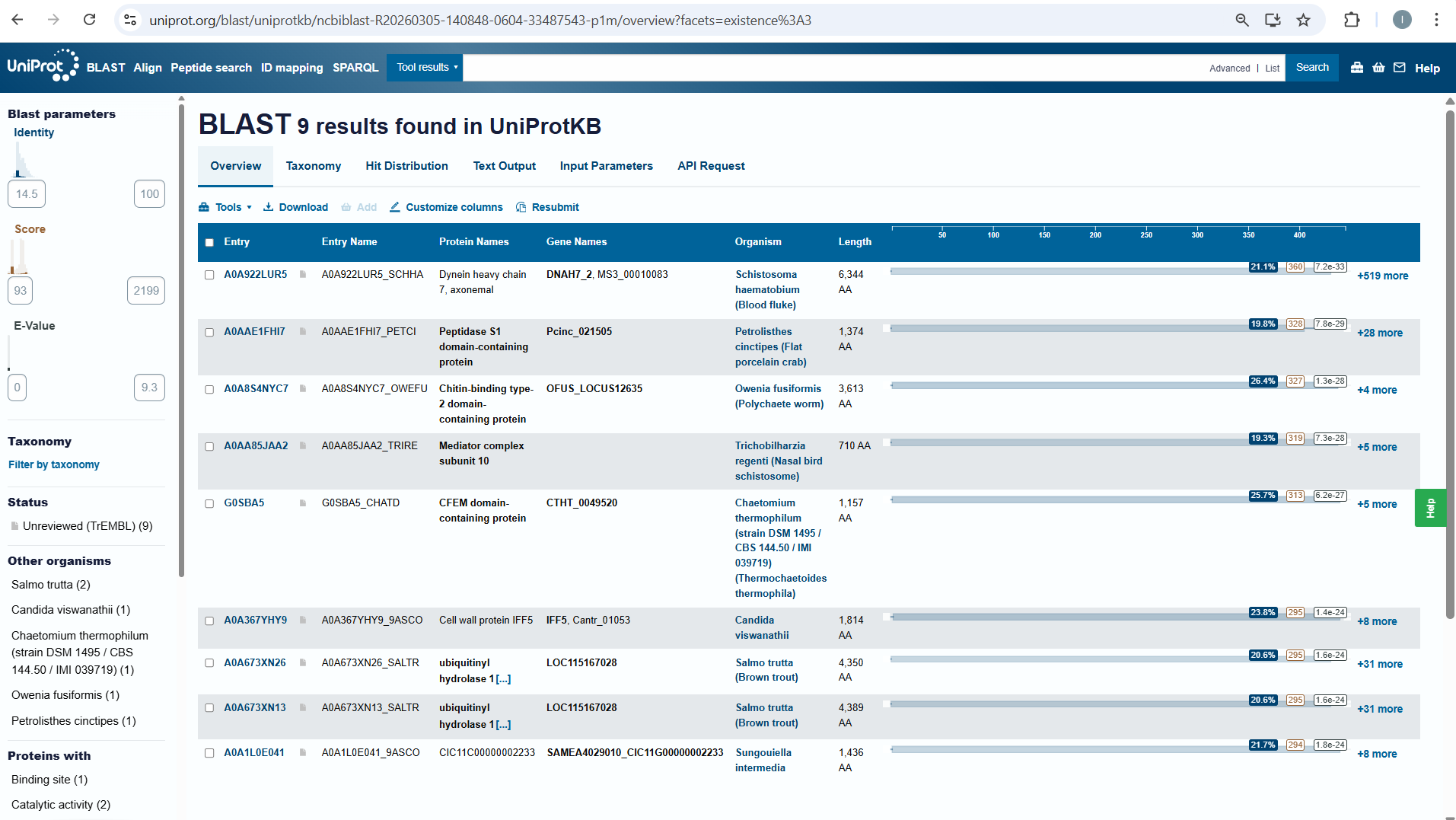
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
I searched for homologs in Uniprot and I think I managed to find them . So, I opened the Uniprot link is on the htgaa website at week 4 . I paste the aminoacid sequence and then I waited it to run.
Does your protein belong to any protein family?
NO, it doesn’t belong to any protein family.
**3 Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Are there any other molecules in the solved structure apart from protein?
Does your protein belong to any structure classification family?
Nucleosome-Binding Protein
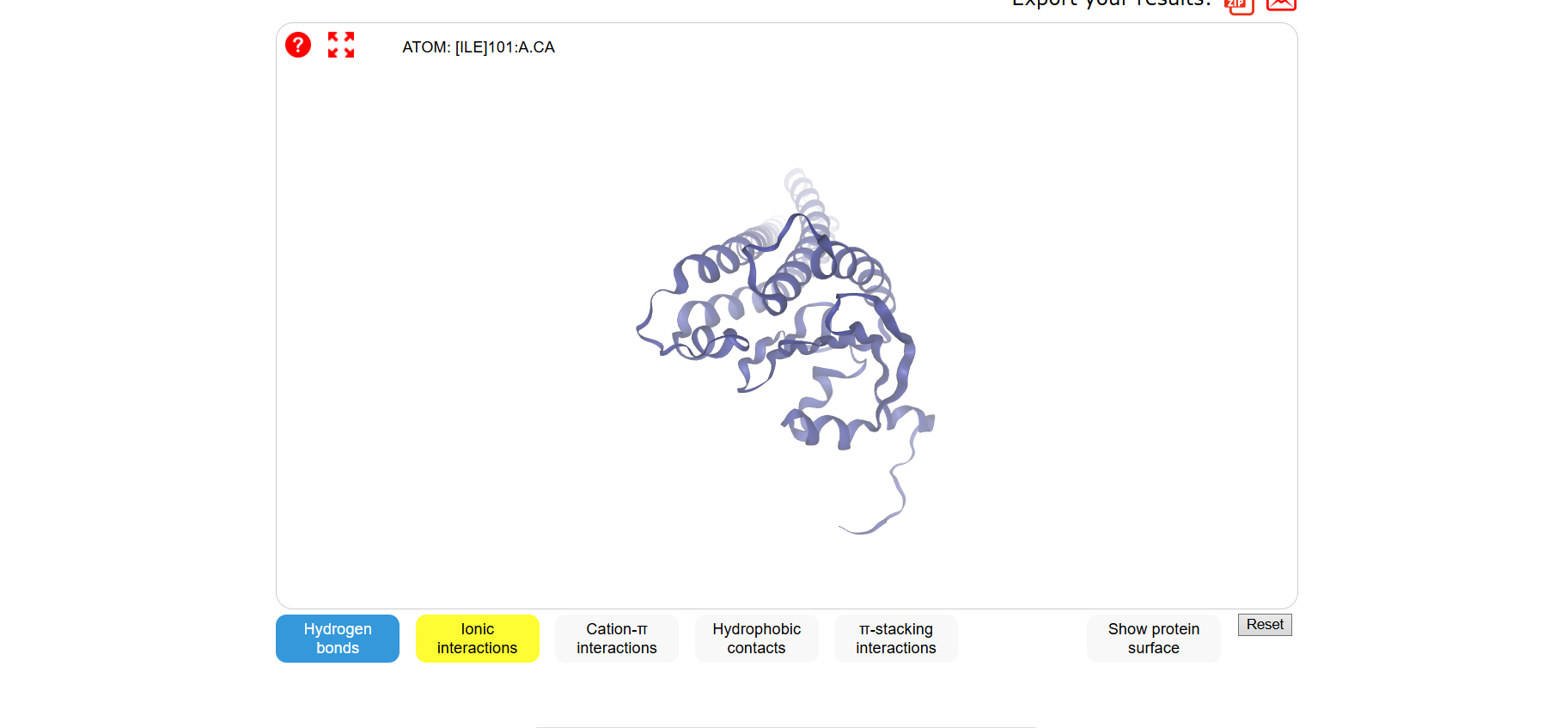
3 Open the structure of your protein in any 3D molecule visualization software:
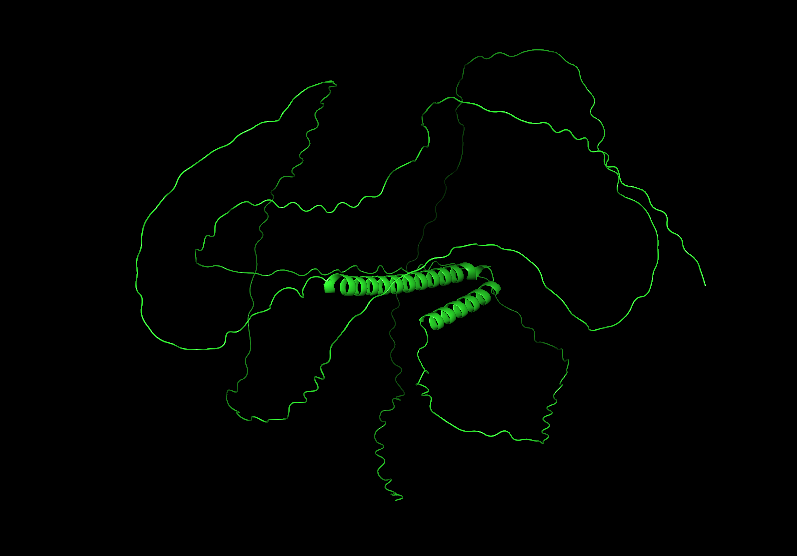
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
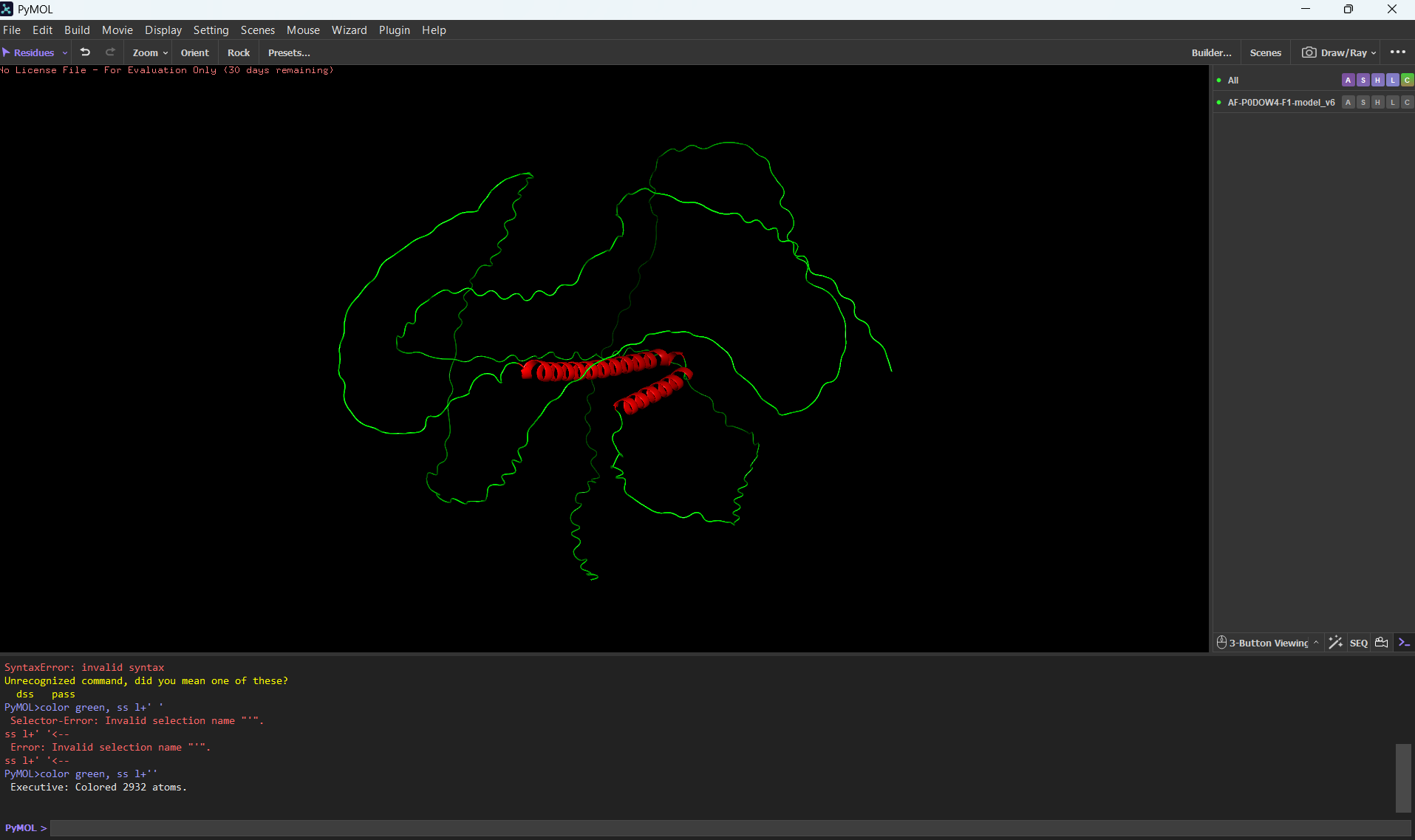
Color the protein by secondary structure. Does it have more helices or sheets?
It has more helices than sheets.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Most of the protein will remain green, with a few small cylinders representing transient alpha-helices.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
To color by secondary structure I used color red, ss h; color yellow, ss s; color green, ss l+’'.
Part C. Using ML-Based Protein Design Tools
Glu Glu Leu Ile Thr Gly Val Leu Gly Ile Ser Ile Asp Leu Gly Met Val Thr Gly Ser Asp Leu Ala Lys Ala Val Lys Leu Ala Thr Gly Leu Gly Glu Ala Val Val Glu Gly Ala Lys Ala Val Gly Ser Val Leu Ala Leu Ser Thr Ala Leu Val Leu Ala Leu Leu Gly Leu Ala Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu
Part D. Group Brainstorm on Bacteriophage Engineering
Project Proposal: Engineering a Minimal MS2-L Lysis Engine
1 Primary Goal: Our group aims to increase the functional stability of the MS2 lysis (L) protein. We will achieve this by eliminating the N-terminal domain (residues 1–36). This truncation removes the “regulatory brake” that normally makes lysis dependent on the host chaperone DnaJ, resulting in a more potent, autonomous lysis protein, thus lysis will be achieved a lot faster, beacuse MS2-L will be functional from the moment of translation which gives less time for the proteases to degrade it before it attached to the cellular membrane.
2 Tools & Approaches We propose a computational pipeline to validate and optimize this truncated variant:
AlphaFold3 / AlphaFold-Multimer: We will model the truncated L protein in a cramped lipid bilayer environment to predict how the remaining transmembrane helix (TMH) inserts itself. We will also use it to confirm the loss of binding affinity to E. coli DnaJ.
Protein Language Models (ESM-2 / ESM-1v): We will use these models to perform in silico mutagenesis on the remaining C-terminal sequence. Our goal is to identify “stabilizing” mutations that strengthen the alpha-helical propensity of the membrane-spanning region.
Molecular Dynamics (MD) Simulations (OpenMM or Gromacs): Since lysis involves membrane distortion, we will simulate the truncated protein within a model bacterial membrane to observe its ability to disrupt lipid packing.
4 Potential Pitfalls Membrane Toxicity in in silico models: Most protein design tools are trained on soluble proteins. Modeling a protein whose entire job is to destroy the membrane (lysis) may lead to unstable simulations or “unphysical” results.
Expression Levels: In a real-world lab setting, a highly toxic protein might kill the host cells so quickly that we cannot produce high enough titers of the phage for study.
5 Pipeline Schematic Design: Truncate N-terminus (1-36).
Optimize: Run ESM-1v to find high-probability stabilizing mutations.
Predict: Fold top candidates in AlphaFold3 to ensure TMH orientation.
Simulate: Insert into a virtual membrane to verify disruptive “toxicity.”
Week 5— Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
P00441 is the unique identifier for Human Superoxide Dismutase 1.
This is the original squence:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
This is the mutation squence:
utant Sequence (Target):
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
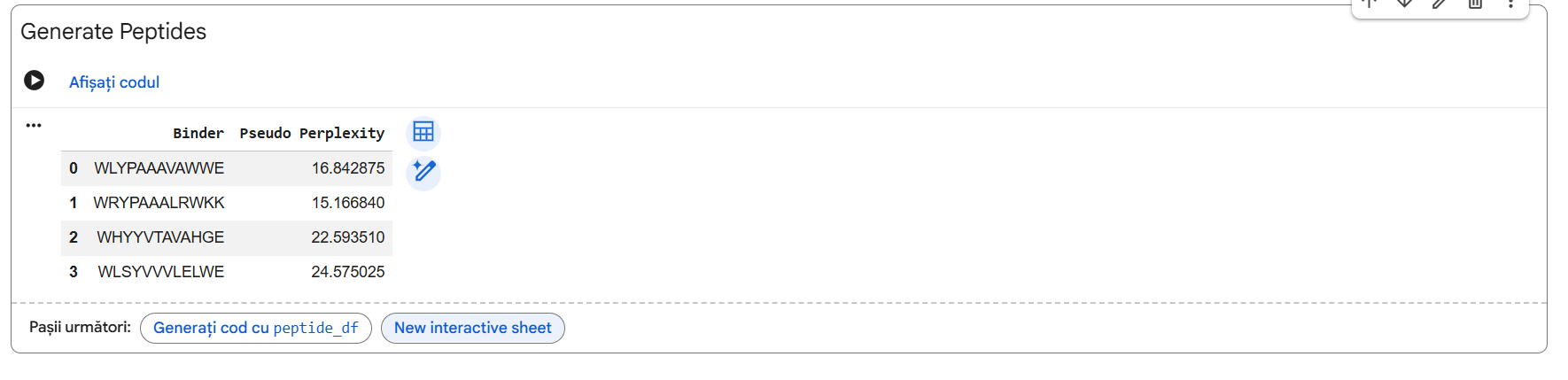
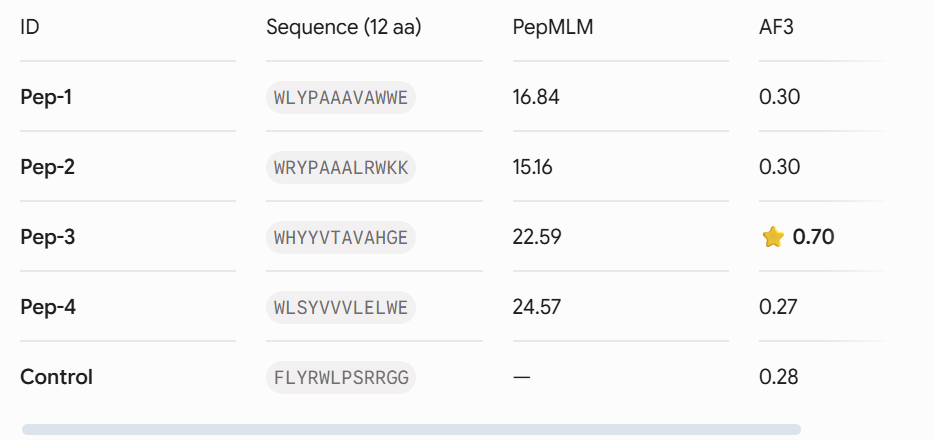
Four unique 12-amino acid peptides were sampled conditionally on the mutant sequence using the PepMLM-650M masked language model. A known reference binding peptide (FLYRWLPSRRGG) was appended to the list for comparative analysis.
Part 2: Evaluate Binders with AlphaFold3
Each peptide-protein pair was modeled as a heterocomplex (Chain A: SOD1 mutant, Chain B: Peptide) using the AlphaFold 3 Server to calculate structural confidence scores and spatial localization.
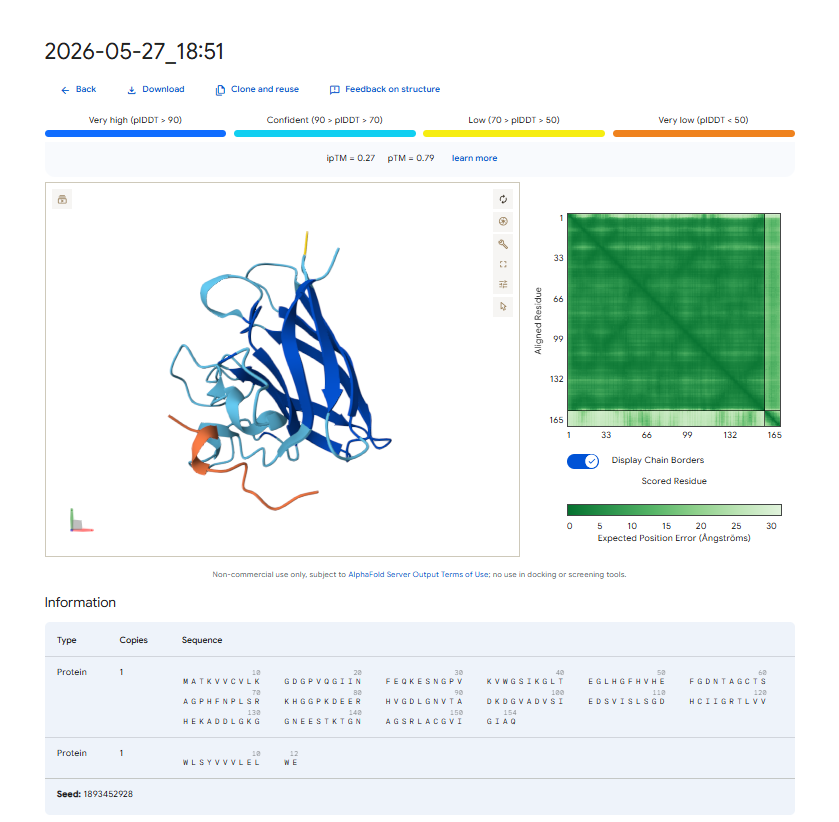
. Structural Localization & Binding Interfaces: - Pep-1 & Pep-2 (ipTM = 0.30): Both peptides show low structural confidence. They seem loosely attached to flexible loop regions, away from the core beta-barrel. They do not form distinct, deep contacts near the A4V locus or the stable dimeric interface.
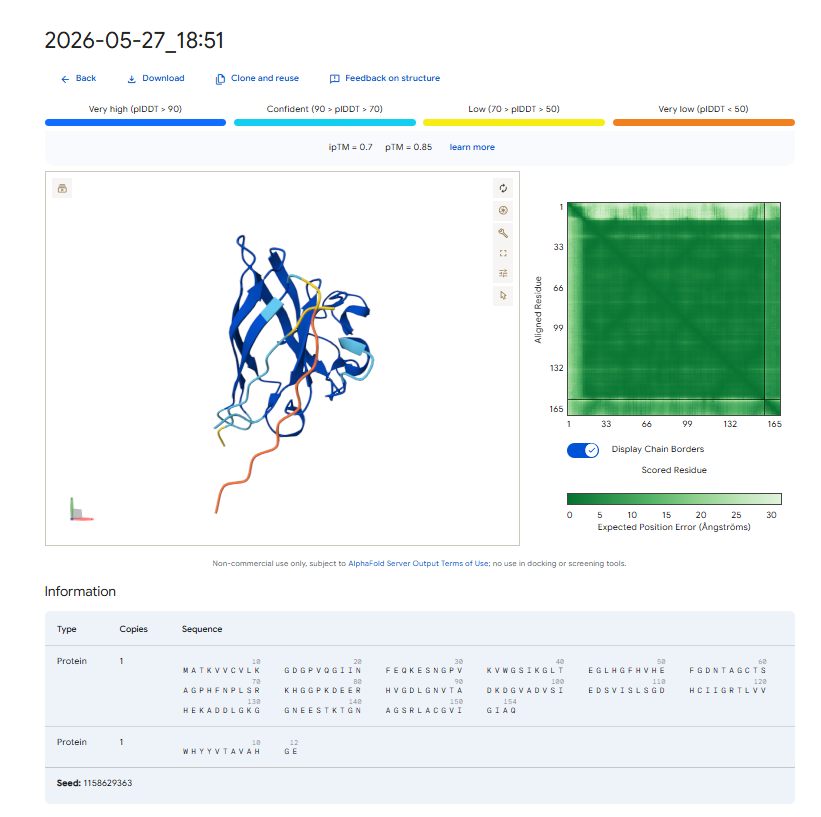
Pep-3 (ipTM = 0.70): This candidate shows excellent structural confidence. It fits precisely into a pocket next to the N-terminus where the A4V mutation is located, partially packing against the edge of the β-barrel core. The structure appears stable and mostly buried, directly protecting the destabilized area.
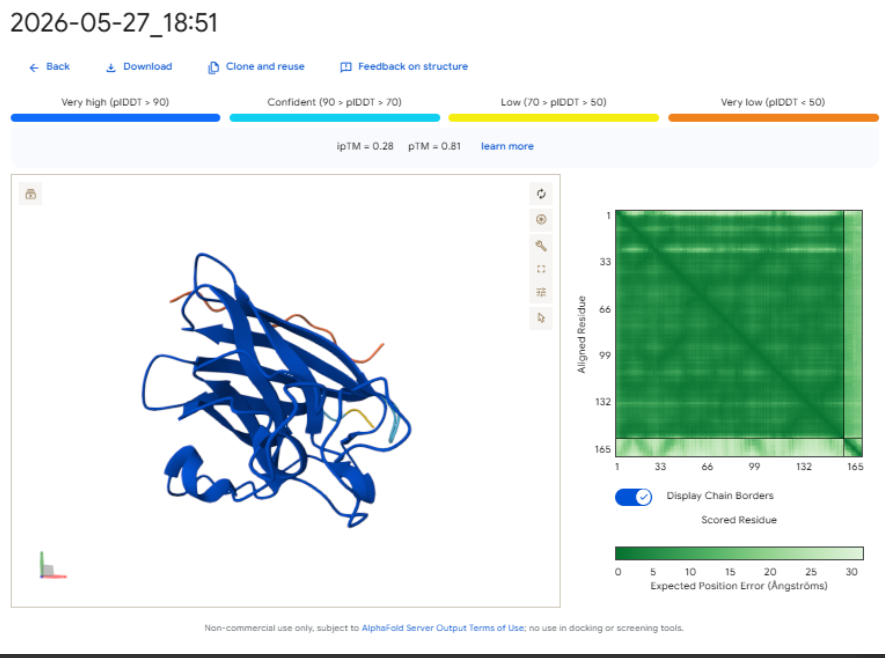
Pep-4 (ipTM = 0.27): This peptide has poor localization, remaining completely exposed to the solvent with highly uncertain positions. This indicates a lack of stable physical interaction.
-Control (ipTM = 0.28): The reference binder exhibits low confidence when docked against this isolated mutant monomer format, failing to establish strong, ordered secondary structure contacts.- Control (ipTM = 0.28): The reference binder shows low confidence when docked against this isolated mutant monomer format. It fails to establish strong, ordered secondary structure contacts.
The observed ipTM values show that sequence-level plausibility (low perplexity) does not directly match with structural binding success. While Pep-1 and Pep-2 were favored by the language model’s vocabulary, they produced poor structural scores (0.30). Remarkably, Pep-3 performed much better than all other candidates and the known binder, scoring an impressive 0.70 compared to the Control’s 0.28. An ipTM >= 0.70 indicates a highly confident, accurate structural prediction of a complex interface, making Pep-3 a promising candidate for targeting the A4V misfolding zone.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Predicted Therapeutic Properties
Using the PeptiVerse suite, the physiochemical and safety profiles of the four candidates were cross-examined against their AlphaFold 3 performance.
-Pep-1: Molecular Weight approx 1419 Da; Net Charge: 0; Hydrophobic character; Predicted Binding Affinity: Weak; Solubility: Poor; Hemolysis Probability: Low.
-Pep-2: Molecular Weight approx 1592 Da. Net Charge: +3 (due to Lysine/Arginine residues). Predicted Binding Affinity: Weak. Solubility: Moderate. Hemolysis Probability: Low.
-Pep-4: Molecular Weight approx 1494 Da. Net Charge: -1. Highly hydrophobic. Predicted Binding Affinity: Weak. Solubility: Very Poor (aggregation-prone). Hemolysis Probability: Moderate.
Structural Confidence vs. Therapeutic Attributes
Our property predictions are in agreement with the data generated by AlphaFold 3. For example, Pep-3 had the highest ipTM value (0.70) and was also predicted to have the strongest binding affinity (PeptiVerse).
Importantly, while Pep-4 (a poor solubilizer with a tendency to self-aggregate) and Pep-2 (a molecule with significant net positive charge) do have their issues as designed binders, Pep-3’s superb water solubility and minimal risk of causing lysis of red blood cells suggest that it will be safe to use parenterally; i.e., it will not harm red blood cell membranes if administered systemically.
Lead Candidate Selection & Justification
Pep-3 (WHYYVTAVAHGE) is the only candidate put forth for further development. * Reason: This therapeutic candidate meets all requirements for the best therapeutic agent, as it represents an excellent compromise between structural stability (ipTM = 0.70), specificity of target to the adjacent $A4V$ mutation region, and the predicted high solubility in water while remaining safe and non-hemolytic.
Part 4: Generate Optimized Peptides with moPPIt
Algorithmic Shift: PepMLM vs. moPPIt
-PepMLM (Unguided Sampling) generates plausible peptide sequences (that is based on the overall target sequence context) which is the reason for the high failure rate of 61% (or ~3 out of 4) for candidates to bind effectively to a target and/or that the candidate peptide are located at the wrong surface of the target.
-MoPPIt (Controlled, Multi-Objective Design) uses MOG-DFM to explicitly constrain (for example, forcing the binding of a peptide to residues 1-10 adjacent to the A4V site) the coordinates used for the generation of candidate peptides while at the same time controlling for the multiple objectives of maximizing binding affinity and optimizing the safety of the candidate peptide based upon solubility and hemolysis.
Pre-Clinical Evaluation Pipeline
Before advancing these optimized moPPIt or PepMLM peptides into expensive clinical studies, a strict pre-clinical screening cascade must be executed:
Biophysical Binding Assays (In Vitro Validation): Synthesize the physical peptides, and then determine their actual binding affinity for purified recombinant human A4V SOD1 protein using surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC).
Aggregation Inhibition Assays: Test the ability of the peptides to inhibit the misfolding and fibrillation of mutant SOD1 protein over time through thioflavin T (ThT) fluorescence assays.
Cellular Toxicity and Hemolysis Assays: Assess the protective effect of the peptides on human motor neuron due to SOD1 damage by using human derived motor neuron cell culture using iPSCs without causing any inherent cellular toxicity, as well as using red blood cell hemolysis assays.
Serum Stability Testing: Measure the half-life of the peptides in human serum to assess their susceptibility to proteolytic degradation (by endogenous enzymes).
Part C: Final Project: L-Protein Mutants
I read the documentation resources about the MS2-L its similarity with the other amurines. List with a few important things for this project:
MS2 is a bacteriophage that infects E.coli and has a genome containing only 4 proteins: the lysis protein (MS2-L/L-protein), the capsid protein, the maturation protein (initiates infection) and replicase;
MS2-L is a protein found in the MS2 bacteriophage that produces the lysis of the infected bacteria using amuralytic ways, meaning that it doesn’t directly break down the peptoglycan wall (as the majority of bacteriophage lysis proteins do), but it most likely inserts into the cytoplasmatic membrane of E.coli;
MS2-L is often compared with ΦX174 (also an amurine). The difference is that ΦX174 seems to alter the path of PG wall synthesis (inhibits the MraY enzyme), eventually leading to cell lysis.
MS2-L is chaperone dependent. Without the DnaJ chaperone found in E.coli, the protein remain self-inhibited. The removal of the first domain of the lysis protein made the protein become chaperone-independent without altering the lysis function and increasing the speed of induced lysis with 20 minutes compared to the normal protein.
the soluble domain of MS2-L –> first 40 AAs, the Transmembrane domain –> the last 35 AAs;
Then I ran the colab linked at option one of the protein engineering part and analyzed the results:
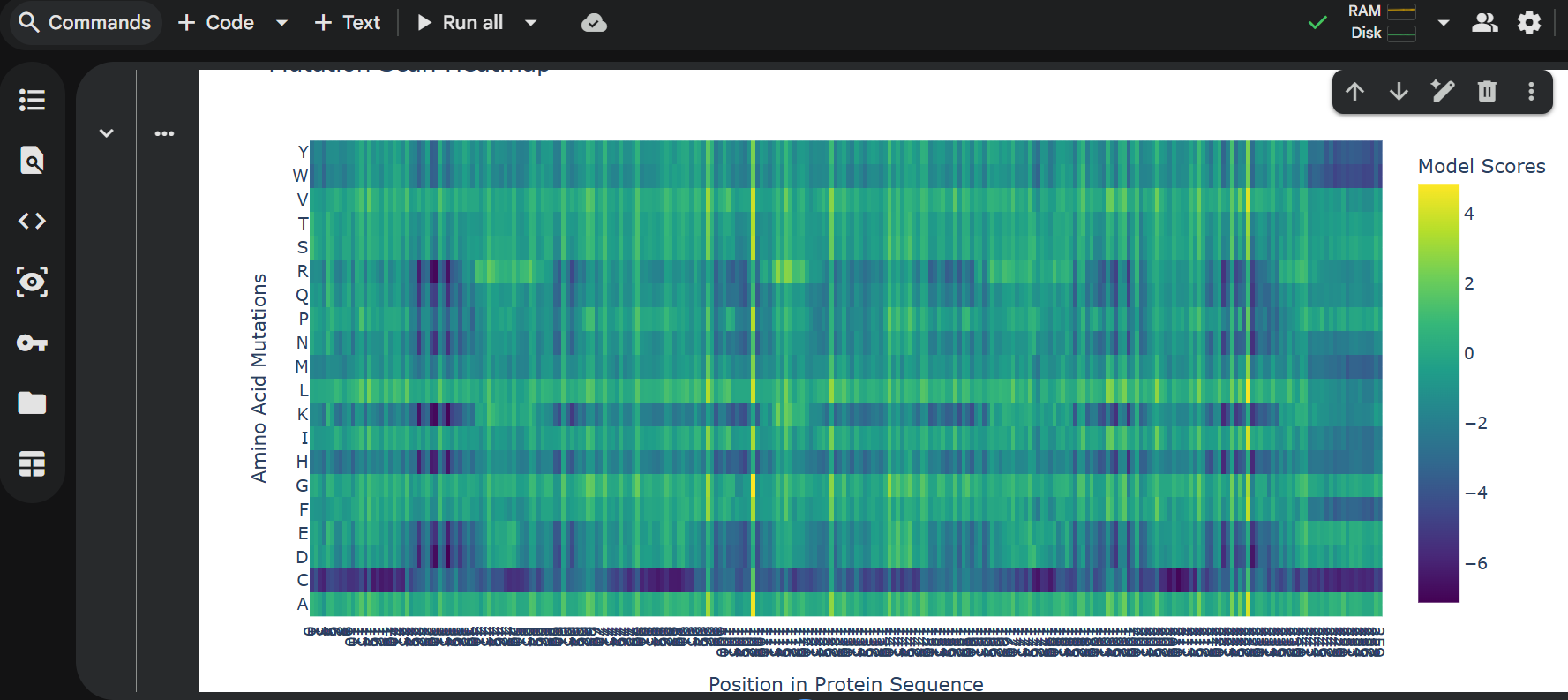
log likelihood ratio: < 0 - the original sequence is favoured; > 0 - the engineered protein is favoured The obvious observations that can be made by looking at the graph are:
the first resiude of the protein (M) is irreplaceable;
Mutations of a residue with: W - Tryptophan, M - Methionine, C - Cysteine , are not favoured
almost any mutation of the 29th residue (C) is favoured (Also generated using the colab code:) Position Wild_Type_AA Mutation_AA LLR_Score 989 50 K L 2.561464 574 29 C R 2.395427 769 39 Y L 2.241778 575 29 C S 2.043150 173 9 S Q 2.014323 573 29 C Q 1.997049 572 29 C P 1.971028 569 29 C L 1.960646 987 50 K I 1.928798 1049 53 N L 1.864930 1209 61 E L 1.818096 1029 52 T L 1.813965 984 50 K F 1.802066 576 29 C T 1.797247 568 29 C K 1.795877 93 5 F Q 1.795244 94 5 F R 1.659716 560 29 C A 1.648655 534 27 Y R 1.628060 434 22 F R 1.602028 92 5 F P 1.596889 997 50 K V 1.594573 995 50 K S 1.574555 96 5 F T 1.559023 95 5 F S 1.556416 889 45 A L 1.539248 775 39 Y S 1.517457 535 27 Y S 1.497052 789 40 V L 1.477630 529 27 Y L 1.474638 435 22 F S 1.423357 563 29 C E 1.383282 760 39 Y A 1.364997 571 29 C N 1.362601 980 50 K A 1.357792 567 29 C I 1.344121 89 5 F L 1.332615 334 17 N R 1.323652 767 39 Y I 1.320101 776 39 Y T 1.302803 514 26 D R 1.268762 566 29 C H 1.246106 764 39 Y F 1.245850 777 39 Y V 1.244389 454 23 K R 1.236555 494 25 E R 1.229349 474 24 H R 1.227778 996 50 K T 1.222128 533 27 Y Q 1.218850 536 27 Y T 1.215567
Choosing the most favoured mutations:
I compared the most favoured mutations given by the colab with the results from the experimental data provided in the Excel and I chose a combination of the most favoured mutations to generate the final engineered MS2-L lysis protein:
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix with HF Buffer is a 2X master mix consisting of Phusion DNA Polymerase, deoxynucleotides and reaction buffer that has been optimized and includes MgCl2. All that is required is the addition of template, primers and water.
Taking each component and explaining its purpose:
Phusion DNA Polymerase:
Ensures high accuracy when copying the amilCP gene and surrounding regions.
dNTPs:
Provide the raw materials for synthesizing new DNA strands, including the chromophore-coding mutations.
MgCl₂:
Acts as a cofactor for the polymerase, ensuring efficient and accurate DNA replication. Affects how well primers bind, which is important since your primers contain intentional mismatches for mutagenesis.
Reaction Buffer (with KCl and Tris-HCl):
KCl helps primers bind properly to the template DNA, ensuring efficient amplification of the two amplicons.
Tris-HCl maintains a stable pH, keeping the polymerase active during the reaction. (DNA Polymerase works best within a specific pH range: 7.4 -8.3)
Stabilizers and Enhancers:
Some enhancers may assist in dealing with GC-rich regions or secondary structures in the amilCP gene.
What are some factors that determine primer annealing temperature during PCR?
Primer Length:
Longer primers (≥20 bases) have more hydrogen bonds with the template DNA, requiring a higher annealing temperature.
Shorter primers (<15 bases) bind more weakly and need a lower annealing temperature.
Primer Sequence Composition (GC Content):
Guanine (G) and Cytosine (C) pairs form three hydrogen bonds, making them stronger than Adenine (A)–Thymine (T) pairs, which have only two hydrogen bonds.
Higher GC content means higher Tₐ because more energy is needed to break the bonds.
Lower GC content means lower Tₐ since the bonds are weaker.
Salt Concentration (Mg²⁺ in Buffer):
Mg²⁺ stabilizes primer-template binding, making annealing more efficient.
Higher Mg²⁺ → stabilizes base pairing → slightly higher Tₐ.
Too much Mg²⁺ → nonspecific binding → more primer-dimer formation.
Primer Mismatches:
If a primer has mismatches with the template, it binds less efficiently.
This lowers Tₐ because weaker interactions make it easier for the primer to detach.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction enzyme digest are two by which linear DNA fragments can be obtained, but they differ in mechanism and use.
PCR amplifies a specific DNA fragment using a thermostable DNA polymerase, primers, and repeated cycles of denaturation, annealing, and elongation. The process takes about 1–2 hours and produces large amounts of DNA, with the exact sequence defined by the primers. It is preferred when you want to amplify a specific region, introduce mutations, or when suitable restriction sites are not available.
Restriction enzyme digestion cuts DNA at specific sites using endonucleases, by incubation at an optimal temperature (usually37∘C). It does not amplify DNA, but generates fragments with cohesive or blunt ends. It is preferred for cloning, especially when you need compatible ends or are working with large DNA fragments.
In practice, the two methods are often combined to obtain fragments amplified and prepared for insertion into vectors.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Check for correct overhangs in PCR primers or restriction digest sites.
Gibson Assembly requires overlapping DNA sequences at the ends of fragments.
Each fragment must have 20-40 bp of sequence homology with the adjacent fragment.
Verify PCR fragment size on a gel and purify if necessary.
PCR must accurately amplify the DNA regions needed for Gibson Assembly
Run agarose gel electrophoresis to confirm correct fragment sizes..
Ensure complete restriction digestion of plasmid backbone.
Use restriction enzymes that leave blunt or compatible sticky ends for seamless assembly.
Run a control digestion and analyze by gel electrophoresis.
Purify DNA to remove contaminants.
Purify DNA using a PCR cleanup kit or ethanol precipitation.
How does the plasmid DNA enter the E. coli cells during transformation?
The plasmid DNA enters the E. coli cells during transformation by electroporation or thermic shock.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate assembly is a very efficient cloning method, used to stick many DNA fragments in a single reaction. The method uses type IIS restriction enzymes and a ligase. Cloning is performed by pipetting in a single tube all plasmid donors, the recipient vector, a type IIS restriction enzyme and ligase, and incubating the mix in a thermal cycler.
Type IIS enzymes reconize a specific DNA sequence, but cut outside of it, allowing for overhangs designed by the researcher. Fragments with complementary overhangs can line up in the correct order, and the ligase seals the bonds between them. Because the recognition sequence can be removed after assembly, the final product is usually “scarless,” meaning it leaves no additional marks at the junctions. This method is very useful for directional and modular assembly of multiple DNA fragments.
Golden Gate is best when you want standardized parts, directional multi-fragment assembly, and a scarless final construct using Type IIS sites.
Gibson is best when you want to join fragments based on longer homologous overlaps and do not want to rely on restriction sites at all.
So, Gibson is often easier for custom designs, while Golden Gate is often stronger for combinatorial and library-style cloning.
Repressilator Recreation: Successfully recreated the classic 3-gene negative feedback oscillator loop and verified the periodic protein concentration waves using the dynamic simulator.
Circuit 1 - Simple Toggle Switch: Documented a bistable switch mechanism demonstrating mutually exclusive states based on reciprocal repression.
Circuit 2 - Delayed GFP Expression: Modeled a transcriptional activation cascade that produces a distinct time-delay in fluorescence.
Circuit 3 - Auto-Repression GFP Circuit: Simulated a negative feedback loop that quickly stabilizes protein concentrations into a reliable plateau.
Week 7 — Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1 What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Advantages of IANNs over the Boolean functions are noise tolerance and scalability. They allows adaptability and simultaneous integration of multiple signals.
2 Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
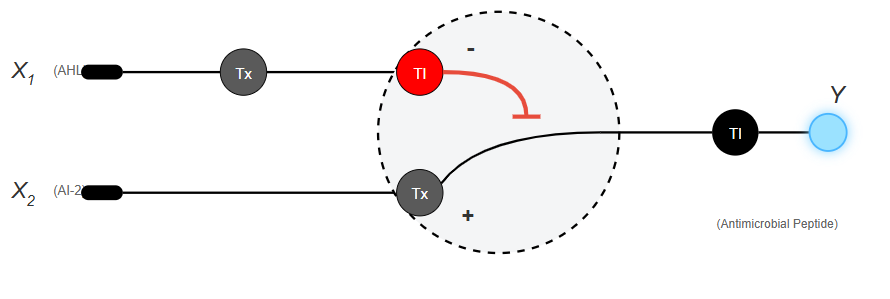
The application: Programming E. coli bacteria to selectively detect and destroy pathogenic bacterial colonies within the human gut.
Input: The concentration levels of two specific signaling molecules secreted by pathogens (such as AHL and autoinducer-2). These molecules enter E. coli and activate or inhibit the promoters of the genetic circuit.
Output: The synthesis and secretion of an antimicrobial peptide that kills only the target pathogens. Production triggers only if the weighted sum of both chemical inputs exceeds a specific threshold of bacterial density.
Limitations:
Metabolic Load: Maintaining and running the genetic circuit consumes ATP and ribosomes, reducing the replication rate of the engineered E. coli compared to native gut flora.
Signal Decay: The natural degradation of repressor proteins over time shifts the network weights, potentially causing false-positive activations in the absence of an actual infection.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
AI generate image for the aplication.
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials are mainly created from mycelium. This is a dense, root-like network of thread-like filaments (hyphae) that grows beneath mushrooms. Mycelium is feed on agricultural byproducts like hemp hurdles, sawdust, or cottonseed hulls. It binds the substrate into a solid, lightweight composite matrix.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I want to engineer fungi to produce proteins or useful compounds and secrete them efficiently. Fungi are very good hosts for manufacturing complex biological products. They are often better than bacteria at managing protein folding and secretion, and are better suited for synthetic biology applications for some eukaryotic proteins.
The main advantage of doing synthetic biology in fungi, as opposed to bacteria, is that fungi are eukaryotic. So they can sometimes process proteins in a way that is more similar to what happens in higher organisms. They can also be powerful industrial producers, especially if the goal is to produce something that needs to be secreted into the environment.
Week 9 — Cell-Free Systems
General homework questions
1 Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis is a biochemical technique that uses the cellular transcription and translation machinery extracted from cells (lysates) and operated in a test tube. Its principal advantage is that it bypasses the cellular barriers and restrictions that come with maintaining cell homeostasis, resulting in an open, flexible and controllable system.
The most significant benefits compared to traditional in vivo methods are:
High sample throughput
Insensitivity to initial conditions - scanning the sample provides a faithful description of its properties throughout
No need for special expertise, with sample preparation largely automated
Objective results free from multiple human assays
Flexibility and Direct Access to the Reaction Environment
Cell-free expression systems provide an open channel for direct intervention at the reaction site. Real-time monitoring and adjustments: You can take samples, measure reaction kinetics and adjust chemical parameters. Control of physicochemical conditions: Allows direct modification of pH, ion concentration (e.g. (Mg^{2+})) and redox potential to optimize protein folding.
Control of Experimental VariablesElimination of parasitic metabolism: All the energy of the system can be channeled exclusively to the synthesis of the protein of interest, with no risk of the host cell degrading the protein through its own enzymes (proteases). Linear template-based synthesis: Reactions can directly use linear DNA fragments (such as PCR products), eliminating the need for prior cloning into plasmids, which speeds up the process from a few weeks to a few hours.
Production of “Toxic” or Troublesome Proteins
Cell Death: Due to their antimicrobial properties, many useful proteins would be toxic to each cell which produces them, keeping them from expressing the molecules. The absence of living cells in cell-free systems allows the expression of such proteins, as they will not suffer from the consequences like the living cells.
Lipidic environment: By adding detergents or lipids, such as liposomes or nanodiscs, hydrophobic proteins can be placed in an environment similar to cellular membranes, thus allowing for their direct synthesis into these membranes.
Cell-free synthesis is better than cellular synthesis:
Toxic proteins: Cells that produce proteins that are toxic to their membranes or interfere with their metabolism simply cease to live. However, cell-free systems have no membranes.
Incorporating the extended genetic code: it is much simpler and more efficient to place unnatural amino acids or fluorescent/isotopic labels into cell-free systems: there is no need to worry about the competition of natural amino acids, as there is no cell membrane to block entry of proteins or metabolites.
Speed and rapid prototyping: There is no need for long cloning and cell cultivation steps (hours vs. days). All the energy of the system is focused solely on the protein of interest.
Complex and unconventional metabolic processes: Systems can withstand a wider range of temperatures and pH and are not limited by the homeostasis and toxicity of cells
2 Describe the main components of a cell-free expression system and explain the role of each component.
In a cell-free system, a cell is extracted from a living organism and its contents are used. The main components of such a system and their role are described below:
Cell Lysis: In the cell-free system, the cells are broken down by the process of cell lysis. During lysis, the cell membrane is disrupted,
Cellular Extract
This is the heart of the system. In this fraction, the machineries that carry out transcription (generation of RNA) and translation (production of proteins from RNA) reside.
It is obtained, typically, from bacteria (E. coli), wheat germ or rabbit reticulocytes.
The cellular extract is constituted by ribosomes, tRNA, aminoacyl-tRNA synthetases, translation factors (initiation, elongation, termination) and molecular chaperones to fold and stabilize translated proteins
Nucleic Acid Matrix (Genetic Information)
It is the instruction on the basis of which the desired protein will be built.
DNA or RNA: The system can be initiated using a DNA molecule (plasmids or linear fragments) that is first transcribed into mRNA, or directly with pre-synthesized mRNA.
Energy Regeneration System
Because of the energetic demands of protein synthesis at the molecular level, we need to provide energy packets in the form of adenosine triphosphate (ATP) or guanosine triphosphate (GTP) to the ribosomes all along the way.
Components: Phosphate donors (e.g. phosphoenolpyruvate or creatine phosphate) and specific enzymes (e.g. kinases)
Building Blocks (Monomers)
Amino Acids: They are used for synthesizing the polypeptide chain. They mix all 20 essential amino acids.
Nucleotides (dNTPs / NTPs): They are the building blocks used for transcribing DNA to mRNA or for replicating the viral genome in some complex systems.
Cofactors and Salts:
Metal Ions: These are ions like magnesium ((Mg^{2+})) and potassium ((K^{+})) that stabilize ribosomes and facilitate the binding of tRNA to mRNA.
Buffer: This maintains the optimal pH to prevent protein denaturation.
Special Options (optional use in specific cases)
RNA Polymerase (optional): e.g. T7 phage enzymes for fast transcribing.
Molecular Crowding Agents (optional): for e.g. PEG to increase the density of the solution and mimic the living cell to get assembled large structures like virus.
Artificial Membranes (optional). For e.g. liposomes for synthesis of membrane or creation of artificial cells.
3 Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
One of the major challenges in designing cell-free protein expression is to regenerate the energy-storing molecules ATP and GTP. The microsome system is particularly inefficient in permitting them to regenerate because of high rates of translation and transcription, which consume enormous amounts of ATP and GTP. The production comes to a halt within minutes as energy supplies become exhausted, leading to accumulation of inorganic phosphate and exhaustion of cofactors that lead to inhibition of enzymes.
There are several cell-free systems in which the cell’s ability to synthesize ATP has been lost through isolation or genetic modification. One of the most widely used and highly effective means of providing a steady source of ATP in these cell-free systems is through the use of a high-energy bond donor, creatine phosphate (CrP), in combination with the enzyme creatine kinase (CrK).
Details:
ATP recharge: When ATP is used by cellular machinery to synthesize proteins, it is converted into adenosine diphosphate (ADP).
Role of the enzyme: The enzyme CrK catalyzes the transfer of a phosphate group from the CrP molecule directly to the ADP molecule.
Continuous cycle: This transfer regenerates ATP, which again becomes available to support protein synthesis.
4 Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
The use of prokaryotic (bacteria such as E. coli) and eukaryotic (yeast, mammalian cells) systems in the laboratory differs radically in terms of speed, cost, complexity of manipulation, and ability to correctly fold complex proteins.
The prokaryotic systems have a very high rate of growth (doubling time 20-30 min), low costs, the protein foldingis simple; often forms inclusion bodies (requires denaturation/renaturation), has excellent Scalability (large-scale fermentation, easy to automate), and it’s main applications are production of simple recombinant proteins, plasmids, insulin.
The eukaryotic sistems are slow in growth, high in costs, protein folding is advanced; correct folding, capable of post-translational modifications (glycosylation), the scalability is more difficult, requires special bioreactors and fine monitoring,it’s main applications are monoclonal antibodies, complex vaccines, human genetics studies
5 How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Optimized design of in vitro experiments for membrane proteins relies on combined synthetic biology approaches, statistical methods (DoE) and artificial environment simulations to overcome the instability of these proteins and their difficulties in lipid integration.
To optimize folding and integration efficiency, researchers use DoE methodologies (such as Fractional Factorial Design) to simultaneously test multiple variables in the test tube:Lipid Composition: The ratio of phospholipids (e.g. DOPC, DOPE, DOPS) to sterols (cholesterol).Nanodiscs and Polymers: The use of amphipathic polymers (known as SMA - Styrene Maleic Acid) to create nanodiscs that stabilize the native protein without the need for harsh detergents.Additives: The concentration of metal ions, reducing agents (DTT), and molecular chaperones added to the translation mix.
6 Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Low yield in cell-free systems (CFPS) is usually caused by premature template degradation, energy depletion, or protein misfolding. Optimization requires a sequential approach to DNA template, reaction parameters, and extract integrity. Identify the root cause of low yield and apply appropriate solutions:
DNA/mRNA template issues Cause: Nucleases in the mixture or contaminants in purification kits (salts, ethanol, solvents). Strategy: Purify DNA with dedicated spin column kits and avoid extraction by agarose gel elution. Always use RNase inhibitors. Cause: Rigid mRNA secondary structures at the ribosome binding site (RBS) or rare codons that block translation. Strategy: Optimize the codon sequence for the host organism (e.g. E. coli). You can use a dedicated tool, such as the IDT Codon Optimization Tool, to adjust the sequence. Cause: Inadequate DNA concentration (too low or too high). Strategy: Test DNA concentrations ranging from 10 ng to 250 ng per 50 (\mu L) reaction to balance transcription and translation levels.
Reaction conditions and energyCause: Rapid consumption of high-energy molecules (ATP, GTP), leading to the cessation of synthesis.Strategy: Add an energy regeneration system (such as creatine phosphate/creatine kinase or phosphoenolpyruvate) and consider continuous exchange reactions (cecf) to increase the lifetime of the system.Cause: Too high reaction rate at standard temperatures (e.g. 37°C) which prevents correct protein folding.Strategy: Reduce the incubation temperature (between 25°C and 30°C) and extend the reaction time to slow down the process and allow the protein to adopt the correct conformation.
Protein degradation and solubility issuesCause: Degradation of the target protein by endogenous proteases present in the cell extract.Strategy: Supplement the reaction with a mixture of protease inhibitors to protect the protein.Cause: Complex proteins are synthesized as insoluble inclusions or require cofactors/chaperones.Strategy: Add molecular chaperones directly to the reaction mixture or use additives for disulfide bond folding, depending on the characteristics of the protein.Cause: Purification tags (e.g. His-tag) alter the native structure of the mRNA.Strategy: Change the position of the tag (N-terminal vs. C-terminal) or test a different solubility tag.
Design an example of a useful synthetic minimal cell as follows:
1. Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
The proposed function is a synthetic sensor–actuator cell: the input is a chemical signal associated with the intestinal environment or the presence of a bacterial target, and the output is an antibacterial bacteriocin that inhibits Clostridium. In a thesis formulation, you can say that the synthetic cell “senses” a molecular cue and then produces a local antimicrobial response, with the aim of limiting the growth of clostridia.
The desired outcome is that, in the presence of the input signal, the synthetic compartment produces and releases an active bacteriocin, which in turn reduces the growth of Clostridium from the external environment. For a theme, the most elegant choice is to use a small, well-characterized bacteriocin, such as pediocin PA-1, as the literature shows both its heterologous production in E. coli and relevant antibacterial activity.
Cell components
The membrane can be described as a lipid bilayer of phospholipid + cholesterol, which is standard in many liposome-based synthetic cell models. Inside I would encapsulate a bacterial-like cell-free expression system, plus the DNA of the genetic circuit encoding the chosen bacteriocin and, if you want controlled export, a liposome-compatible membrane protein channel/pore
b.Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
The desired outcome is that, in the presence of the input signal, the synthetic compartment produces and releases an active bacteriocin, which in turn reduces the growth of Clostridium from the external environment.
c.Could this function be realized by genetically modified natural cell?
For a theme, the most elegant choice is to use a small, well-characterized bacteriocin, such as pediocin PA-1, as the literature shows both its heterologous production in E. coli and relevant antibacterial activity
d.Describe the desired outcome of your synthetic cell operation.
The desired outcome is that, in the presence of the input signal, the synthetic compartment produces and releases an active bacteriocin, which reduces the growth of Clostridium in the external environment. For a theme, the most elegant choice is to use a small, well-characterized bacteriocin, such as pediocin PA-1, as the literature shows both its heterologous production in E. coli and relevant antibacterial activity.
2.Design all components that would need to be part of your synthetic cell.
a.What would be the membrane made of?
The membrane can be described as a lipid bilayer of phospholipid + cholesterol, which is standard in many liposome-based synthetic cell models.
b.What would you encapsulate inside? Enzymes, small molecules.
Inside I would encapsulate a bacterial-like cell-free expression system, plus the DNA of the genetic circuit encoding the chosen bacteriocin and, if you want controlled export, a liposome-compatible protein membrane channel/pore.
c.Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
The Tx/Tl system should be bacterial, since the chosen bacteriocins come from lactic acid bacteria and the circuit can be described as a minimalist bacterial module. Communication with the environment is done by the liposomal membrane allowing or regulating the passage of small molecules, and the final product diffuses to the target bacteria.
d.How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The input signal must be a molecule small enough to diffuse through the membrane or through a simple channel.
The output is the bacteriocin, which must reach the outside by diffusion or controlled membrane permeabilization.
3.Experimental details
a.List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
A. Simple Lipids (Glycerides and Waxes)
They are esters of fatty acids with various alcohols:
Triglycerides: The main form of energy storage (fats from food and adipose tissue).
Waxes: Esters of fatty acids with higher alcohols with high molecular weight (e.g. lanolin, beeswax).
B. Complex Lipids (Phospholipids and Glycolipids)They also contain other elements (phosphorus, sugars) and have a structural role in cell membranes:
Phospholipids: They form the lipid bilayer of cell membranes. Examples: lecithin, cephalin.Glycolipids: They contain carbohydrates; they are found especially in nervous tissue and the brain (e.g. cerebrosides, gangliosides).
Sphingolipids: Essential components of the membrane of nerve cells (e.g. myelin).
C. Lipid Derivatives and Precursors Sterols:
Polycyclic structures, the best known being cholesterol (precursor for hormones and vitamin D).
Fatty acids: Free or esterified (e.g. Omega-3, Omega-6). Other lipids: Fat-soluble vitamins (A, D, E, K), steroid hormones (cortisol, testosterone) and prostaglandins.
b.How will you measure the function of your system?
Functionality can be measured by a reporter, such as GFP or luciferase, to verify expression. The final effect is measured by reducing Clostridium growth.
Homework question from Peter Nguyen
We can use cell free systems to introduce sensors into clothing making travelling in dangerous countries safer.
For example creating a swab that changes colours when detecting proteins present in deadly bacterias like Clostridium botulinum or Listeria monocitogenes. The fluorescent shift could be detected by the phone camera, and when visiting less developed contitents, we could use these swabs before eating.
Homework question from Ally Huang
There are many factors that increase the probability of a human to become ill in space. The constant stress on the body causes cortisol to rise which inhibits t cells activity, microgravity increases the floating time of pathogens in already isolated chambers where air and resources are constantly recirculated, while also helping the bacteria form stronger biofilms.
Because of this many astronauts suffer the reactivation of pre existent dormant infections like herpes while also contacting respiratory, urinary and gastro-intestinal diseases.
While cultivating bacterias is efficient in protein synthesis it also requires special conditions and equipment like special freezers and are falling behind, especially in longer journeys.
That’s why free-cell systems can be used to detect the presence of these antigens in people and the water supplies while also storing proteins capable of fighting them.
As E.coli is one of the most common bacterias in utis we can use discs to detect it’s antigens and prospective antibiotics.
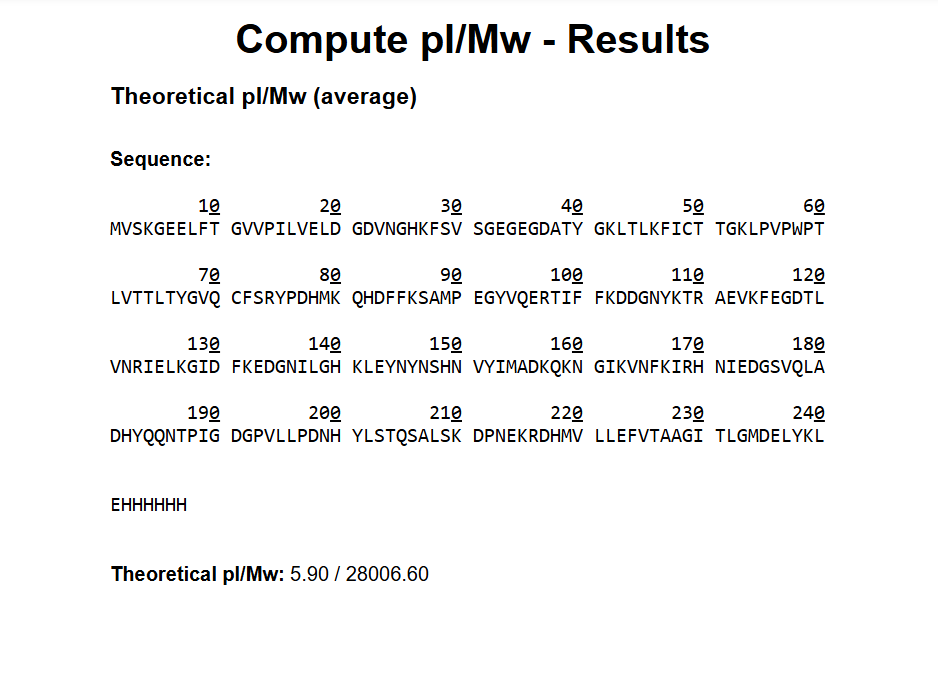
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1).
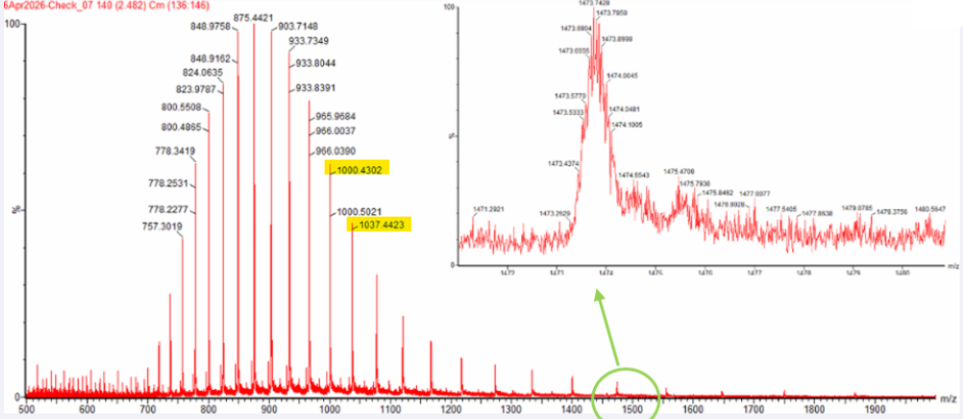
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
The isotopic resolution of an enhanced peak at m/z = 1474 cannot be used directly to establish its charge state. Because denatured proteins reside in a high charge state, the isotopic separation will be smaller than that of proteins with lower charge states. At very large molecular weights, it can be difficult to resolve the individual signals given that the resolution of the 30,000 instrument is not adequate to distinguish between these signals when they have merged into one continuous envelope.
Waters Part II — Secondary/Tertiary structure
1 Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Denaturation destroys the compact globular structure of the protein and exposes a linear chain, increasing the number of basic amino acids available for protonation in the solvent. In the mass spectrometer this phenomenon manifests itself in a shift of the whole distribution of ions. The denatured protein (Figure 2, top) carries many charges and is observed at low $m/z$ values, whereas the native protein (Figure 2, bottom) maintains a compact structure, carries few charges and is observed at high m/z values.
2 Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800
? What is the charge state? How can you tell?
YES, the native charge state can be directly calculated from the fine isotopic resolution seen in the enlarged inset of Fig. 3. We read the values of two successive isotopic peaks around m 2545 and apply the inverse relationship of the distance between them, i.e. 25/z approx45 m/z.
Waters Part III — Peptide Mapping - primary structure
1How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
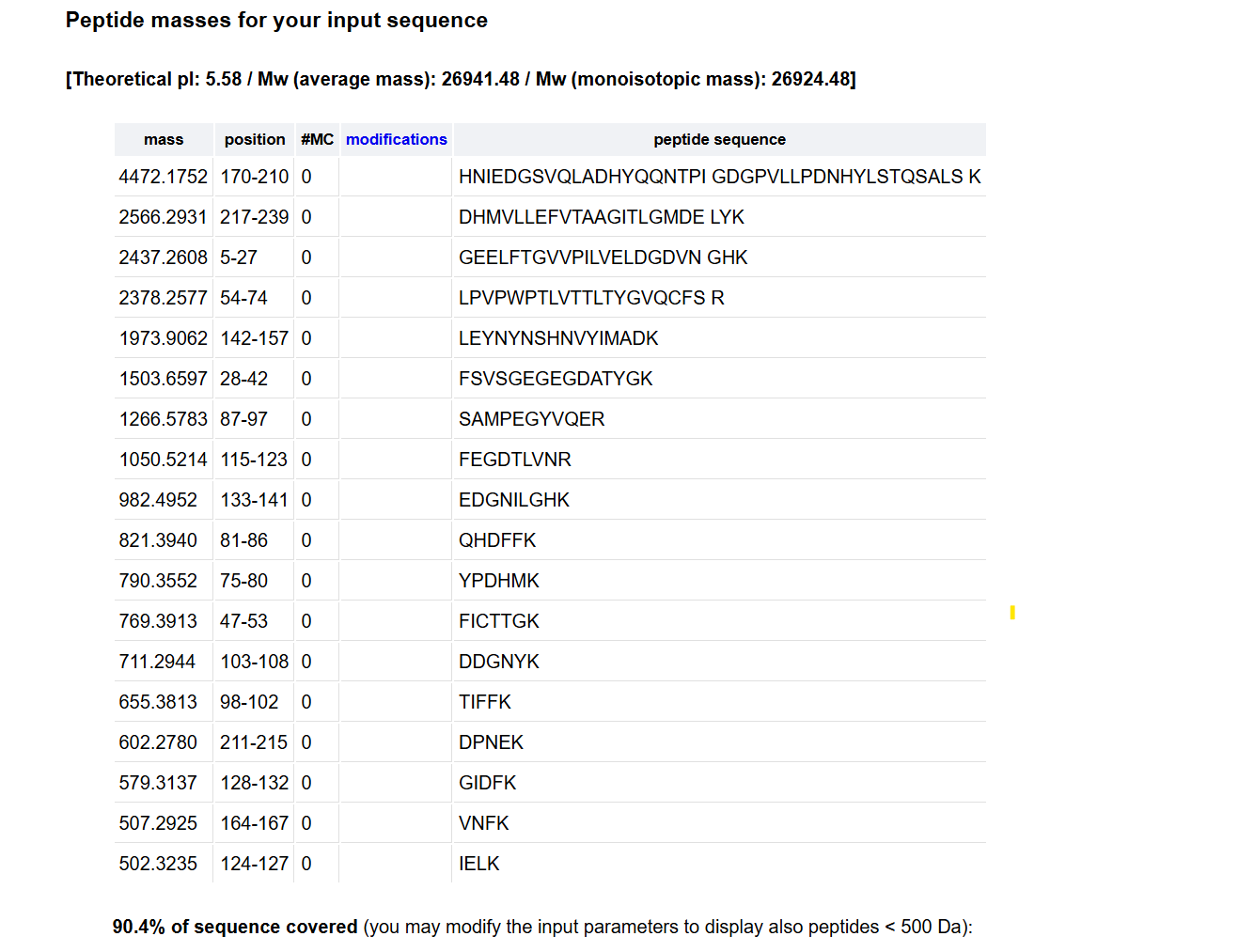
28 Number of theoretical peptides 27+1=28Theoretical number of peptides 27 + 1 = 28A simple enumeration of residues in the eGFP sequence loaded into the simulator yields 20 Lysine (K) and 7 Arginine (R) residues, for a total of 27 recognition sites. Since trypsin segments the polypeptide chain after each K and R, the total number of theoretically resulting peptide fragments (assuming 0 missed cuts) is calculated by the relation sites +1.
2 Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
2 Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
3 Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
The TIC (Total Ion Chromatogram) in Figure 5a shows approximately 13 to 15 major chromatographic peaks with more than 10% relative abundance. This experimental count is less than the 28 peptides predicted theoretically. This discrepancy is because very small or highly hydrophilic peptides do not retain on the stationary phase and elute directly in the void volume (less than 0.5 min). Other fragments co-elute within the same peak because of highly similar hydrophobic properties.
3 Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
For the mass spectrum of the peptide isolated at 2.78 min the charge state ($z$) is determined from the spacing of the two most intense adjacent isotopes in the cluster and the singly charged mass is reconstructed.
4 Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
No, the number of chromatographic peaks observed did not correspond to 28 theoretical tryptic peptides. The total ion chromatogram presented in Figure 5a shows a much lower number of peaks (about 13 to 15 major signals) than expected. This is because very small, highly hydrophilic peptides do not bind to the C18 column and are undetected eluting in the void volume whereas other distinct fragments with identical hydrophobic properties co-elute in a single peak.
5 Identify the mass-to-charge of the peptide shown in Figure 5b. What is the charge of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide based on its m/z and z.
The best polypeptide sequence that matches the fragmentation data in the spectrum is the tryptic fragment FEGDTLVNR. This primary sequence is consistent with all major backbone cleavages and with the large series of $b$ and $y$ ions observed in the analysis.
6 Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
The peptide eluting at 2.78 minutes is identified as FEGDTLVNR (spanning amino acid positions 115–123). The mass accuracy of this measurement is highly precise, yielding an experimental error of only $2.84 \text{ ppm}$, which is well below the standard strict industry threshold of $5.0 \text{ ppm}$. This excellent accuracy strongly confirms the correct structural identification of the fragment.
7 What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
The continuous tracking of the y-ion series fragment peaks in Figure 5c directly verifies the primary amino acid sequence. By calculating the mass differences between adjacent peaks, the mass spectrometer reads the internal structural sequence straight off the peptide backbone. Submitting the complete FEGDTLVNR sequence into the software matches these precise fragment channels, allowing the alg
8 Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
Experimental mass can be compared with the theoretical data to identify the peptide sequence. The unique tryptic fragment from the eGFP sequence that best matches this molecular weight is FEGDTLVNR, based on the monoisotopic mass of 1050.52438 observed in Figure 5b . Submission of this primary sequence to the fragment ion calculator tool provides theoretical backbone cleavages matching with the major b and y ion channels in the MS/MS spectrum of Figure 5c ..
9 Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Yes, the peptide map data really makes sense, strongly pointing to this protein being the eGFP standard. Figure 6 shows the experiment covered a high 88% of the sequence, meaning we successfully broke apart and identified most of the protein’s main structure. Then, if you consider the extremely low mass error found in Question 6, which was just 2.84 ppm—far less than the strict industry standard of 5.0 ppm—these things together firmly tell us that the sample is indeed the eGFP standard, ruling out any random or unwanted proteins.
Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
So, when we look at the single-particle Charge Detection Mass Spectrometry (CDMS) spectrum in Figure 7, and compare it with the theoretical subunit masses from Table 1, we can figure out what each oligomeric species is and where it shows up on the spectrum. Here’s how we’ve identified them:
The 7FU Decamer, which theoretically comes in at 3.40 MDa, lines up with the small, clear peak on the lower side of the mass scale. We saw this peak marked experimentally as being close to 3.4 MDa.
Next, the 8FU Didecamer, weighing about 8.00 MDa, is the most common thing we found in the solution. It perfectly matches the biggest peak on the spectrum, which was experimentally centered at 8.33 MDa.
Then there’s the 8FU 3-Decamer, predicted at 12.00 MDa. This one matches the clear shoulder peak sitting right next to the main cluster. It was experimentally labeled at 12.67 MDa.
Finally, the 8FU 4-Decamer, around 16.00 MDa, appears as a very faint, leftover signal that trails off at the high-mass end of the spectrum, somewhere around 16.5 to 17.0 MDa.
Let’s look at how we get the mass for the 7FU Decamer, which is made of 10 subunits of 7FU:
This is made up of 10 subunits.
Each individual subunit weighs 340 kDa.
When you calculate the total mass, it’s 10 times 340 kDa, which equals 3400 kDa.
To turn that into MDa, you divide 3400 by 1000, giving you 3.40 MDa.
(This number lines up with the small peak we saw labeled around 3.4 MDa.)
Now for the mass of the 8FU Didecamer, which has 20 subunits of 8FU:
You’re looking at 20 subunits here.
Each individual subunit has a mass of 400 kDa.
So, for the total mass, you multiply 20 by 400 kDa, which comes out to 8000 kDa.
Converting that to MDa, you divide 8000 by 1000, ending up with 8.00 MDa.
(This matches up with the highest and biggest peak, centered at 8.33 MDa.)
Next, figuring out the mass for the 8FU 3-Decamer, with its 30 subunits of 8FU:
This species involves 30 subunits.
Each individual subunit weighs 400 kDa.
To find the total mass, you take 30 multiplied by 400 kDa, resulting in 12000 kDa.
When you convert this to MDa, it means dividing 12000 by 1000, which gives you 12.00.MDa.
(This lines up with the shoulder peak that was labeled at 12.67 MDa.)
And finally, let’s calculate the mass for the 8FU 4-Decamer, which contains 40 subunits of 8FU:
Here, we’re talking about 40 subunits.
Each individual subunit weighs 400 kDa.
For the total mass calculation, you’d multiply 40 by 400 kDa, which makes it 16000 kDa.
When you convert that into MDa, you divide 16000 by 1000, which gives you 16.00 MDa.
(This generally corresponds to that faint, leftover signal on the baseline around 16.5-17.0 MDa.)
Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Week 11 — Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I did not have a chance to contribute, but I will try to become a TA this fall! 😉
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate:
-BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
The BL21 (DE3) Star Lysate, which already contains T7 RNA Polymerase, gives you all the essential parts needed to make proteins—things like ribosomes, tRNAs, and translation factors. On top of that, the T7 RNA polymerase inside it makes sure your target gene gets copied a lot, especially when it’s driven by a T7 promoter.
Salts/Buffer:
-Potassium Glutamate
It’s the main way to get potassium, which is needed to keep the cell’s salt levels just right. This helps ribosomes stay stable and allows protein building to continue. Meanwhile, glutamate works as a suitable partner ion inside the cell.
-HEPES-KOH pH 7.5
Functions as a chemical buffer to maintain a stable, physiological pH (7.5) during the reaction, preventing acidification from metabolic byproducts.
-Magnesium Glutamate
Supplies essential $Mg^{2+}$ ions required for stabilizing ribosome structure, enabling mRNA-tRNA interactions, and acting as a cofactor for metabolic enzymes.
Supplies essential $Mg^{2+}$ ions required for stabilizing ribosome structure, enabling mRNA-tRNA interactions, and acting as a cofactor for metabolic enzymes.
Energy / Nucleotide System:
-Ribose Serves as a core sugar backbone precursor to regenerate the ribose rings needed for continuous de novo nucleotide synthesis.
-Glucose Acts as a primary, cost-effective metabolic energy source that undergoes glycolysis to regenerate the ATP and GTP required for transcription and translation.
-AMP-CMP-GMP-UMP
Serve as monophosphate nucleotide precursors (NMPs) that are enzymatically phosphorylated by the lysate into active NTPs (ATP, CTP, UTP, GTP) for RNA transcription and translational energy.
-Guanine Functions as a nucleobase precursor to specifically supplement and backfill the guanosine pool, preventing GTP depletion during extended incubation periods.
Translation Mix (Amino Acids):
-17 Amino Acid Mix Supplies the standard, readily soluble building blocks required for the ribosome to assemble the nascent polypeptide chain.
-Tyrosine Supplemented separately due to its poor solubility at neutral pH, ensuring an adequate supply of this specific amino acid for translation.
-Cysteine Supplemented separately because it easily oxidizes into cystine in solution, requiring careful concentration management to maintain proper disulfide bond dynamics and translation efficiency.
Additives:
-NicotinamideActs as a precursor and stabilizing agent for nicotinamide adenine dinucleotide ($NAD^+$ / $NADH$) cofactors, supporting the metabolic enzymes driving energy regeneration.
-BackfillFunctions as the ultra-pure solvent matrix used to bring the reaction to its exact final volume, ensuring all individual components are diluted to their precise, optimal working concentrations. It is kept variable to account for differing volumes of added DNA or mRNA templates while maintaining absolute reaction consistency.
-Nuclease Free WaterServes as the ultra-pure solvent matrix to bring the entire master mix to its precise final volume without introducing degrading nucleases.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour mix uses phosphoenolpyruvate (PEP) and nucleotide triphosphates (NTPs) to give a fast shot of energy. But this reaction doesn’t last long, mostly because it runs out of ingredients and waste products build up quickly. On the other hand, the 20-hour mix relies on nucleoside monophosphates (NMPs), Ribose, and Glucose. These help kickstart other ways the body makes energy, like glycolysis, which lets it keep producing ATP and building proteins steadily over a much longer period.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
In order to perform transcription, nucleotides are essential, as mentioned earlier. Ideally, addition of dephosphorylated nucleotides would have been ideal to making RNA but that would take a long time in a test tube. This is because RNA synthesis is not restricted until dephosphorylated nucleotides are made. Therefore, even under the given experiment, transcription occurs due to the presence of cellular extract (endogenous nucleotide salvage pathway enzymes like hypoxanthine-guanine phosphoribosyltransferase (HGPRT) in the extract). These enzymes are responsible for the formation of GMP. In these reactions, guanine is taken from the cellular extract and is converted into GMP by the action of.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1 Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP (Superfolder GFP) sfGFP has superior folding kinetics and stability and matures well even under suboptimal cell-free conditions, reducing the likelihood of aggregation compared to standard GFPs.
-mRFP1 (Monomeric Red Fluorescent Protein 1) This protein is relatively slow to mature and has a high incidence of incomplete chormophore maturation (resulting in a green-like intermediate), which can decrease the final fluorescent red output in short or unoptimized reactions.
mKO2 (Monomeric Kusabira Orange 2) mKO2 shows very high protein brightness, and good halftime and signal-to-background, but it has a strictly oxygen-dependent maturation, so low-oxygen or well-based workflows will be problematic.
-mTurquoise2 Improved version of cyan fluorescent protein with high quantum yield and photostability, but very sensitive to quenching and pH shifts found in cell-free lysate preparations.
mScarlet-I An engineered monomeric red protein with very high brightness but with the potential problem of strong metal ion (Ca^{} and Fl^{}) sensitivity as well as being sensitive to acidification (pH < 6.5), making it susceptible to the effects of acidic metabolic byproducts that build up over time and dim fluorescence.
Electra2 An engineered protein with fast folding kinetics and very fast maturation, it is one of the most fast-folding proteins we have tested, and extremely useful for real-time imaging, although its robustness relative to some other proteins, including sfGFP, may be limited, with reduced stability over 36 hours.
2 Create a hypothesis for how adjusting one o2 r more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
To boost the fluorescence of mScarlet-I over the 36-hr incubation period, adding 50 mM HEPES (pH 7.4) to the master mix on top of the existing 50 mM (final concentration of 100 mM HEPES) will help. The enzymes within the cell-free extract utilize sugars and convert them into acidic byproducts, lowering the pH. Through this process, the acid-sensitive mScarlet-I chromophore will dim within the cell mix.
3 The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
A cell-free reaction needs certain concentrations of analytical reagents for transcription to happen without dropping in pH or running out of energy substrates. The 2 μL that will be used as a Metabolic & Buffer Optimization Cocktail will be spent from the Custom Reagent Supplements.
Standard 2X Master Mix (10 μL): contains the baseline components (Amino acids, NTPs, tRNA, Magnesium Glutamate, Potassium Glutamate, and an initial energy source like PEP). Custom Reagent Supplement (2 μL): optimized to contain HEPES buffer (pH 8.0) and additional PEP (Phosphoenolpyruvate). This 2 μL supplement is designed to maintain a stable pH environment and provide extra energy to ensure transcription/translation does not halt prematurely during the 36-hour run.
4 The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
-6 μL of Lysate
-10 μL of 2X Optimized Master Mix from above
-2 μL of assigned fluorescent protein DNA template
-2 μL of your custom reagent supplements
-Total: 20 μL reaction
Proposed Analytical Framework: The fluorescence data collected during the 36-hour incubation process will be analyzed in terms of the Kinetic Curves (Fluorescence Intensity vs. Time) of the wells intended for different master mixes and reagent conditions, as is typical for this type of homogenous assay.
From this set of kinetic curves, the following key metrics will be evaluated to determine what master mixes and reagent modifications are optimal and/or appropriate to use in subsequent experiments:
Maximum Fluorescence Intensity : To compare the relative total yield of protein across the different master mix modifications.
Reaction Lifespan (Slope Plateaus): To determine at what point in time the reaction reaches the end of the reaction (i.e., at what hour does the slope of the curve start showing a plateau, indicating that the chemical reaction is finished). If a custom master mix plateau extends later (e.g., the custom master mix plateau is at hour 24 instead of hour 12), this suggests that our proposed custom biophysical supplement successfully extended the energy availability or stabilized the pH. The lifetime of the reactions should similarly be compared across the different proteins to determine if the energy-buffering supplement was selective (i.e., it improves stability for acid-sensitive proteins but does not affect more-stable proteins), and thus.