Week 4 HW: Protein Design Part I
A
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
A Dalton is equal to 1/12 atom of carbon, so aprox. 1.66x10-27kg. I will chose beef , because the type isn’t specified . 100g of beef has aporx. 25g protein, so 500g has 125g protein. 1g=6.022x1023 atomic mass units =>125g=7,5275 x 10^25 Dalton
2.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans eat beef but do not become a cow, eat fish but do not become fish, because these foods are decomposed in substances that are used or throw away . They don’t have an impact on our DNA so we don’t become cows or fishes.
3. Why are there only 20 natural amino acids?
There are only 20 natural amino acids because like that they are more stable and it can provide a big variety of proteins.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, it is possible , but I won’t do it.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before enzymes that make them, and before life started amino acids come from abiotic synthesis. Simple inorganic molecules reacted under high-energy conditions.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
While natural L-amino acids form right-handed alpha-helices, a polymer made of D-amino acids would adopt a left-handed conformation.
7. If you make an α-helix using D-amino acids , there will be a left handedness.
Yes, they have been discovered as rare but stable variants. The 3-helix is more tightly wound and elongated, appearing at the termini of regular helices. The pi-helix is wider and shorter, it is found near functional sites in enzymes.
8. Why are most molecular helices right-handed?
Most molecular helices are right-handed because that direction keeps the amino acid side chains from crashing into the backbone.
9. Why do β-sheets tend to aggregate? • What is the driving force for β-sheet aggregation?
Beta-sheets aggregate because of the hydrophobic effect, where water molecules force non-polar side chains together to hide them from the aqueous environment, making the sheets into stacks. Once they meet, unsatisfied hydrogen bonds along the edges act like a chemical glue, locking the strands into stable fibers called amyloids.
10. Why do many amyloid diseases form β-sheets? • Can you use amyloid β-sheets as materials?
Sheet aggregation is Mostly governed by strong, repetitive hydrogen bonding that occurs between the backbone NH and C=O groups of neighboring strands. The role of dehydration and burial of the hydrophobic side chains is to assist this process. Besides this, stacking of aromatic rings and dipole interactions contribute by further stabilizing the structure. As a result, aligned -sheets are not only energetically favorable but are also very likely to form larger aggregates.
11. Design a β-sheet motif that forms a well-ordered structure.
For amyloid diseases, misfolded proteins form cross- fibrils due to trapping the exposed aggregation-prone regions in stable -sheet conformations that stack to form very long, persistent fibrils. Besides the pathological implications, the same stability and ability of self-assembly feature of amyloid fibrils can be exploited for generating nanofibers, hydrogels, and scaffolds used in tissue engineering and drug delivery. Forced folding of the -sheet motif in a -hairpin form with the sequence VIVTYGGYTVIV is a good example of this. Here the strands VIVTY and YTVIV are joined through the GG turn. One hydrophobic stacking face and one soluble face are produced by hydrophobic (Val Ile Tyr) and polar (Thr, Tyr) residues alternating along the strand. This sort of arrangement is that of a compact, ordered two-stranded -sheet.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
1Briefly describe the protein you selected and why you selected it.
I chose the same protein that I used in homework two, because Dsup is very interseting and I like very uch tardigrades.
2 Identify the amino acid sequence of your protein.
MASTHQSSTEPSSTGKSEETKKDASQGSGQDSKNVTVTKGTGSSATSAAIVKTGGSQGKDSSTTAGSSSTQGQKFSTTPTDPKTFSSDQKEKSKSPAKEVPSGGDSKSQGDTKSQSDAKSSGQSQGQSKDSGKSSSDSSKSHSVIGAVKDVVAGAKDVAGKAVEDAPSIMHTAVDAVKNAATTVKDVASSAASTVAEKVVDAYHSVVGDKTDDKKEGEHSGDKKDDSKAGSGSGQGGDNKKSEGETSGQAESSSGNEGAAPAKGRGRGRPPAAAKGVAKGAAKGAAASKGAKSGAESSKGGEQSSGDIEMADASSKGGSDQRDSAATVGEGGASGSEGGAKKGRGRGAGKKADAGDTSAEPPRRSSRLTSSGTGAGSAPAAAKGGAKRAASSSSTPSNAKKQATGGAGKAAATKATAAKSAASKAPQNGAGAKKKGGKAGGRKRK
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
THe protein ha 445 aminoacids.The most frequent aminoacid is Serine. It apears 84 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
I searched for homologs in Uniprot and I think I managed to find them . So, I opened the Uniprot link is on the htgaa website at week 4 . I paste the aminoacid sequence and then I waited it to run.
Does your protein belong to any protein family?
NO, it doesn’t belong to any protein family.
**3 Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
Nucleosome-Binding Protein
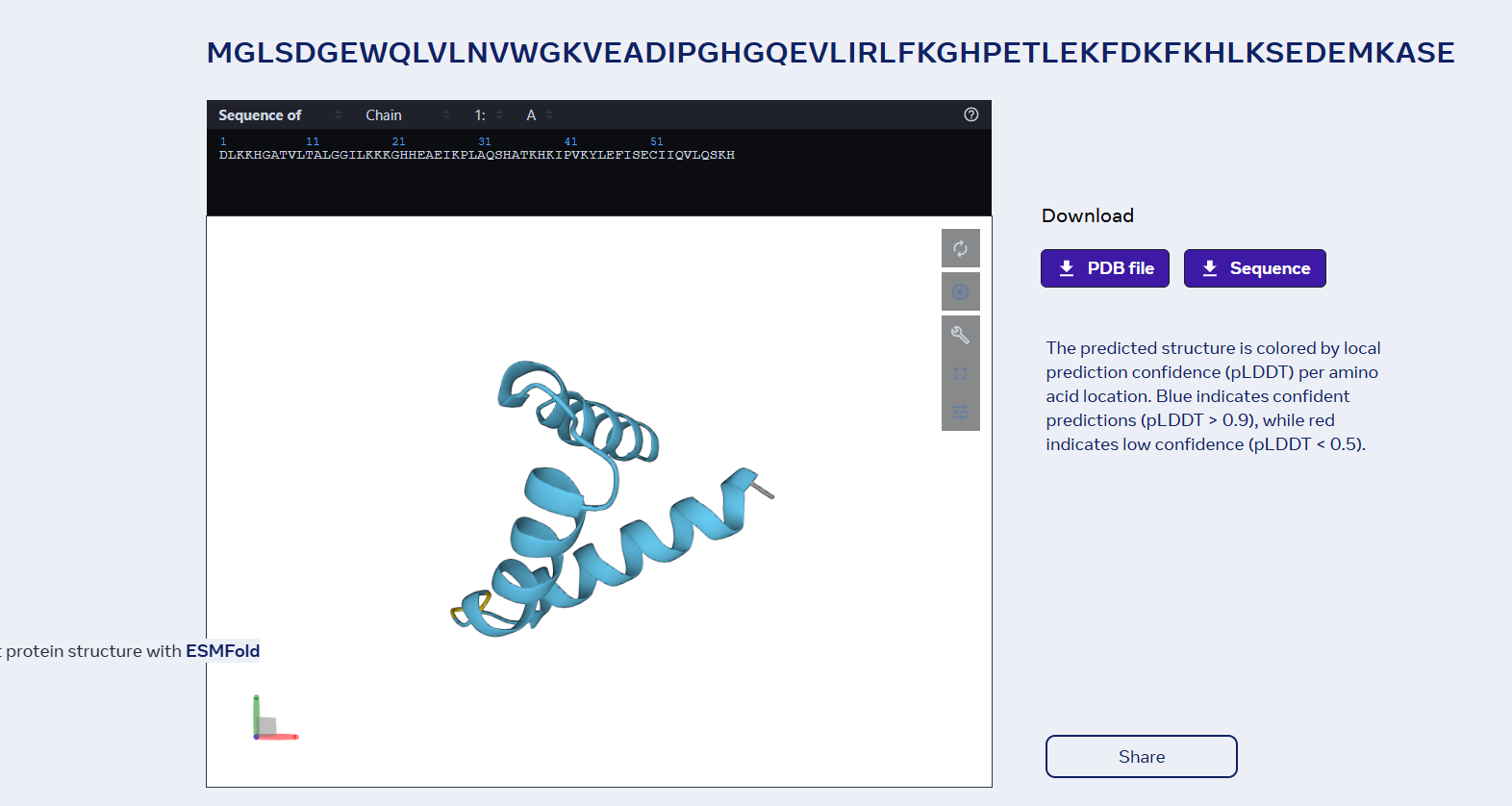
3 Open the structure of your protein in any 3D molecule visualization software: Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
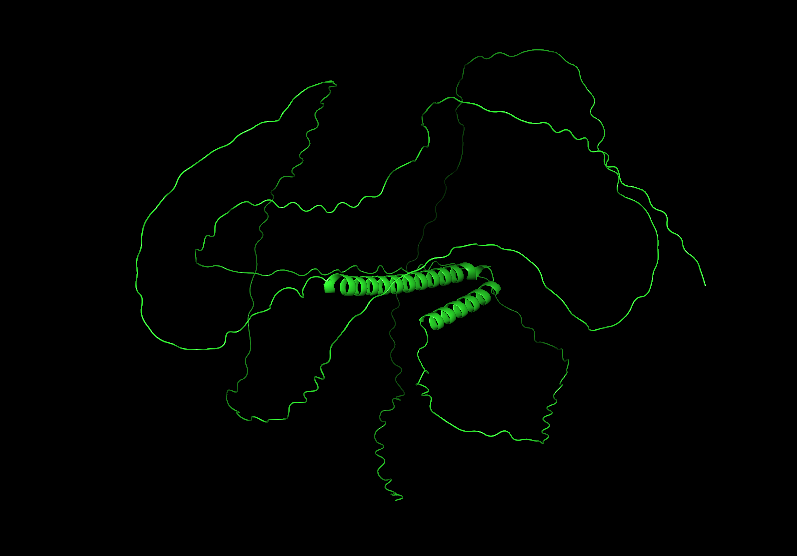
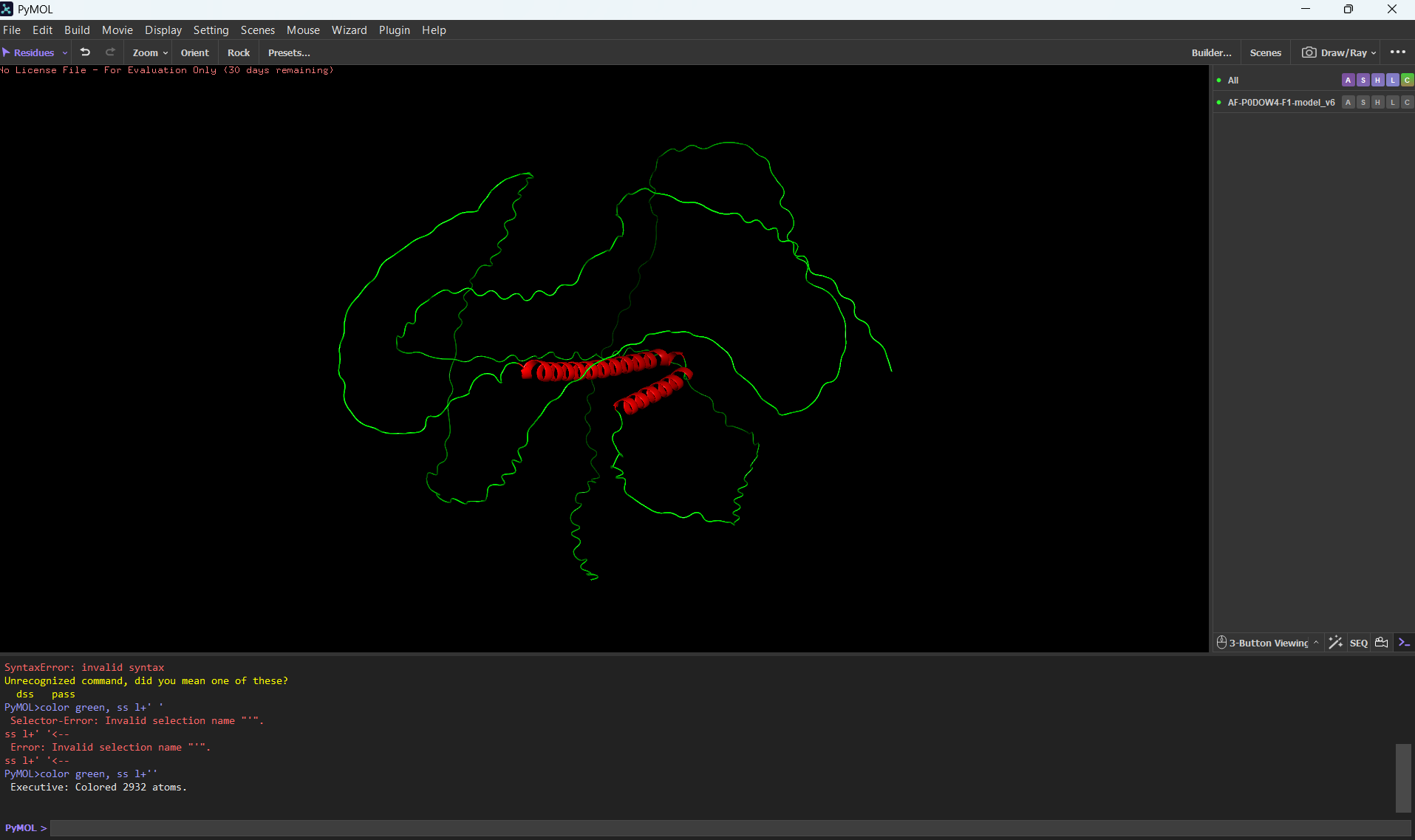
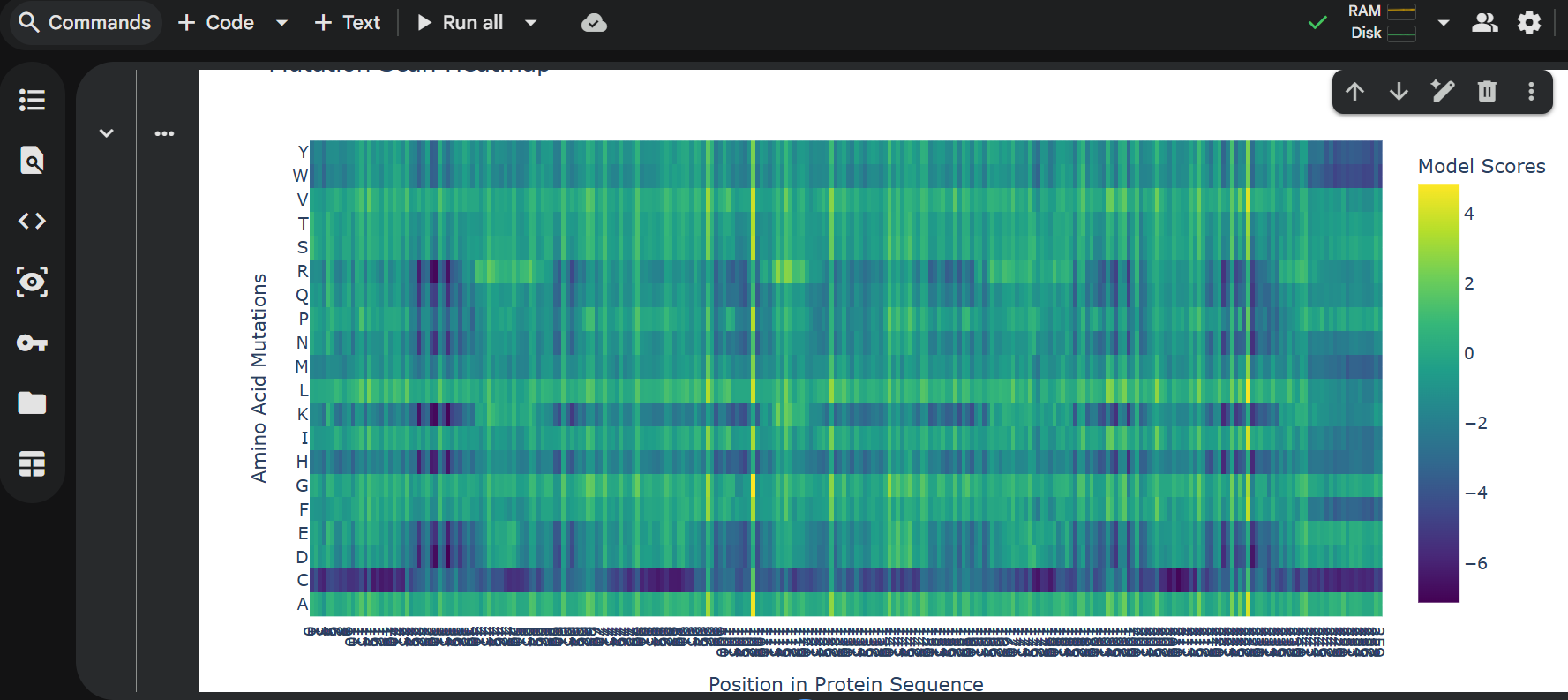
Color the protein by secondary structure. Does it have more helices or sheets?
It has more helices than sheets.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Most of the protein will remain green, with a few small cylinders representing transient alpha-helices.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
To color by secondary structure I used color red, ss h; color yellow, ss s; color green, ss l+’'.
Part C. Using ML-Based Protein Design Tools
Glu Glu Leu Ile Thr Gly Val Leu Gly Ile Ser Ile Asp Leu Gly Met Val Thr Gly Ser Asp Leu Ala Lys Ala Val Lys Leu Ala Thr Gly Leu Gly Glu Ala Val Val Glu Gly Ala Lys Ala Val Gly Ser Val Leu Ala Leu Ser Thr Ala Leu Val Leu Ala Leu Leu Gly Leu Ala Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu
sp|P02144|MYG_HUMAN Myoglobin OS=Homo sapiens OX=9606 GN=MB PE=1 SV=2 MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASE DLKKHGATVLTALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKH PGDFGADAQGAMNKALELFRKDMASNYKELGFQG
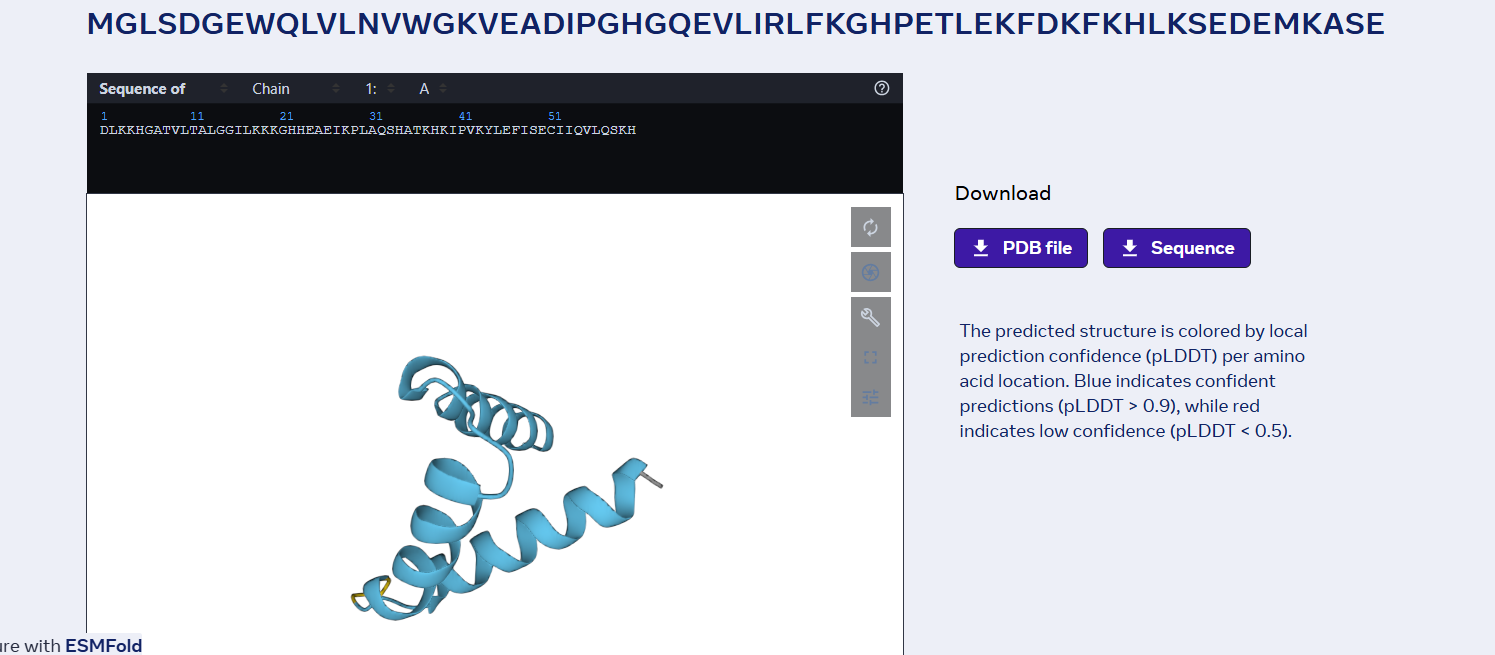
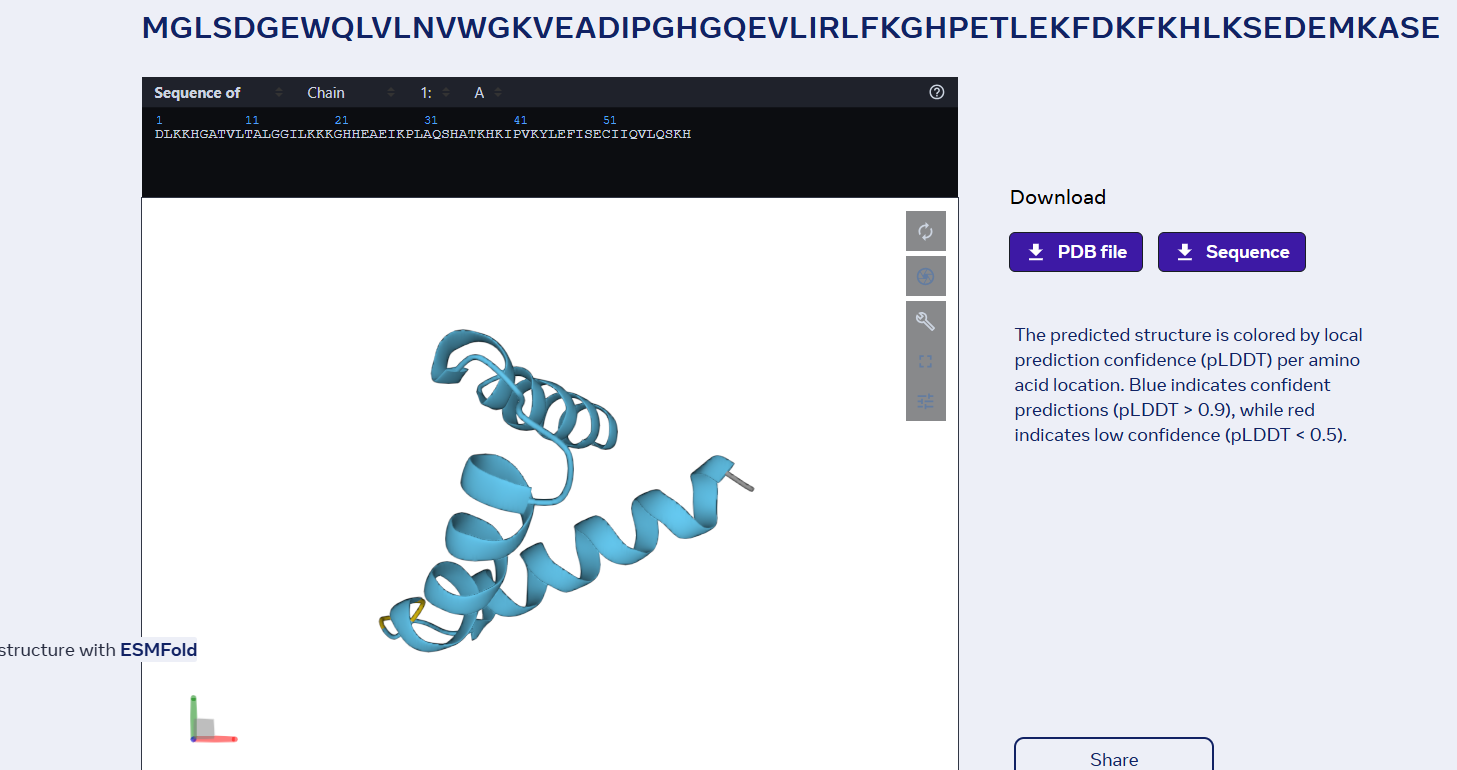
Part D. Group Brainstorm on Bacteriophage Engineering
Project Proposal: Engineering a Minimal MS2-L Lysis Engine
1 Primary Goal: Our group aims to increase the functional stability of the MS2 lysis (L) protein. We will achieve this by eliminating the N-terminal domain (residues 1–36). This truncation removes the “regulatory brake” that normally makes lysis dependent on the host chaperone DnaJ, resulting in a more potent, autonomous lysis protein, thus lysis will be achieved a lot faster, beacuse MS2-L will be functional from the moment of translation which gives less time for the proteases to degrade it before it attached to the cellular membrane.
2 Tools & Approaches We propose a computational pipeline to validate and optimize this truncated variant:
AlphaFold3 / AlphaFold-Multimer: We will model the truncated L protein in a cramped lipid bilayer environment to predict how the remaining transmembrane helix (TMH) inserts itself. We will also use it to confirm the loss of binding affinity to E. coli DnaJ.
Protein Language Models (ESM-2 / ESM-1v): We will use these models to perform in silico mutagenesis on the remaining C-terminal sequence. Our goal is to identify “stabilizing” mutations that strengthen the alpha-helical propensity of the membrane-spanning region.
Molecular Dynamics (MD) Simulations (OpenMM or Gromacs): Since lysis involves membrane distortion, we will simulate the truncated protein within a model bacterial membrane to observe its ability to disrupt lipid packing.
4 Potential Pitfalls Membrane Toxicity in in silico models: Most protein design tools are trained on soluble proteins. Modeling a protein whose entire job is to destroy the membrane (lysis) may lead to unstable simulations or “unphysical” results. Expression Levels: In a real-world lab setting, a highly toxic protein might kill the host cells so quickly that we cannot produce high enough titers of the phage for study.
5 Pipeline Schematic Design: Truncate N-terminus (1-36). Optimize: Run ESM-1v to find high-probability stabilizing mutations.
Predict: Fold top candidates in AlphaFold3 to ensure TMH orientation.
Simulate: Insert into a virtual membrane to verify disruptive “toxicity.”