Week 4 HW: Protein Design - Part 1
Part A: Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
On average, if we assume that 25% of meat is protein, then we would be taking 500*0.25 = 125 g of protein intake. With an average of 100 Da per amino acid as it’s molecular weight, the total number of moles of amino acids become 1.25 moles. Multiplying with the Avagadro’s number, we get roughly seven hundred fifty-two sextillion (7.528 * 10^23) molecules !!
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we eat beef or fish, our body does not absorb the protein in their native format. It degrades them to the lowest possible level: amino acids. These amino acids are what are absorbed and then reused to construct other new proteins that our body needs. Thus, we do not become what we eat, rather we become what we construct from what we eat.
3. Why are there only 20 natural amino acids?
These set of 20 amino acids are what are used by majority of the organisms on the planet. Though there isn’t a set rule, these 20 amino acids make possible all the proteins and their conformations that are required to sustain life by having & conferring suitable properties. Probably, we can argue that changing this “perfect set” of amino acids might be harmful/lethal to the organism, which would’ve been why only 20 amino acids are present.
4. Can you make other non-natural amino acids? Design some new amino acids.
We can! And this area of study is termed as Xenobiology.
For example, we can design amino acids with different side chain groups such as cyano (-CN) or a boron (-B) groups/atoms. These would enable the amino acids to bind to certain substrates more effectively, repel others, act as sensors, and open up a plethora of unique functions.
5. Where did amino acids come from before enzymes that make them, and before life started?
This is explained by the Miller-Urey experiment of abiotic synthesis. They showed that, in the “primordial soup” of early Earth, energy from lightnings, hydrothermal vents and other sources, reacted with gases such as methane, ammonia, etc. to form simple organic molecules, which could have been the earliest source of amino acids.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
D-amino acids are the “mirrored” versions of the L-amino acids. So, the steric hindrances present in the L-amino acid helices (which has a right handedness) would flip, conferring a left handedness to the D-amino acid helix.
7. Can you discover additional helices in proteins?
Yes of course! Some of them include: the 3(10)helix, which contains tighter and thinner helices; the pi helix, which is wider and shorter relative to the normal alpha helix.
8. Why are most molecular helices right-handed?
Most biological helices are right-handed due to the chirality of their L-amino acids (proteins), which cause steric clashes in their left-handed forms.The right-handed alpha-helices avoid side-chain and backbone repulsions, thus optimizing stability.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
The primary driving force for aggregation of beta sheets are hydrogen bonding between the backbones and the hydrophobic effects involved. Since the edges of the sheets have available hydrogen bond donors and acceptors, they tend to form aggregates by grabbing onto other beta sheets.
Part B: Protein Analysis and Visualization
My protein of choice is the DnaA protein from Mycobacterium tuberculosis. It acts as a replication initiator, aiding in recognizing, binding and unwinding the oriC region to kick-start replication. It is absolutely essential for replication in M.tuberculosis, and without it, the bacterium will not be able to initiate replication.
I chose this particular protein, because I am currently working on it, especially with it’s DNA Binding domain (Domain 4), to screen and identify inhibitors against it. It is also a biologically sound target, since it is essential for replication and also has no eukaryotic homologs.
The amino acid sequence details:
Sequence:
sp|P9WNW3|DNAA_MYCTU Chromosomal replication initiator protein DnaA OS=Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) OX=83332 GN=dnaA PE=1 SV=1 MTDDPGSGFTTVWNAVVSELNGDPKVDDGPSSDANLSAPLTPQQRAWLNLVQPLTIVEGF ALLSVPSSFVQNEIERHLRAPITDALSRRLGHQIQLGVRIAPPATDEADDTTVPPSENPA TTSPDTTTDNDEIDDSAAARGDNQHSWPSYFTERPHNTDSATAGVTSLNRRYTFDTFVIG ASNRFAHAAALAIAEAPARAYNPLFIWGESGLGKTHLLHAAGNYAQRLFPGMRVKYVSTE EFTNDFINSLRDDRKVAFKRSYRDVDVLLVDDIQFIEGKEGIQEEFFHTFNTLHNANKQI VISSDRPPKQLATLEDRLRTRFEWGLITDVQPPELETRIAILRKKAQMERLAVPDDVLEL IASSIERNIRELEGALIRVTAFASLNKTPIDKALAEIVLRDLIADANTMQISAATIMAAT AEYFDTTVEELRGPGKTRALAQSRQIAMYLCRELTDLSLPKIGQAFGRDHTTVMYAQRKI LSEMAERREVFDHVKELTTRIRQRSKR
Length: 507 residues
The most frequent amino acid: Alanine (54 times), Leucine (46 times), threonine (42 times). (Using given colab notebook)
Number of protein sequence homologs: 250 homologs found, from using the Uniprot BLAST Tool.
Belonging to a protein family: Yes, belongs to the DnaA family.
Protein Structure in RCSB:
PDB ID: 3PVV
Resolved in: 2011
Resolved by: Tsodikov, et al..
Link to the article: Here
Resolution: 2 Angstroms (It is a good quality structure.)
The solved structure contains the protein’s DNA Binding Domain (also called as the Domain 4), bound to a MtDnaA box, that is, bound to a DNA helix.
Note: The PDB structure only has the domain 4, and not the entire protein’s structure. However, the full structure of the protein is available in the Alphafold database.
Through the search in SCOP Database, I found that the protein indeed belongs to the structure classification family of “Chromosomal replication initiation factor DnaA C-terminal domain IV”. This family currently consists of DnaA proteins from three bacteria: E.coli, M.tuberculosis and A.aeolicus.
3D visualization:
Chosen Software: PyMol
Note: Only the PDB structure has been used in this homework for visualization.
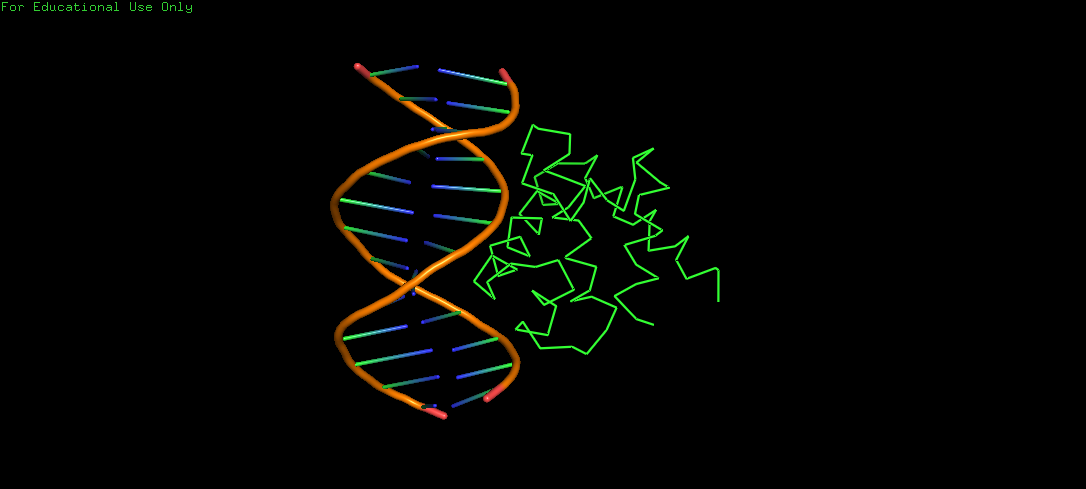
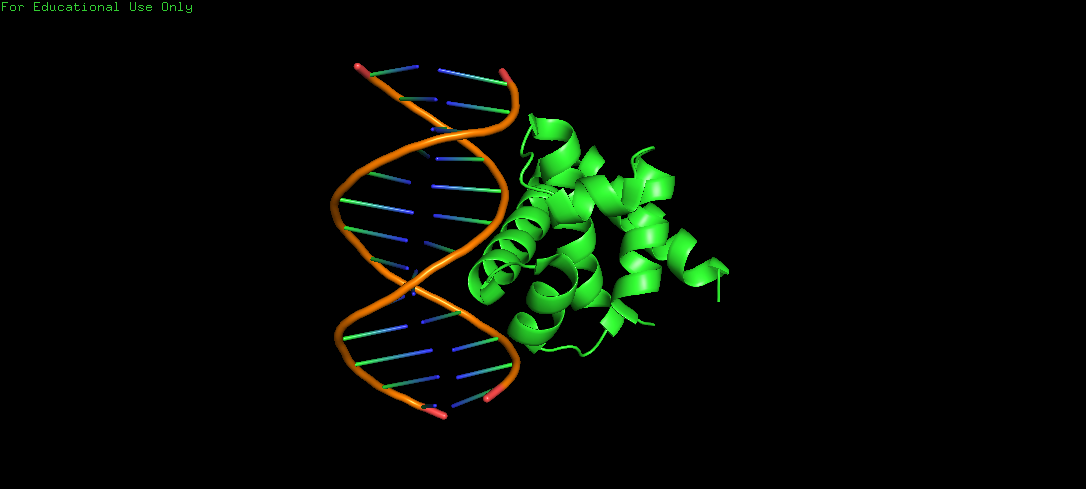
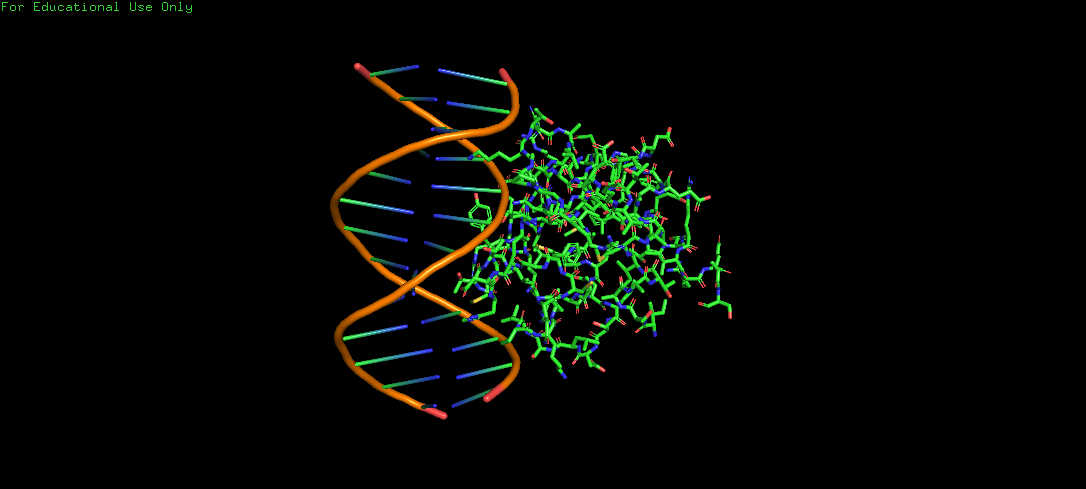
Protein Structure visualized as a Ribbon, Cartoon and Ball and Sticks.
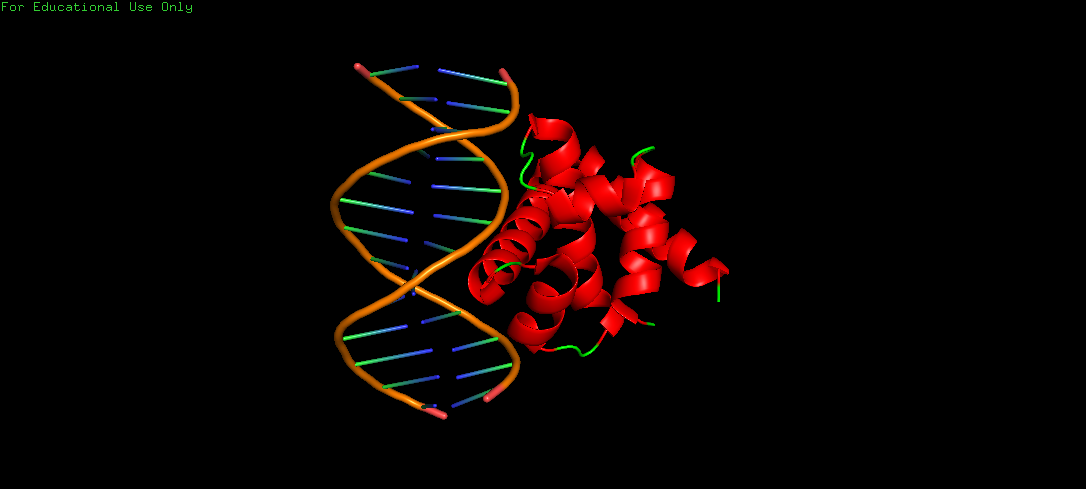
Upon colouring by secondary structure it is found to have a lot of helices. No beta sheets/strands were seen.
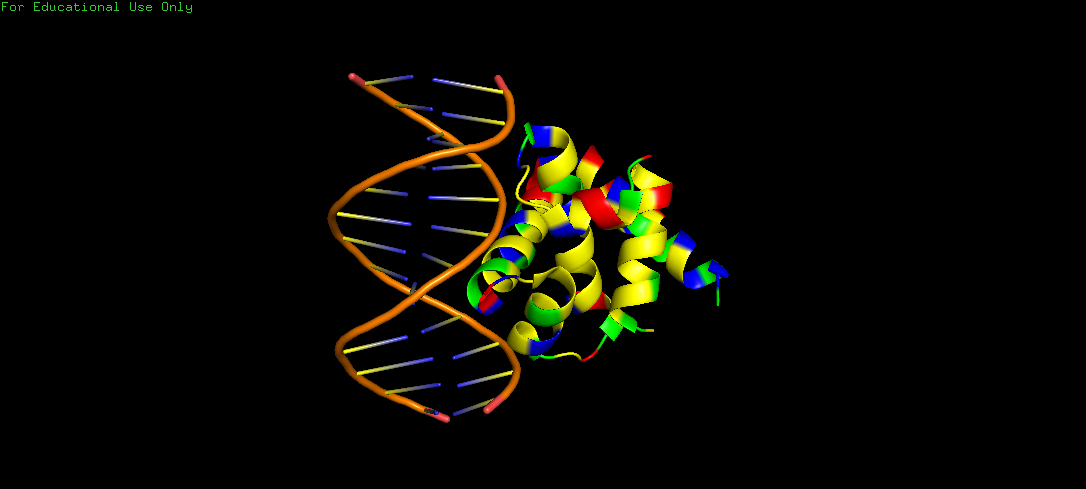
The DNA-interacting of the protein is likely enriched in hydrophilic/basic residues, enabling electrostatic attraction to the negatively charged phosphate backbone. These interactions are predominantly polar and ionic, not hydrophobic, since DNA is highly water-exposed and also charged. Whereas, hydrophobic residues are probably buried within the protein core, stabilizing its folded alpha helical structure.
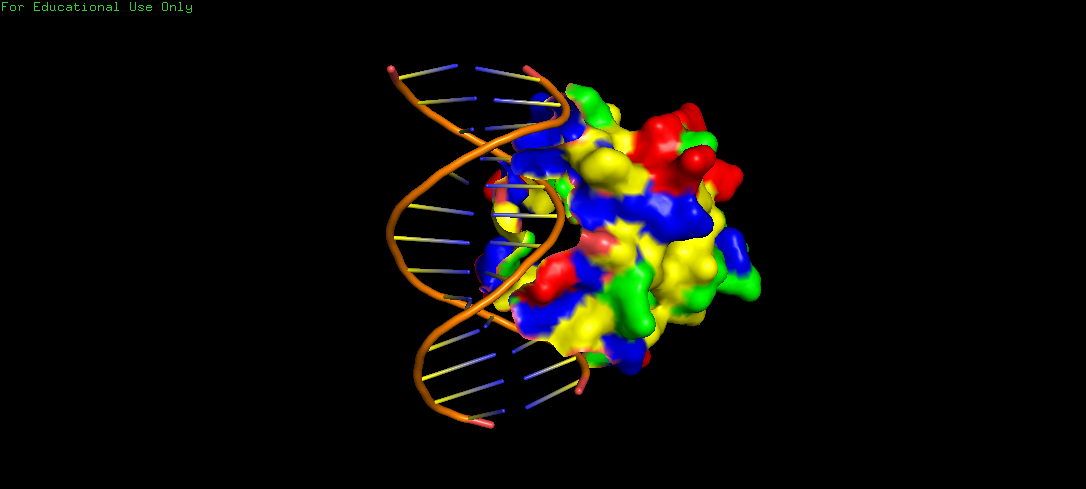
The surface representation shows visible grooves and cavities at the protein–DNA interface, suggesting potential binding pockets. These pockets appear partially recessed and contoured, which are consistent with nucleotide accommodation. Also, the clustering of varied residues around these areas indicate chemically diverse environments, suitable for specific binding to the DNA. However, visualizing binding pockets through the Alphafold structure might be more effective, since here, we can mistake the start and end of the protein sequence to be a binding pocket, when in fact, it is just the sequence and structure that has been determined.
Part C: Using ML-Based Protein Design Tools
C1: Protein Language Modeling
1. Deep Mutational Scans: a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. b. Can you explain any particular pattern? (choose a residue and a mutation that stands out) c. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
2. Latent Space Analysis a. Use the provided sequence dataset to embed proteins in reduced dimensionality. b. Analyze the different formed neighborhoods: do they approximate similar proteins? c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
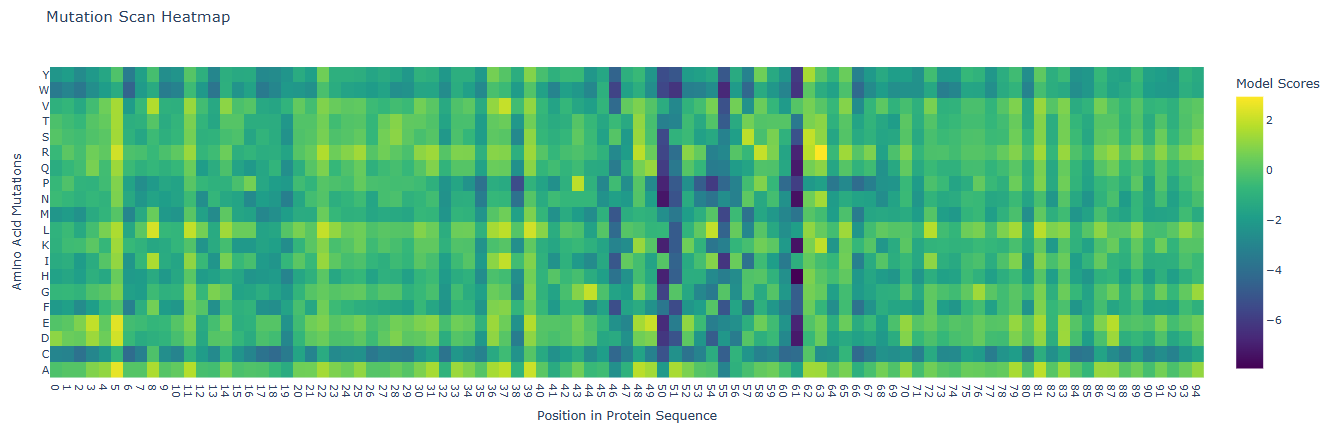
Deep Mutational Scans
Positions 5, 11, 22, 37, 39 and some others: These positions are highly bright or yellow across almost all amino acid substitutions. This indicates that these residues might be highly solvent-exposed or non-critical surface residues, where mutating the native amino acid to almost any other amino acid does not destabilize the domain. These include residues such as isoleucine (5), glutamic acid (11), arginine (22), alanine (37), and tyrosine (39), among others.
Positions 50, 51, 55, 61: These columns have vertical dark purple columns representing regions where almost any mutation is detrimental to the protein’s score. Because Domain 4 of DnaA is the specific DNA-binding domain (with the helix-turn-helix motif that recognizes the DnaA box), these negative zones typically correspond to buried hydrophobic core residues, which are essential for structural folding, or residues involved in direct base-contacting or phosphate-backbone-stabilizing of the DNA. They include residues of proline (50), lysine (51), alanine (55) and threonine (61), which are residues present along the active site pocket, contributing indirectly to the binding process.
Latent Space Analysis
- The 3D t-SNE plot successfully compresses the high-dimensional protein dataset into three latent components (TSNE1, TSNE2, TSNE3). The color gradient strongly correlates with TSNE3, representing a dominant structural or functional feature.
- Yes, neighborhoods approximate similar proteins. t-SNE preserves local similarities, meaning tightly packed points share sequence or functional traits. Notably, the dark blue cluster on the bottom right represents a distinct, isolated protein subfamily.
- By placing my protein by finding its TNSE-1,2,3 coordinates, I analyzed it. If fell near a cluster, thus indicating it shares high biochemical or evolutionary similarity with the immediate neighbors.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?

- ESMFold matches the experimental structure because its transformer model accurately captures the stable, evolutionary conserved alpha-helical topology of DnaA.
- Point Mutations: It is resilient on the surface, fragile in the core. Surface mutations are easily tolerated. However, single mutations in the hydrophobic core or the DNA-binding helix-turn-helix motif disrupt critical stabilizing interactions, causing local misfolding.
- Large Segments: Deleting or replacing large segments destroys the cooperative folding network. ESMFold shows a collapsed structure indicating disordered loops instead of the original fold.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. Input this sequence into ESMFold and compare the predicted structure to your original.
New Sequence: MTAELIAEVTAEFFGTTVEELKGSKREPKLALARRIAMYLCVELTDLSQEEIGKAFNISPETVTKAVEWIRKKMEESEEVREAVEELKERILARAG
- High Confidence (Yellow spots): Driven by strict geometric constraints (e.g., highly packed hydrophobic core positions, rigid turns at ~23 and ~53).
- High Entropy (Purple regions): Surface-exposed regions showing high tolerance for sequence variation without disrupting the backbone fold.
- Alignment & Sequence Comparison
Original: ISAATIMAATAEYFDTTVEELRGPGKTRALAQSRQIAMYLCRELTDLSLPKIGQAFGRDHTTVMYAQRKILSEMAERREVFDHVKELTTRIRQRSK :..: :..::.******* : . ::*** ..:. . .** .* .:: .::..** *::. : .. Predicted: MTAELIAEVTAEFFGTTVEELKGSKREPKLALARRIAMYLCVELTDLSQEEIGKAFNISPETVTKAVEWIRKKMEESEEVREAVEELKERILARAG
- The central structural core (TTVEEL, AMYLC) and the essential flexible Glycine turn (GPGK to GSKR) are strictly preserved.
- N-terminus: ISAATIM changed to MTAELIA optimizes surface charge/stability.
- C-terminus: Redesigned to an amphipathic alpha-helix signature (AVEELKERILARAG) to stabilize the terminal helix of Domain 4.
ESMFold VS Original Structure Comparison
- Topology Preservation: The generated structure perfectly maintained the native all-alpha helical bundle topology. ProteinMPNN successfully navigated alternative sequence space while obeying the exact spatial constraints of the Mtb DnaA backbone.
- Key Features: The crucial DNA-binding Helix-Turn-Helix (HTH) motif is resolved. Changing the flexible loop from GPGK to GSKR successfully preserved the tight hinge orientation required for major groove DNA docking.
- Discrepancies: Minor angstrom-level variations are limited strictly to the dynamic, surface-exposed N- and C-termini due to redesigned local electrostatic networks.
Thus, the inverse folding cycle is a success and ESMFold independently validated that the newly designed sequence folds back into the native conformation.
Group Brainstorm on Bacteriophage Engineering
Form a group of ~3–4 students and read through the Phage Reading material listed under “Reading & Resources”. Review the Bacteriophage Final Project Goals for engineering the L Protein, which includes - Increased stability (easiest), Higher titers (medium), Higher toxicity of lysis protein (hard). Now, choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”). Write a 1-page proposal (bullet points or short paragraphs) describing: a. Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”). b. Why do you think those tools might help solve your chosen sub-problem? c. Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”). d. Include a schematic of your pipeline. Also, include your group’s short plan for engineering a bacteriophage
Project Proposal: Engineering the L Protein for Enhanced Lysis
Main Goals
- To improve the robustness of the L protein, ensuring it remains functional under varied environmental conditions during manufacturing and therapy.
- To enhance the ability of the L protein to disrupt the bacterial cell wall, leading to more “robust killing” of the target pathogen.
Tools and Approaches
- I propose using PLMs (such as ESM-2) to perform in silico mutagenesis. These models can predict which amino acid substitutions are likely to increase the structural stability of the L protein based on evolutionary patterns.
- Alphafold Multimer can be utilized to model the L protein’s interaction with host components, such as E. coli DnaJ. I will use it to check the structural integrity of the mutated variants and to identify specific residues that can be altered to disrupt inhibitory interactions with host chaperones.
- Once stable candidates are identified, Molecular Dynamics simulations will be used to verify their thermal stability over time, ensuring they do not denature during storage.
Why These Tools Help
- Streamlining Development: Identifying the right phage components is often described as finding a million keys for a million locks. AI and computational tools accelerate this by filtering out ineffective mutations before any wet-lab work begins.
- Optimizing Interactions: By using AlphaFold-Multimer, we can move beyond trial-and-error to design phages that specifically target bacterial components more effectively. Understanding host-phage interactions at a molecular level is crucial for engineering optimized phages that outperform naturally occurring ones.
Potential Pitfalls
- Limited Host-Interaction Data: A significant hurdle is that we may lack enough training data on phage–bacteria interactions for specific lysis proteins like L, which could limit the accuracy of the structural models.
- Premature Lysis: Increasing toxicity too much could cause the host cell to burst before the phage has finished replicating, which might inadvertently lead to lower titers and a less effective phage soup.
Schematic of Pipeline
- Sequence Input: Wild-type L protein sequence is processed.
- Mutant Generation: PLMs to generate a library of candidates with predicted high stability.
- Interaction Modeling: AlphaFold-Multimer predicts the binding affinity of candidates to E. coli DnaJ.
- Stability Validation: MD simulations confirm thermal resilience.
- Final Output: Optimized sequence ready for synthetic genome assembly.
Short Plan for Engineering a Bacteriophage
To engineer a complete bacteriophage, we could focus on creating a synthetic variant of a P. aeruginosa phage designed for hospital-acquired infections.
- Payload Integration: Using CRISPR-Cas3 technology, we can engineer the phage to deliver a payload that irreparably shreds the bacterial DNA, ensuring high-efficiency killing.
- Genome Assembly: The modified genome can be built using yeast-based assembly or in-vitro assembly of PCR-amplified fragments.
- Rebooting: To avoid contamination from bacterial toxins (endotoxins) during the initial production, the synthetic genome can be “rebooted” using a cell-free transcription-translation (TXTL) system.
- Host Range Expansion: We could use BRED (Bacteriophage Recombineering of Electroporated DNA) to modify tail fiber genes, allowing the engineered phage to target a broader range of clinical strains.
Thank You!