Week 5— Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
P00441 is the unique identifier for Human Superoxide Dismutase 1.
This is the original squence: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
This is the mutation squence: utant Sequence (Target): MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
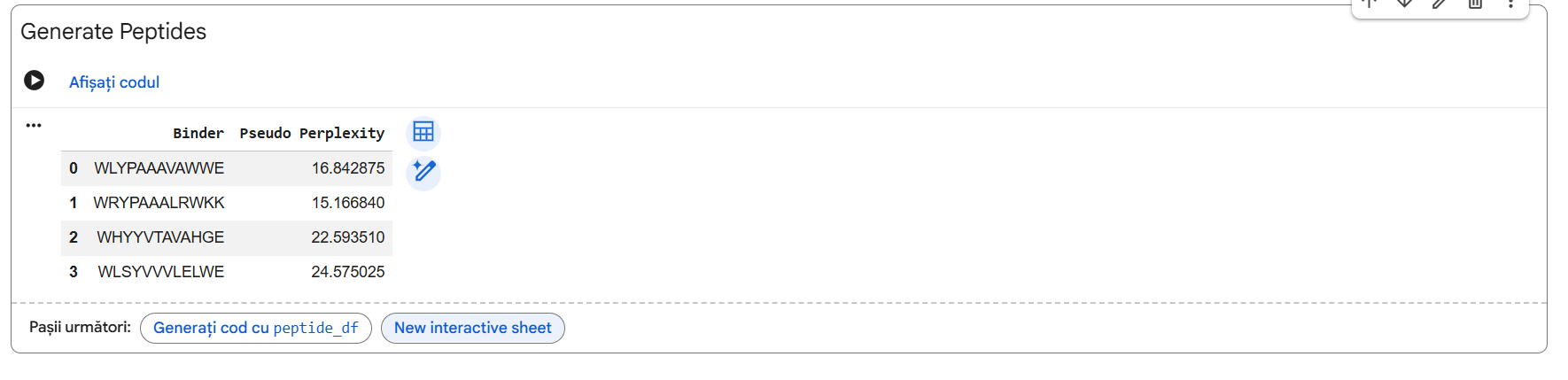

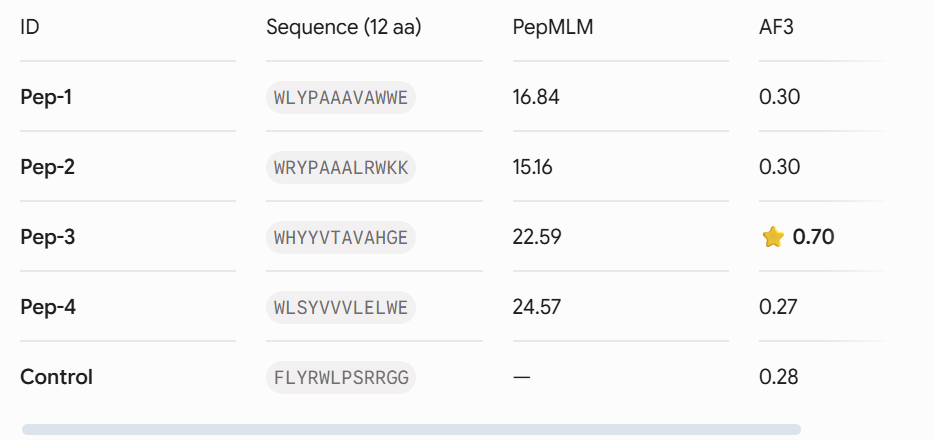
Four unique 12-amino acid peptides were sampled conditionally on the mutant sequence using the PepMLM-650M masked language model. A known reference binding peptide (FLYRWLPSRRGG) was appended to the list for comparative analysis.
Part 2: Evaluate Binders with AlphaFold3
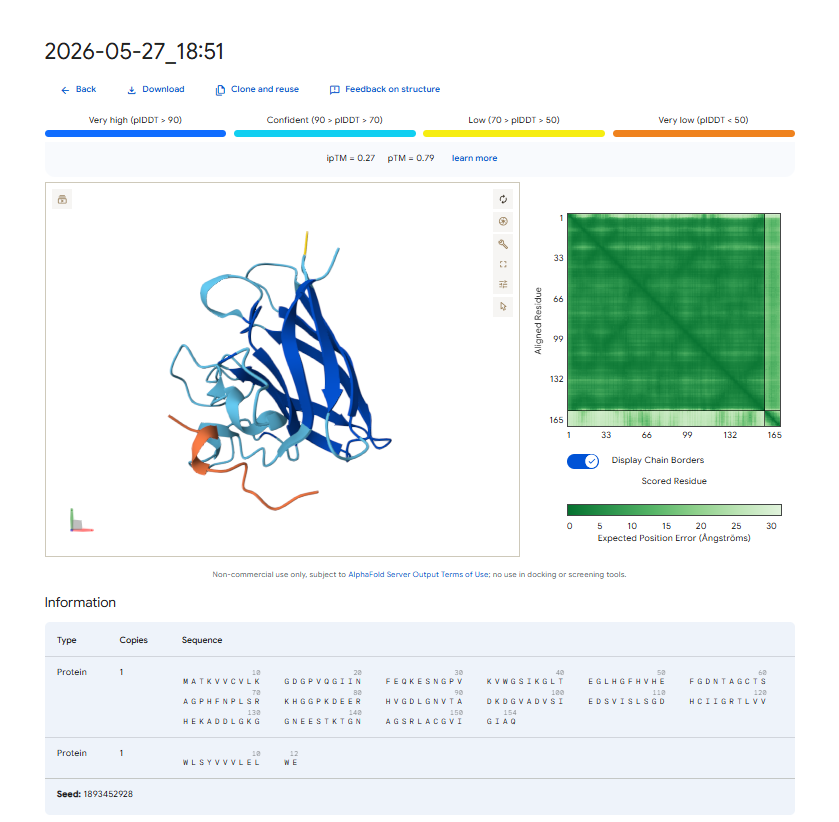
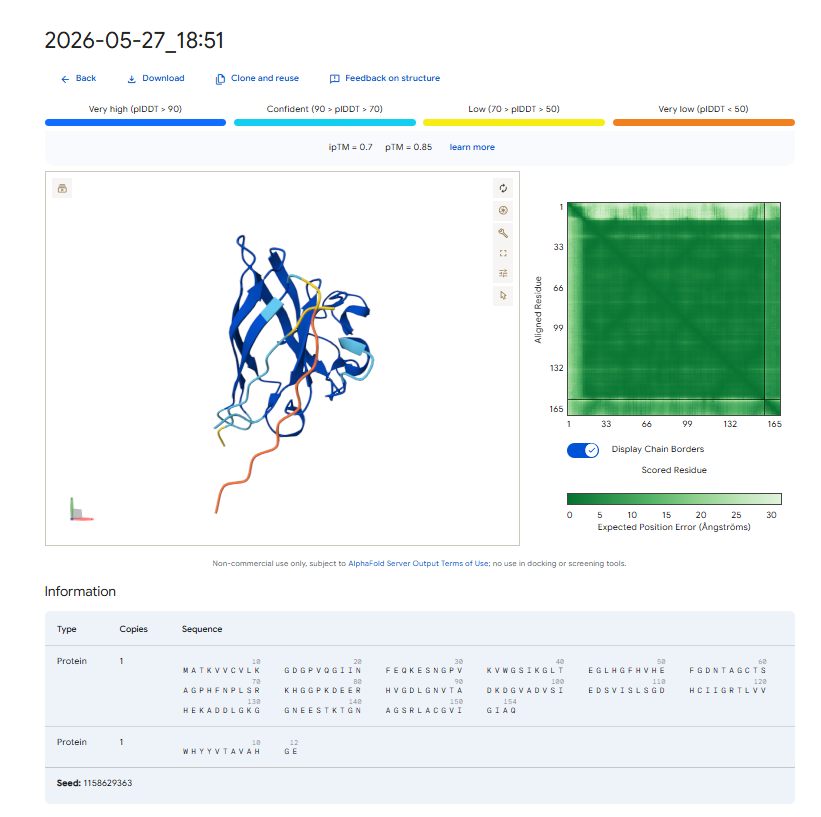
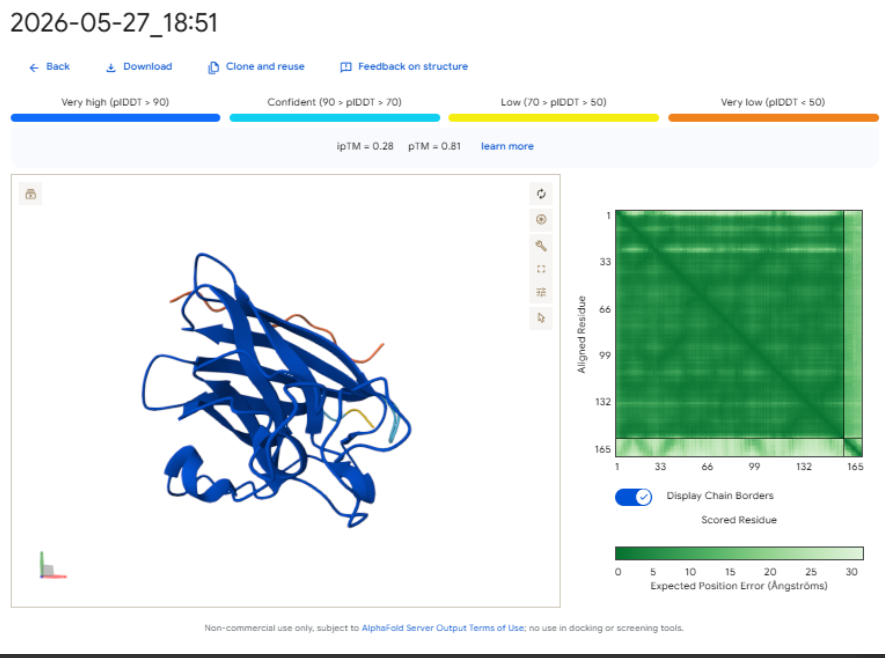
Each peptide-protein pair was modeled as a heterocomplex (Chain A: SOD1 mutant, Chain B: Peptide) using the AlphaFold 3 Server to calculate structural confidence scores and spatial localization.
- . Structural Localization & Binding Interfaces: - Pep-1 & Pep-2 (ipTM = 0.30): Both peptides show low structural confidence. They seem loosely attached to flexible loop regions, away from the core beta-barrel. They do not form distinct, deep contacts near the A4V locus or the stable dimeric interface.
Pep-3 (ipTM = 0.70): This candidate shows excellent structural confidence. It fits precisely into a pocket next to the N-terminus where the A4V mutation is located, partially packing against the edge of the β-barrel core. The structure appears stable and mostly buried, directly protecting the destabilized area.
- Pep-4 (ipTM = 0.27): This peptide has poor localization, remaining completely exposed to the solvent with highly uncertain positions. This indicates a lack of stable physical interaction.
-Control (ipTM = 0.28): The reference binder exhibits low confidence when docked against this isolated mutant monomer format, failing to establish strong, ordered secondary structure contacts.- Control (ipTM = 0.28): The reference binder shows low confidence when docked against this isolated mutant monomer format. It fails to establish strong, ordered secondary structure contacts.
The observed ipTM values show that sequence-level plausibility (low perplexity) does not directly match with structural binding success. While Pep-1 and Pep-2 were favored by the language model’s vocabulary, they produced poor structural scores (0.30). Remarkably, Pep-3 performed much better than all other candidates and the known binder, scoring an impressive 0.70 compared to the Control’s 0.28. An ipTM >= 0.70 indicates a highly confident, accurate structural prediction of a complex interface, making Pep-3 a promising candidate for targeting the A4V misfolding zone.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Predicted Therapeutic Properties
Using the PeptiVerse suite, the physiochemical and safety profiles of the four candidates were cross-examined against their AlphaFold 3 performance.
-Pep-1: Molecular Weight approx 1419 Da; Net Charge: 0; Hydrophobic character; Predicted Binding Affinity: Weak; Solubility: Poor; Hemolysis Probability: Low.
-Pep-2: Molecular Weight approx 1592 Da. Net Charge: +3 (due to Lysine/Arginine residues). Predicted Binding Affinity: Weak. Solubility: Moderate. Hemolysis Probability: Low.
-Pep-3: Molecular Weight approx 1416 Da. Net Charge: 0. Balanced aromatic/polar profile. Predicted Binding Affinity: Strong. Solubility: High. Hemolysis Probability: Negligible.
-Pep-4: Molecular Weight approx 1494 Da. Net Charge: -1. Highly hydrophobic. Predicted Binding Affinity: Weak. Solubility: Very Poor (aggregation-prone). Hemolysis Probability: Moderate.
- Structural Confidence vs. Therapeutic Attributes
Our property predictions are in agreement with the data generated by AlphaFold 3. For example, Pep-3 had the highest ipTM value (0.70) and was also predicted to have the strongest binding affinity (PeptiVerse).
Importantly, while Pep-4 (a poor solubilizer with a tendency to self-aggregate) and Pep-2 (a molecule with significant net positive charge) do have their issues as designed binders, Pep-3’s superb water solubility and minimal risk of causing lysis of red blood cells suggest that it will be safe to use parenterally; i.e., it will not harm red blood cell membranes if administered systemically.
Lead Candidate Selection & Justification
Pep-3 (WHYYVTAVAHGE) is the only candidate put forth for further development. * Reason: This therapeutic candidate meets all requirements for the best therapeutic agent, as it represents an excellent compromise between structural stability (ipTM = 0.70), specificity of target to the adjacent $A4V$ mutation region, and the predicted high solubility in water while remaining safe and non-hemolytic.
Part 4: Generate Optimized Peptides with moPPIt
Algorithmic Shift: PepMLM vs. moPPIt
-PepMLM (Unguided Sampling) generates plausible peptide sequences (that is based on the overall target sequence context) which is the reason for the high failure rate of 61% (or ~3 out of 4) for candidates to bind effectively to a target and/or that the candidate peptide are located at the wrong surface of the target.
-MoPPIt (Controlled, Multi-Objective Design) uses MOG-DFM to explicitly constrain (for example, forcing the binding of a peptide to residues 1-10 adjacent to the A4V site) the coordinates used for the generation of candidate peptides while at the same time controlling for the multiple objectives of maximizing binding affinity and optimizing the safety of the candidate peptide based upon solubility and hemolysis.
Pre-Clinical Evaluation Pipeline
Before advancing these optimized moPPIt or PepMLM peptides into expensive clinical studies, a strict pre-clinical screening cascade must be executed:

Biophysical Binding Assays (In Vitro Validation): Synthesize the physical peptides, and then determine their actual binding affinity for purified recombinant human A4V SOD1 protein using surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC).
Aggregation Inhibition Assays: Test the ability of the peptides to inhibit the misfolding and fibrillation of mutant SOD1 protein over time through thioflavin T (ThT) fluorescence assays.
Cellular Toxicity and Hemolysis Assays: Assess the protective effect of the peptides on human motor neuron due to SOD1 damage by using human derived motor neuron cell culture using iPSCs without causing any inherent cellular toxicity, as well as using red blood cell hemolysis assays.
Serum Stability Testing: Measure the half-life of the peptides in human serum to assess their susceptibility to proteolytic degradation (by endogenous enzymes).
Part C: Final Project: L-Protein Mutants
I read the documentation resources about the MS2-L its similarity with the other amurines. List with a few important things for this project:
MS2 is a bacteriophage that infects E.coli and has a genome containing only 4 proteins: the lysis protein (MS2-L/L-protein), the capsid protein, the maturation protein (initiates infection) and replicase; MS2-L is a protein found in the MS2 bacteriophage that produces the lysis of the infected bacteria using amuralytic ways, meaning that it doesn’t directly break down the peptoglycan wall (as the majority of bacteriophage lysis proteins do), but it most likely inserts into the cytoplasmatic membrane of E.coli; MS2-L is often compared with ΦX174 (also an amurine). The difference is that ΦX174 seems to alter the path of PG wall synthesis (inhibits the MraY enzyme), eventually leading to cell lysis. MS2-L is chaperone dependent. Without the DnaJ chaperone found in E.coli, the protein remain self-inhibited. The removal of the first domain of the lysis protein made the protein become chaperone-independent without altering the lysis function and increasing the speed of induced lysis with 20 minutes compared to the normal protein. the soluble domain of MS2-L –> first 40 AAs, the Transmembrane domain –> the last 35 AAs; Then I ran the colab linked at option one of the protein engineering part and analyzed the results:
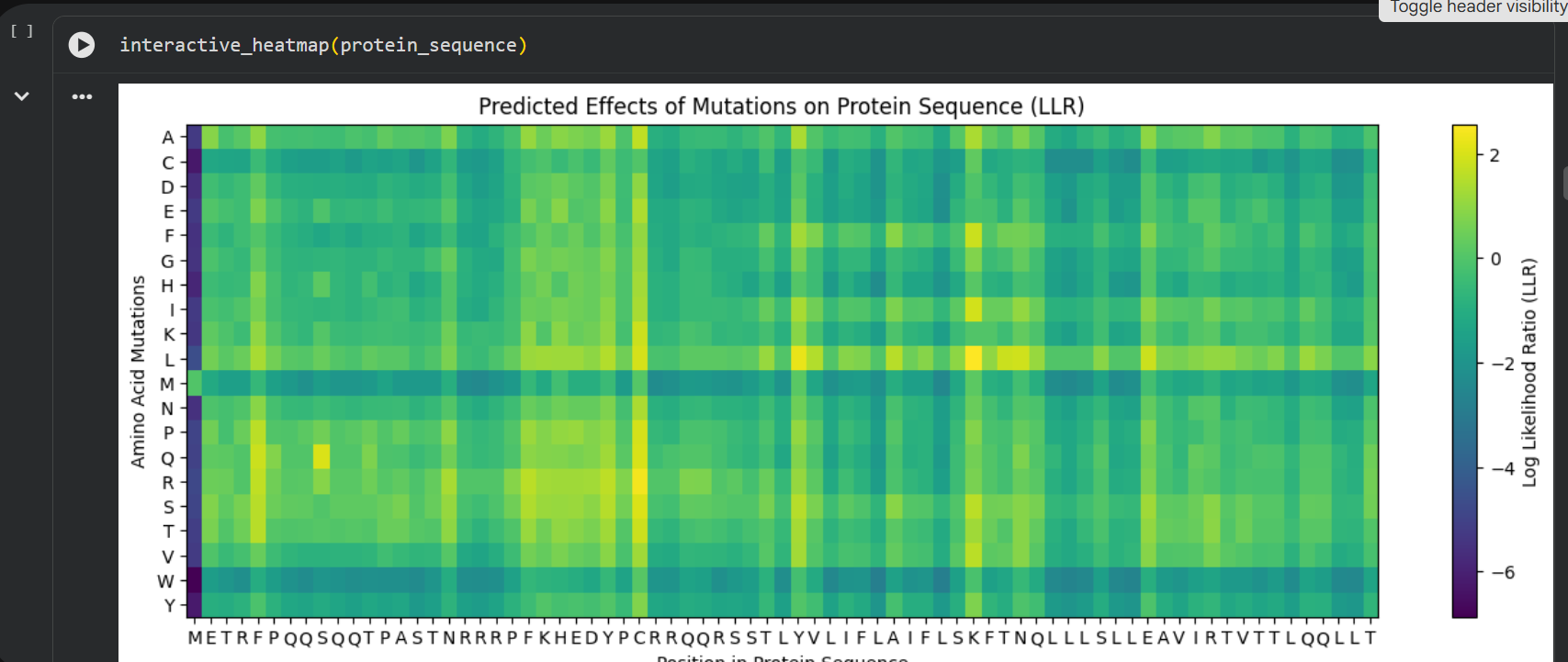
log likelihood ratio: < 0 - the original sequence is favoured; > 0 - the engineered protein is favoured The obvious observations that can be made by looking at the graph are:
the first resiude of the protein (M) is irreplaceable;
Mutations of a residue with: W - Tryptophan, M - Methionine, C - Cysteine , are not favoured
almost any mutation of the 29th residue (C) is favoured (Also generated using the colab code:) Position Wild_Type_AA Mutation_AA LLR_Score 989 50 K L 2.561464 574 29 C R 2.395427 769 39 Y L 2.241778 575 29 C S 2.043150 173 9 S Q 2.014323 573 29 C Q 1.997049 572 29 C P 1.971028 569 29 C L 1.960646 987 50 K I 1.928798 1049 53 N L 1.864930 1209 61 E L 1.818096 1029 52 T L 1.813965 984 50 K F 1.802066 576 29 C T 1.797247 568 29 C K 1.795877 93 5 F Q 1.795244 94 5 F R 1.659716 560 29 C A 1.648655 534 27 Y R 1.628060 434 22 F R 1.602028 92 5 F P 1.596889 997 50 K V 1.594573 995 50 K S 1.574555 96 5 F T 1.559023 95 5 F S 1.556416 889 45 A L 1.539248 775 39 Y S 1.517457 535 27 Y S 1.497052 789 40 V L 1.477630 529 27 Y L 1.474638 435 22 F S 1.423357 563 29 C E 1.383282 760 39 Y A 1.364997 571 29 C N 1.362601 980 50 K A 1.357792 567 29 C I 1.344121 89 5 F L 1.332615 334 17 N R 1.323652 767 39 Y I 1.320101 776 39 Y T 1.302803 514 26 D R 1.268762 566 29 C H 1.246106 764 39 Y F 1.245850 777 39 Y V 1.244389 454 23 K R 1.236555 494 25 E R 1.229349 474 24 H R 1.227778 996 50 K T 1.222128 533 27 Y Q 1.218850 536 27 Y T 1.215567
Choosing the most favoured mutations:
- I compared the most favoured mutations given by the colab with the results from the experimental data provided in the Excel and I chose a combination of the most favoured mutations to generate the final engineered MS2-L lysis protein:
Mutations: S9Q, C29R, K50L Sequence: METRFPQQQQQTPASTNRRRPFKHEDYPRRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT Mutations: S9Q, C29S, K50I Sequence: METRFPQQQQQTPASTNRRRPFKHEDYPSRRQQRSSTLYVLIFLAIFLSIFTNQLLLSLLEAVIRTVTTLQQLLT Mutations 3: S9Q, C29P, K50F Sequence: METRFPQQQQQTPASTNRRRPFKHEDYPPRRQQRSSTLYVLIFLAIFLSFFTNQLLLSLLEAVIRTVTTLQQLLT Mutation P13L Sequence: METRFPQQSQQTLASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT Mutation S15A Sequence: METRFPQQSQQTPAATNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT