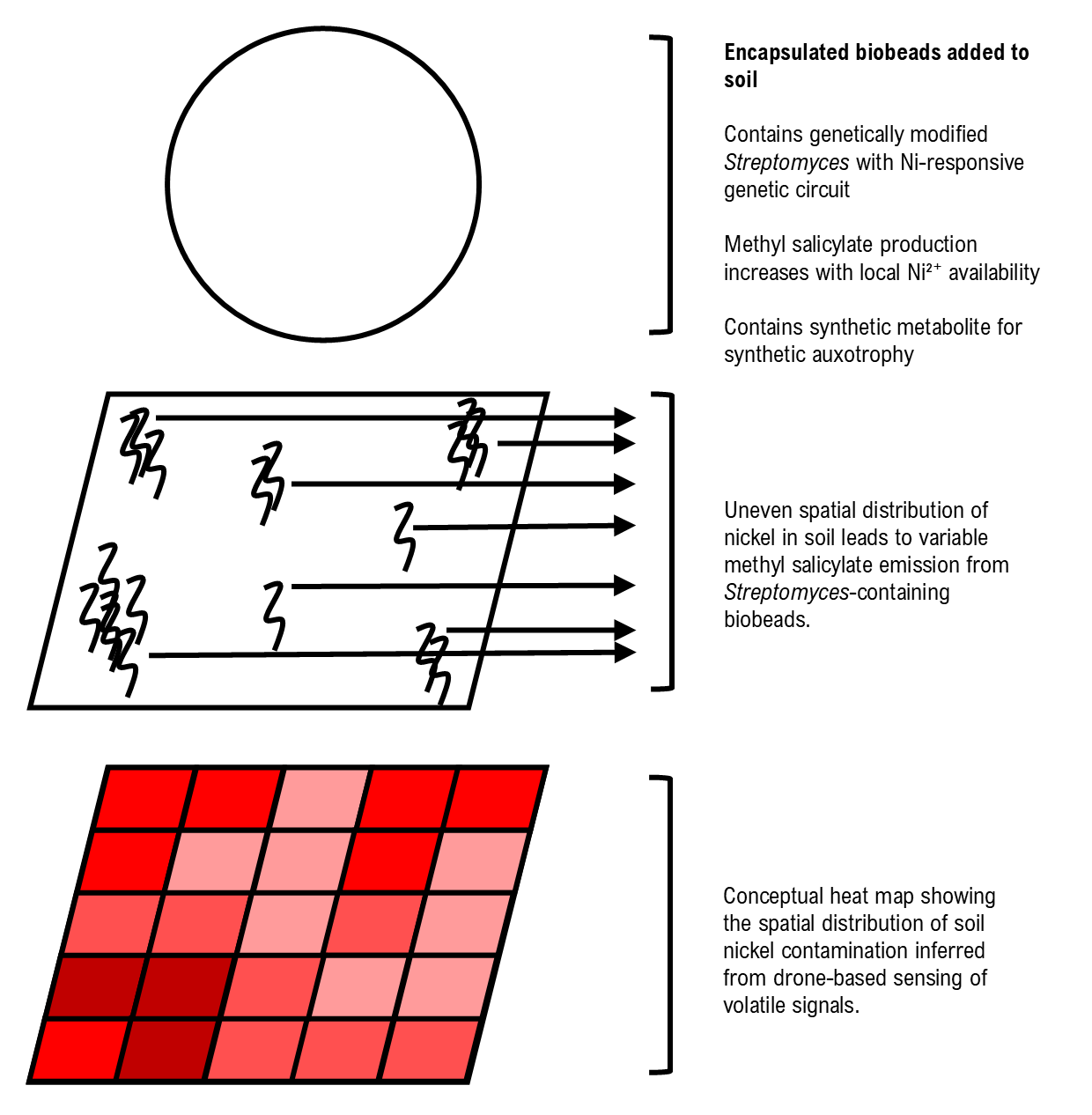
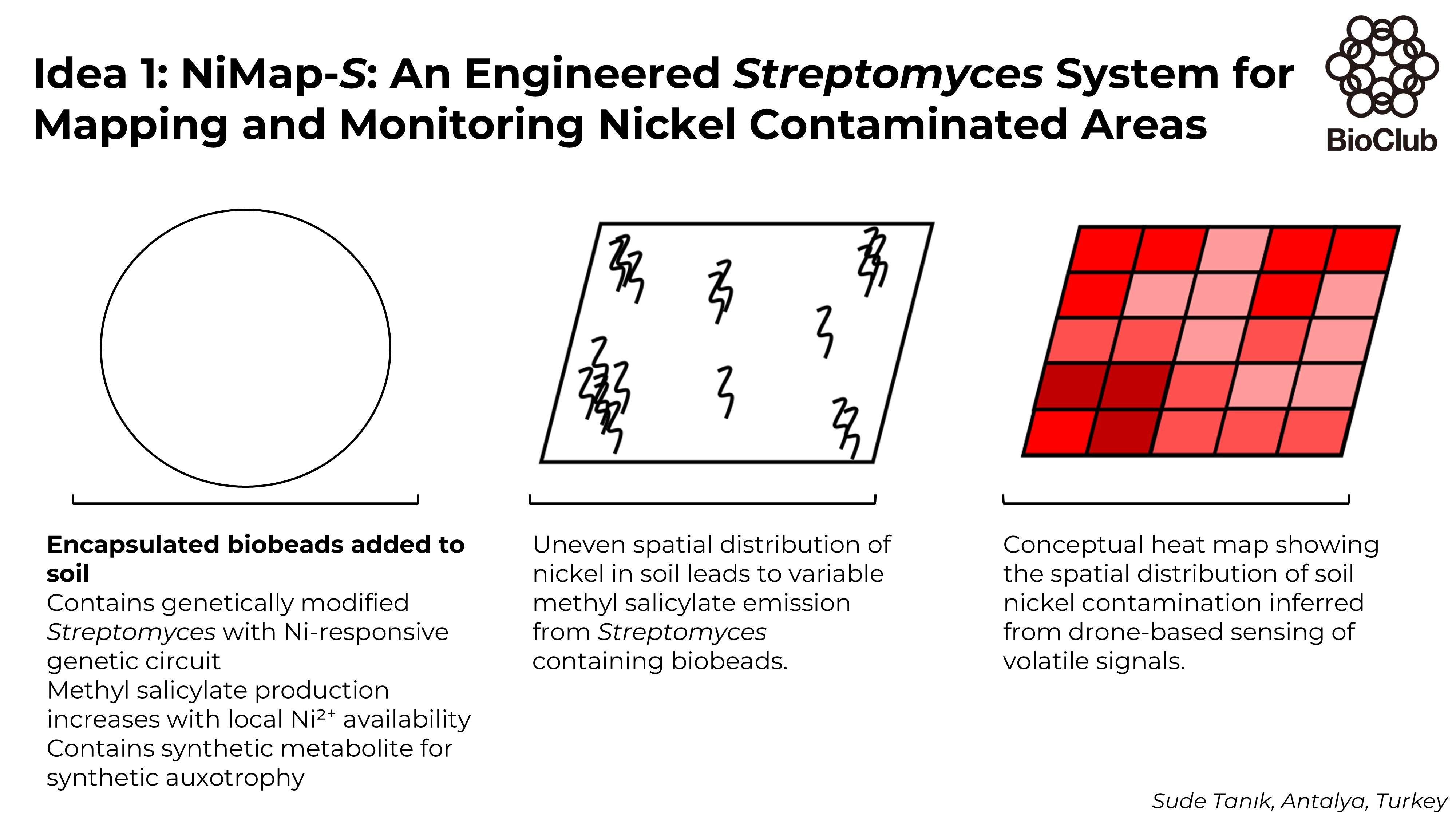
NiMap-S: An Engineered Streptomyces System for Mapping and Monitoring Nickel Contaminated Areas In this project I propose NiMap-S, a synthetic microbial biosensor system designed to identify local patterns of nickel contamination in soil. The system is based on a genetically modified, soil native Streptomyces strain that functions as a biological reporter. Streptomyces is selected because it is already well adapted to soil environments and because its rich secondary metabolism allows sensing and reporting functions to be integrated without introducing a completely foreign organism into the soil ecosystem. The engineered bacteria are intended to be physically contained within biodegradable biobeads, allowing controlled interaction with the surrounding soil while limiting dispersal.
In NiMap-S, bioavailable nickel ions (Ni2+) are detected through a nickel-responsive genetic circuit in which metal sensing is coupled to regulated expression of a methyl salicylate biosynthesis pathway. As local nickel availability increases, the circuit drives a corresponding increase in the production and release of methyl salicylate (C8H8O3), a volatile compound commonly involved in plant signalling. This chemical output is chosen intentionally, as it enables nondestructive sensing and creates a functional link between microbial detection and existing plant-soil communication processes.
One of the main limitations of phytoremediation is limited information on how contaminants are distributed across an area. Soil contamination is spatially heterogeneous, yet existing analysis methods rely on destructive sampling, slow laboratory workflows, limited information, and high costs when applied at field scale. As a result, remediation strategies are often applied uniformly, even when contamination varies sharply across short distances.
NiMap-S is therefore conceived as a premapping system that operates before phytoremediation plants, such as sunflowers, are introduced. The system generates a chemical heat map of bioavailable nickel hotspots across the field. These signals can be detected using low-altitude drone platforms equipped with gas-sensing instruments that are already used in environmental monitoring. Importantly, the choice of methyl salicylate avoids reliance on speculative or custom built detection hardware, grounding the system by utilizing approaches that are already in practical use.
NiMap-S makes it possible to adjust remediation inputs with greater precision, for example by applying chelators only where and in the amounts needed, selecting appropriate plant varieties for specific locations, reducing unnecessary inputs, and overall remediation costs. Conceptual overview of the NiMap-S system
Part 1: Gel Art I searched for the lambda DNA sequence registered in NCBI using its RefSeq accession number and clicked import.
Then I selected the enzymes listed on the HTGAA site and added them to the tool.
After clicking Run digest, a lot of restrictions appeared and the sequences were too short. I realized I was not supposed to select all enzymes at once because the system used all at the same time, so I ran each digestion separately.
Part 1: Python Script for Opentrons Artwork First, I created my design on the website and drew a bacteriophage
Then, I copied the coordinates of my design
I also published my design to the gallery.
After that, I went to colab
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Most red meat contains about 20% protein, so 500 g of meat contains roughly 100 g of protein. Since 1 Da=1.66×10-24 g, an amino acid with a mass of about 100 Da has a mass of 1.66×10-22 g. Dividing 100 g by 1.66×10-22 g gives approximately 6×1023 amino acids.
SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM Colab link: https://colab.research.google.com/drive/1_l-gF1EFDOHIyetFlJT4wAGmYJr-raXB
Sequence (non-mutated): MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Sequence (A4V): MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
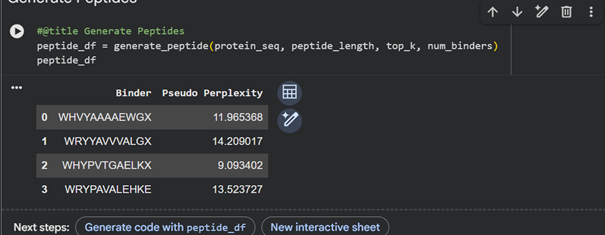
Peptide Sequence Perplexity Peptide 1 WHHVYAAAAEWG 11.965 Peptide 2 WRYYAVVVALGA 14.209 Peptide 3 WHYPVTGAELKA 9.093 Peptide 4 WRPYAVALEHKE 13.523 Known peptide FLYRWLPSRRGG - Part 2: Evaluate Binders with AlphaFold3
Subsections of Homework
Week 1 HW: Principles and Practices
NiMap-S: An Engineered Streptomyces System for Mapping and Monitoring Nickel Contaminated Areas
In this project I propose NiMap-S, a synthetic microbial biosensor system designed to identify local patterns of nickel contamination in soil. The system is based on a genetically modified, soil native Streptomyces strain that functions as a biological reporter. Streptomyces is selected because it is already well adapted to soil environments and because its rich secondary metabolism allows sensing and reporting functions to be integrated without introducing a completely foreign organism into the soil ecosystem. The engineered bacteria are intended to be physically contained within biodegradable biobeads, allowing controlled interaction with the surrounding soil while limiting dispersal.
In NiMap-S, bioavailable nickel ions (Ni2+) are detected through a nickel-responsive genetic circuit in which metal sensing is coupled to regulated expression of a methyl salicylate biosynthesis pathway. As local nickel availability increases, the circuit drives a corresponding increase in the production and release of methyl salicylate (C8H8O3), a volatile compound commonly involved in plant signalling. This chemical output is chosen intentionally, as it enables nondestructive sensing and creates a functional link between microbial detection and existing plant-soil communication processes.
One of the main limitations of phytoremediation is limited information on how contaminants are distributed across an area. Soil contamination is spatially heterogeneous, yet existing analysis methods rely on destructive sampling, slow laboratory workflows, limited information, and high costs when applied at field scale. As a result, remediation strategies are often applied uniformly, even when contamination varies sharply across short distances.
NiMap-S is therefore conceived as a premapping system that operates before phytoremediation plants, such as sunflowers, are introduced. The system generates a chemical heat map of bioavailable nickel hotspots across the field. These signals can be detected using low-altitude drone platforms equipped with gas-sensing instruments that are already used in environmental monitoring. Importantly, the choice of methyl salicylate avoids reliance on speculative or custom built detection hardware, grounding the system by utilizing approaches that are already in practical use.
NiMap-S makes it possible to adjust remediation inputs with greater precision, for example by applying chelators only where and in the amounts needed, selecting appropriate plant varieties for specific locations, reducing unnecessary inputs, and overall remediation costs.
Conceptual overview of the NiMap-S system
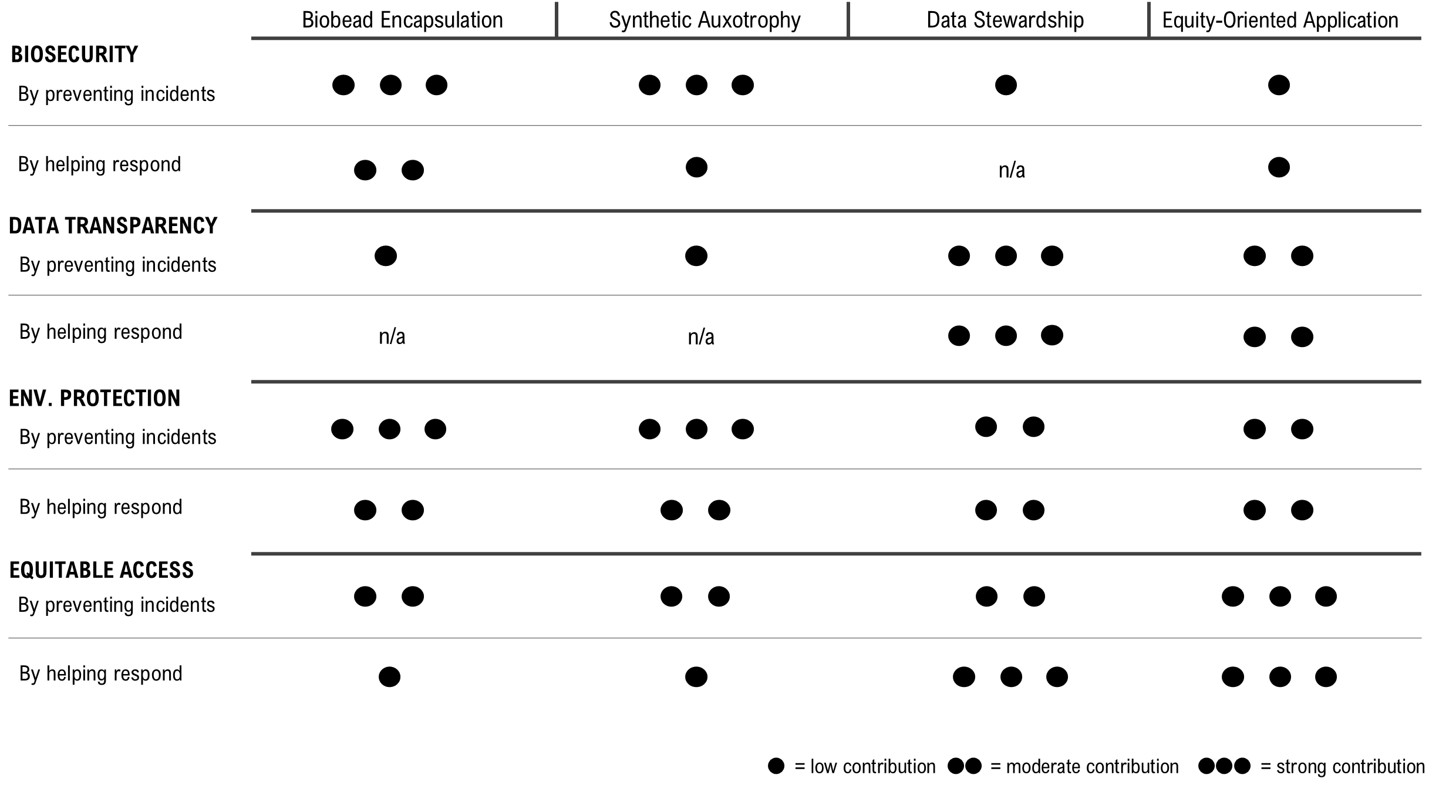
Governance Goals
Goal 1: Providing Biosecurity
The first policy goal is to prevent biological and ecological harm resulting from the environmental use of engineered microorganisms. Since NiMap-S is intended to operate outside controlled laboratory settings, biosafety cannot depend solely on correct use or post application control. Instead, safety needs to be built directly into the biological design of the organism and the material design of the system.
Sub-goal 1.1: Limiting persistence
Engineered microorganisms should not remain active beyond their intended use. NiMap-S is therefore designed with the assumption that environmental presence should be temporary, so that microbial activity naturally ends once sensing objectives have been achieved.
Sub-goal 1.2: Preventing uncontrolled spread and evolution
The system should not spread beyond its area of application or adapt in ways that increase ecological risk. This subgoal focuses on containment strategies that restrict dispersal and reduce opportunities for long-term evolution in open environments.
Sub-goal 1.3: Ensuring reversibility
Environmental application should remain reversible. If unexpected effects occur, the system should allow for intervention.
Goal 2: Data Transparency and Responsible Governance
The second policy goal addresses how environmental data generated by NiMap-S is managed and used. Because such data can shape land-use application choices and remediation strategies, data governance is not only a technical issue but also an ethical one.
Sub-goal 2.1: Decision support with transparency
Data produced by NiMap-S should support informed actions, not surveillance or enforcement. Measurement goals, uncertainty, and technical limits should be communicated clearly to public institutions and local stakeholders.
Sub-goal 2.2: Shared access and control
Environmental data should remain accessible to public agencies, municipalities, and communities. Governance structures should avoid exclusive control by large corporate actors and instead support broader access to environmental knowledge.
Goal 3: Environmental Protection and Ecosystem Integrity
The third policy goal is to ensure that NiMap-S contributes directly to environmental recovery while respecting ecosystem integrity. Here, sensing is treated not as an end in itself, but as a tool to support remediation efforts that reduce contamination without creating additional ecological disturbance. Here, environmental protection is understood not just as avoiding harm, but as actively enabling soil systems to recover.
Sub-goal 3.1: Supporting effective remediation through phytoremediation
NiMap-S should strengthen phytoremediation by improving how contamination is identified and spatially mapped. By providing information on bioavailable nickel at appropriate scales, the system allows remediation efforts to be applied more efficiently, increasing cleanup effectiveness while reducing unnecessary use of water, chemicals, and land.
Sub-goal 3.2: Protecting ecosystem integrity during intervention
Remediation should reduce pollution without introducing new ecological risks. This includes limiting disturbance to surrounding soil, plants, and microbial communities, and ensuring that technological elements of the system remain compatible with ecosystem protection. In this sense, environmental protection overlaps with biosecurity, as containment and reversibility also help safeguard broader ecosystem functioning.
Goal 4: Equitable Access, Shared Benefits, and Collaborative Use
The fourth policy goal is to ensure that NiMap-S benefits not only the environment, but also the people whose land and livelihoods are affected by soil contamination. In agricultural contexts, pollution can reduce productivity in ways that are not always obvious, and these losses are often attributed to other causes. By improving access to environmental information, the system aims to support environmental recovery and agricultural viability together.
Sub-goal 4.1: Linking environmental recovery with agricultural productivity
NiMap-S should help farmers and local land managers recognise when contamination contributes to yield loss and use this information to guide remediation, crop choice, and land use. By enabling earlier and more targeted phytoremediation, the system supports environmental improvement while also helping maintain or restore agricultural productivity and local economic resilience.
Sub-goal 4.2: Supporting coordinated action in practice
NiMap-S is intended to operate within real world soil management and remediation practices involving researchers, farmers, local authorities, and oversight bodies. Rather than functioning as a stand alone technology, the system supports collaboration at the field level, where scientific knowledge, local experience, and regulatory oversight intersect. This subgoal emphasises shared use and responsibility.
Actions
Action 1: Physical Encapsulation via Biobeads for Environmental Application
Purpose
This action proposes requiring biodegradable biobead encapsulation as a condition for environmental use, limiting microbial dispersal while still allowing interaction with the surrounding soil.
Design
Biosafety regulators would define biobead encapsulation as part of environmental approval processes. Researchers and organisations applying NiMap-S would integrate bead-compatible designs early on, while materials scientists establish standards for bead composition, degradation behaviour, and diffusion properties.
Assumptions
This action assumes that encapsulation meaningfully limits dispersal under field conditions such as rainfall and soil movement, and that bead degradation can be reasonably predicted across environments. It also assumes that physical confinement does not substantially reduce sensing performance.
Risks of Failure and Success
Failure could occur if beads degrade faster than expected or if physical containment leads to reduced attention to genetic safeguards. At the same time, widespread success could normalise environmental microbial applications before broader governance frameworks fully mature.
Action 2: Synthetic Auxotrophy
Purpose
This action introduces synthetic auxotrophy as a licensing requirement, ensuring that engineered organisms cannot persist without an added metabolite.
Design
Regulatory bodies would define acceptable synthetic metabolites and testing standards, while academic laboratories and companies engineer dependencies into essential metabolic pathways. Funding agencies could reinforce this approach by prioritising projects that demonstrate stable auxotrophy.
Assumptions
This action assumes that evolutionary escape remains rare and that synthetic metabolites are not present in natural environments.
Risks of Failure and Success
Failure could occur through genetic bypass or unintended environmental availability of the metabolite. Successful implementation may also reduce incentives to develop additional containment strategies or disadvantage smaller research groups.
Action 3: Public Interest Data Stewardship
Purpose
Environmental biosensing data is often controlled by the organisation developing the system, which limits transparency, public oversight, broader access to environmental information. This action reframes NiMap-S data as a shared public tool that supports learning, wide implementation, transparency, and accountability.
Design
Environmental approval or public funding would be linked to compliance with open data standards. Researchers and organisations would document uncertainty, calibration assumptions, and known limitations of the measurements. Public agencies or municipalities would act as data stewards, ensuring that data remains accessible to other researchers, local authorities, and communities, and that it is presented in formats suitable for nonspecialist use as well as expert analysis.
Assumptions
This action assumes that wider access to environmental data helps reduce power imbalances, supports more informed decisions, and enables environmental protection beyond individual application sites. It also assumes that public institutions have, or can develop, the capacity to manage, contextualise, and communicate complex biological data responsibly.
Risks of Failure and Success
Failure could result in poorly documented or misinterpreted data, undermining trust or leading to inappropriate conclusions. At the same time, success may lead to data being overinterpreted, selectively reused, or applied outside its original context. These risks make clear metadata, uncertainty reporting, and active stewardship essential.
Action 4: Equity-Oriented Application and Incentives
Purpose
Environmental sensing technologies are often used where financial return is highest, not where environmental need is most urgent. In agricultural contexts, soil contamination can quietly reduce yields without farmers clearly identifying pollution as the cause. This action focuses on prioritising deployment in polluted or underserved areas, where both ecological damage and hidden productivity losses are likely, and where access to monitoring tools is limited.
Design
Public funding agencies and local authorities would steer application through targeted incentives. Municipalities and land managers would help identify relevant sites, while developers adapt the system to work under low-resource agricultural conditions. Direct involvement of farmers ensures that the system supports common implementation.
Assumptions
This action assumes that delayed intervention is often driven by lack of information, and that farmers and local actors are more likely to engage when monitoring is clearly connected to soil health and productivity.
Risks of Failure and Success
Incentives might not be strong enough to compete with profit goals, and experiments could be placed in sensitive areas without good supervision. However, if the system works properly, it can detect contamination earlier and help protect both the environment and farming.
Scoring of Each Governance Action Against Rubric of Policy Goals
Prioritisation of Governance Actions
Based on my scoring matrix, I prioritise some governance actions as a package. NiMap-S only makes sense if biosafety and data practices are considered together. In this framing, Actions 1 and 2 define how the system can be used safely in the environment, while Action 3 shapes how the system and its data are made transparent and accountable beyond the laboratory.
Actions 1 and 2 should be implemented together from the outset. Biobead encapsulation limits physical spread, while synthetic auxotrophy reduces the risk of longterm persistence. Together, they lower the chance that a single failure leads to lasting ecological harm. The tradeoff is additional technical work and validation, which may slow early deployment but is necessary in open soil environments where engineered microorganisms raise legitimate concern. The key assumption here is that these biosafety features continue to work under real environmental conditions, including stable biobead encapsulation and effective auxotrophy, without rapid breakdown or unexpected behaviour in soil.
Action 3 focuses less on biological design and more on how information produced by NiMap-S is handled. Being clear about how the system works, its limits, its outputs, and remaining uncertainties helps justify the environmental use of engineered bacteria, while open and shared access to information on nickel contamination and NiMap-S outputs supports wider use and informed decision making by regulators, implementers, local authorities, and the public. This level of transparency can also lower barriers to environmental approval. The tradeoff is coordination effort, as preparing data and documentation for public use requires time and institutional capacity. This action assumes that the use of NiMap-S is approved by relevant public institutions and that transparency helps maintain this approval. Transparency also requires careful data stewardship to avoid misuse or unintended consequences.
These recommendations can be directed to national environmental and agricultural agencies, together with municipal authorities in contaminated regions. These actors are best positioned to align biosafety rules, shared data practices, and incentive structures, and to turn NiMap-S from a laboratory-based conceptual system into an applicable environmental tool.
Thought Process
I initially considered using a luminescence-based reporter, since optical signals are common in microbial biosensing. However, I moved away from this option because soil depth and opacity make luminescence difficult to recover without disturbing the soil. To overcome this, I briefly explored a retrievable bead system, where luminescent beads could be pulled out of the soil for external reading. While this addressed signal visibility, it raised practical concerns such as mechanical fragility, risk of damage during retrieval, and the need for repeated manual inspection. I therefore abandoned luminescence as a reporting strategy, while keeping biobeads as a containment and application element in the final system. I also considered electrochemical reporting, but did not pursue it further because I could not clearly conceptualise a reliable setup for heterogeneous soil environments within the scope of this project. Finally, I decided to use a volatile chemical signal. I first considered geosmin, a naturally produced Streptomyces compound that could be placed under nickel-dependent regulation. However, because many native soil bacteria already produce geosmin, I was concerned about background signal and ambiguity. I therefore selected methyl salicylate, which provides a stronger and more distinguishable signal and can be more clearly linked to nickel-induced stress. An additional reason for this choice was the possibility that methyl salicylate release could support systemic acquired resistance (SAR) in remediation plants during phytoremediation.
AI Statement
I used Chat GPT 5.2 to check the technical feasibility of my ideas and to summarize some reading documents for the assignment. I used it to ask specific questions about the practicality of biobeads in soil, drone sensor sensitivity (like ppb-level detection of methyl salicylate), and wind interference. It was also helpful for reviewing my governance and actions framework, checking if my goals were consistent and if the roles for different actors were realistic. While using AI, I asked for reasoning or references when needed to check whether the answers made sense.
AI was used for grammar corrections, technical terminology, especially in the governance goals and actions sections, and sometimes as a writing support tool based on outlines and sentence fragments, which I prepared derived from my ideas.
DNA Read, Write, and Edit
Faculty Questions
Homework Questions from Professor Jacobson:
Q1: Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Human genome length is 3.2 Gbp (Slide 10). Since 1 Gbp=109 bp, the human genome is 3.2×109 bp. Because DNA polymerase can make mistakes approximately every 106 bp (Slide 8), this makes the human genome prone to thousands of potential mistakes (3×109/106≃3000) during replication. In order to deal with this discrepancy, mismatch repair systems (Slide 14) detect and fix replication errors that escape polymerase proofreading.
Q2: How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The number of average base pairs in a human protein is around 1036 bp (Slide 6), which corresponds to approximately 1036/3≃345 amino acids. Depending on the amino acid, each one can be coded by more than one codon (for example, leucine and serine have 6 codons, alanine and glycine have 4 codons, while methionine and tryptophan have only 1 codon). Because of this, depending on the amino acid composition of the protein, there can be many different DNA coding combinations for the same protein sequence. In practice, not all coding sequences work because different DNA sequences can have very different physical properties. Factors such as GC content (Slide 39) can interfere with DNA synthesis and proper expression by affecting secondary structure and minimum free energy. For this reason, coding sequences often need to be optimized for the host organism, since each organism has its own requirements in this respect.
Homework Questions from Dr. LeProust:
Q1: What’s the most commonly used method for oligo synthesis currently?
Modern oligonucleotide synthesis is most commonly based on phosphoramidite method (Slide 11; Hoose et al. 2023).
Q2: Why is it difficult to make oligos longer than 200nt via direct synthesis?
Since the elongation efficiency is not 100%, errors accumulate with each nucleotide addition cycle. Consequently, beyond 200 nt, the system becomes dominated by incorrectly synthesized DNA fragments. Moreover, as the chain length increases, the risks of depurination, strand breakage, and stabilized hairpin formation significantly reduce the final yield of pure DNA (Hoose et al., 2023).
Q3: Why can’t you make a 2000bp gene via direct oligo synthesis?
Even oligonucleotide fragments longer than 200 nt accumulate a high proportion of incorrectly synthesized DNA for the reasons discussed in Question 2. This means that synthesizing a 2000 bp sequence directly is even more difficult, as errors accumulate at a much higher rate and strand breakage and hairpin formation become much more likely. To build a 2000 bp gene, shorter oligonucleotide fragments must be assembled using methods such as Gibson assembly (Hoose et al., 2023)
Homework Question from George Church:
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Essential amino acids for all animals are: arginine, histidine, isoleucine, leucine, methionine, phenylalanine, threonine, tryptophan, valine, lysine. According to https://jurassicpark.fandom.com/wiki/Lysine_contingency, “lysine contingency” is a method of inserting a faulty enzyme gene in dinosaurs so that lysine metabolism is disrupted and dinosaurs are unable to biosynthesize the lysine they need. This was intended as a containment strategy, preventing them from surviving outside the island, because they are supposedly dependent on lysine rich plants found there. However, this logic is flawed, because in reality, like all animals, dinosaurs would not have been able to synthesize lysine in the first place. Instead, they would have obtained lysine from dietary sources. Thus, lysine dependency is a natural condition in animals, including humans, rather than an effective synthetic containment mechanism.
References
Hoose, A., et al. (2023). DNA synthesis technologies to close the gene writing gap. Nature Reviews Chemistry, 7, 144-161.
AI was used for grammatical corrections and paraphrasing ideas.
Week 2 HW: DNA Read, Write & Edit
Part 1: Gel Art
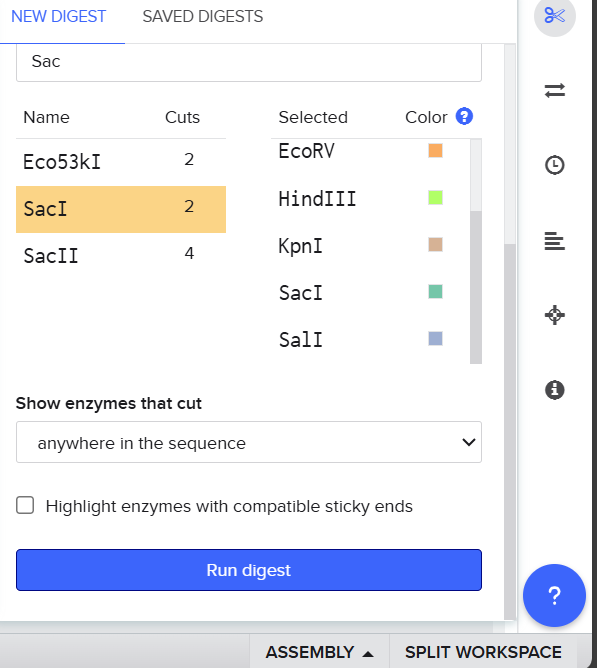
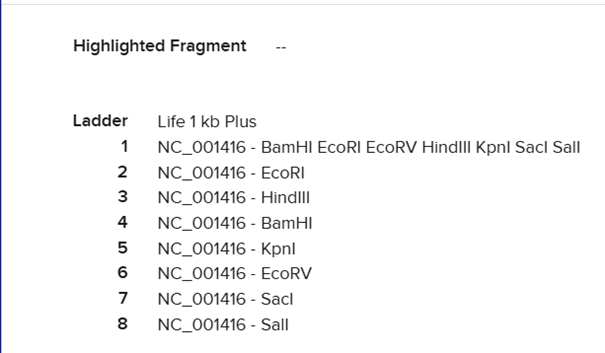
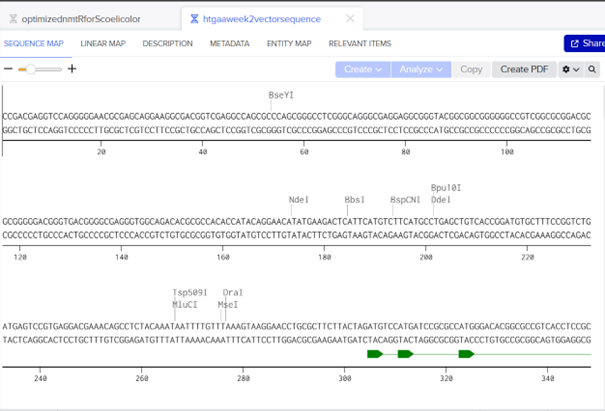
I searched for the lambda DNA sequence registered in NCBI using its RefSeq accession number and clicked import.
Then I selected the enzymes listed on the HTGAA site and added them to the tool.
After clicking Run digest, a lot of restrictions appeared and the sequences were too short. I realized I was not supposed to select all enzymes at once because the system used all at the same time, so I ran each digestion separately.
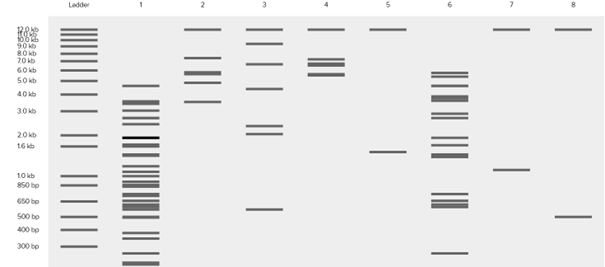
For the trident design, I planned the order as: EcoRI on the left, then KpnI, then EcoRV, followed again by KpnI and EcoRI. After that I moved to Ronan Donovan’s website to construct the gel image.
Based on these fragments, I want to create Poseidon’s trident. In Ronan Donovan’s website, I first added a ladder. According to the ladder, the gel ranges from 100 bp (bottom) to 15 kb (top). The pattern looked like this.
It does not look exactly like a trident at first, but considering that this is a gel image and that we can crop the upper part, we can say that it resembles a trident 🙂
Part 3: DNA Design Challenge
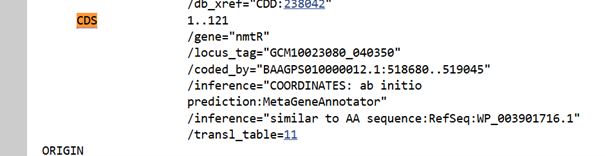
I selected NmtR (Nickel-Metal Transporter Regulator) from Streptomyces:
NmtR is a Ni/Co sensor and a transcriptional regulator (Kim et al., 2015),
In the absence of metal, it binds to DNA and represses transcription,
When Ni binds, it dissociates from DNA, allowing target genes to be expressed, with expression levels changing depending on Ni concentration,
I chose it because it is compatible with a Streptomyces chassis and minor adjustments are enough for optimization since my target organism is also a Streptomyces,
All these features make this protein suitable for the vector design I plan to build for Streptomyces coelicolor, and these selections are directly connected to the NiMap-S tool I designed in Week 1 HW
I performed my search in the NCBI database and decided to use the sequence from Streptomyces pseudoechinosporeus:
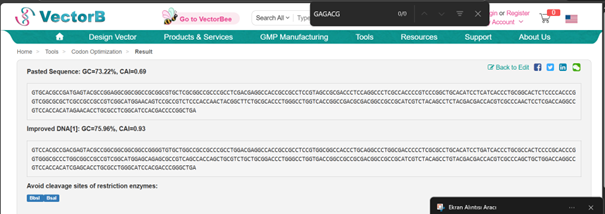
For optimization, I performed codon optimization for Streptomyces coelicolor A3(2), which is the model organism for the Streptomyces genus, using en.vectorbuilder.com. I used this platform because I forgot my IDT password and the reset email did not arrive, and I was also unable to complete the process on TWIST, I manually changed the start codon because the tool didnot recognize GTG as a start codon, which is common in Streptomyces
Optimized DNA sequence:
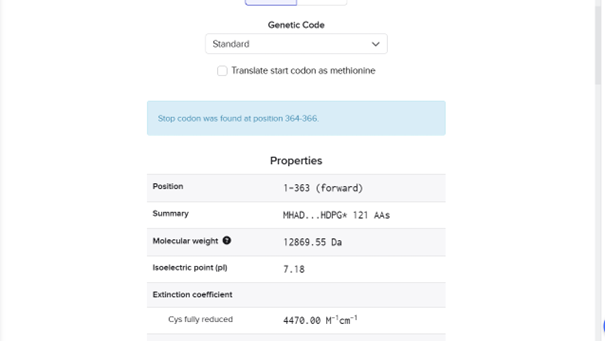
ATGCACGCCGACGAGTACGCCGGCGGCGGCGGCCGGGGTGTGCTGGCCGCCGCCCGCCTGGACGAGGCCACCGCCGCCTCCGTGGCGGCCACCCTGCAGGCCCTGGCGACCCCCTCGCGCCTGCACATCCTGATCACCCTGCGCCACTCCCCGCACCCGGTGGGCGCCCTGGCGGCCGCCGTCGGCATGGAGCAGAGCGCCGTCAGCCACCAGCTGCGTCTGCTGCGGACCCTGGGCCTGGTGACCGGCCGCCGCGACGGCCGCCGCATCGTCTACAGCCTGTACGACGACCACGTCGCCCAGCTGCTGGACCAGGCCGTCCACCACATCGAGCACCTGCGCCTGGGCATCCACGACCCGGGCTGA
DNA sequence:
ATGCACGCCGACGAGTACGCCGGCGGCGGCGGCCGGGGTGTGCTGGCCGCCGCCCGCCTGGACGAGGCCACCGCCGCCTCCGTGGCGGCCACCCTGCAGGCCCTGGCGACCCCCTCGCGCCTGCACATCCTGATCACCCTGCGCCACTCCCCGCACCCGGTGGGCGCCCTGGCGGCCGCCGTCGGCATGGAGCAGAGCGCCGTCAGCCACCAGCTGCGTCTGCTGCGGACCCTGGGCCTGGTGACCGGCCGCCGCGACGGCCGCCGCATCGTCTACAGCCTGTACGACGACCACGTCGCCCAGCTGCTGGACCAGGCCGTCCACCACATCGAGCACCTGCGCCTGGGCATCCACGACCCGGGCTGA
AA sequence:
MHADEYAGGGGRGVLAAARLDEATAASVAATLQALATPSRLHILITLRHSPHPVGALAAAVGMEQSAVSHQLRLLRTLGLVTGRRDGRRIVYSLYDDHVAQLLDQAVHHIEHLRLGIHDPG
Part 4: Twist Vector Order
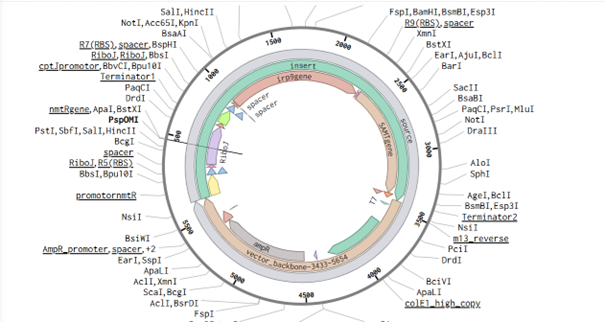
I frequently used NCBI, vectorbuilder, and the article by Bai et al. (2015) while designing this vector. The main idea is to build a vector for S. coelicolor so that it can produce increasing amounts of methyl salicylate (MeSA) according to different nickel levels in the soil when inoculated with the biobeads described in the Week 1 homework.
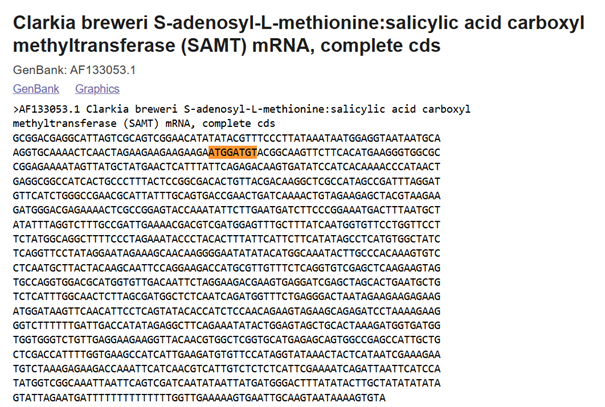
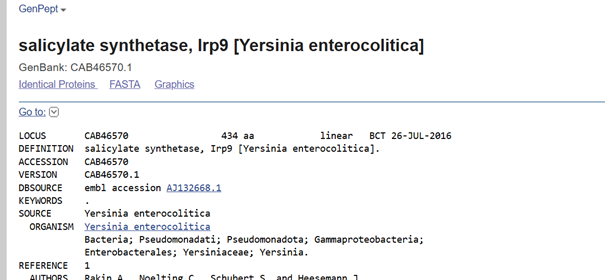
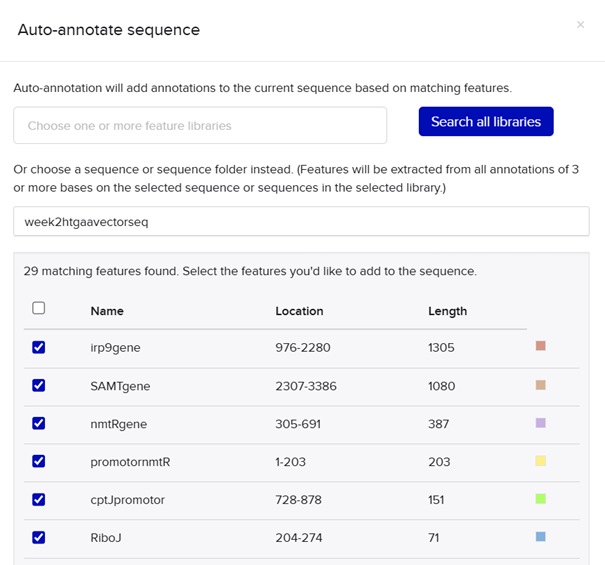
In plants, MeSA is synthesized from salicylic acid (SA) by the enzyme SAMT, which bacteria do not have. Therefore, the SAMT gene in the vector was taken from Clarkia breweri, because its SAMT is highly selective for SA (Ross et al., 1999; Ward et al., 2022). In addition, although some bacteria can produce SA (Bakker et al., 2014), Streptomyces can not, so the gene for salicylate synthase (Irp9) was obtained from Yersinia enterocolitica (Kerbarh et al., 2005). NCBI was used to retrieve the sequences, and they were optimized for Streptomyces coelicolor using vectorbuilder.
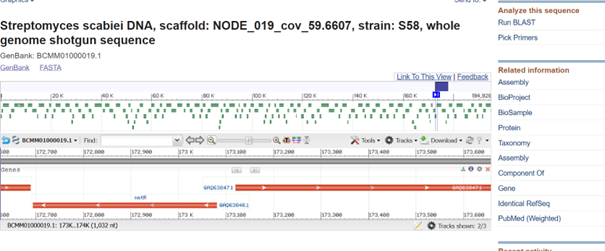
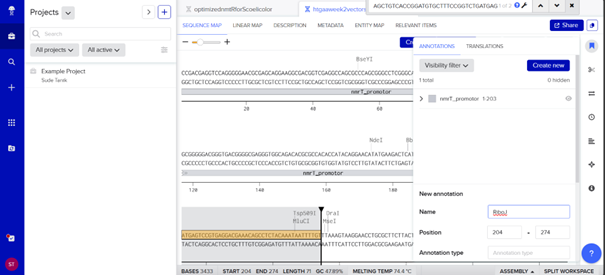
One of the key features of NiMap-S from week 1 HW is increasing MeSA production in an analog manner, so a graded transcriptional response is required. This is why NmtR was selected (Busenlehner et al., 2003). For the protein itself, I had chosen another Streptomyces in Part 3, but I was not confident about its promoter. Therefore, I searched for a Streptomyces species with a divergent operon showing NmtR regulated expression and selected Streptomyces scabiei (Osman & Cavet, 2010). To place two separate expression cassettes, I included 200 bp from the ATG start sites of both oppositely oriented genes, covering the intergenic region, to avoid missing any promoter elements.
However, I have some concerns because the intergenic region in this divergent operon is very short, so the promoter regions overlap with the complementary sequences of the opposing CDS, which could lead to unintended expression from the opposite strand. Perhaps the intergenic region alone would be sufficient, but since NmtR is a repressor that binds as a homodimer and sigma factors will also bind in this region, it seemed too short. If I continue developing this project, I will investigate this further and may look for alternative promoters that provide more reliable, nickel dependent graded expression.
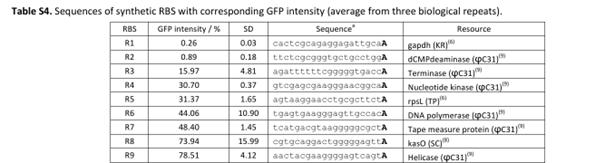
My RBS regions and RiboJ sequences were also chosen based on Bai et al. (2015). I used the R5, R7, and R9 RBS sequences from their supplementary data for nmtR, irp9, and SAMT, respectively, from weakest to strongest (Bai et al., 2015).
RBS sequences of varying strengths from Bai et al. (2015).
Ross, J. R., Nam, K. H., D’Auria, J. C., & Pichersky, E. (1999). S-Adenosyl-L methionine:salicylic acid carboxyl methyltransferase, an enzyme involved in floral scent production and plant defense. Archives of Biochemistry and Biophysics
Ward, L. C., McCue, H. V., & Carnell, A. J. (2020). Carboxyl methyltransferases: Natural functions and potential applications in industrial biotechnology. ChemCatChem
Bakker, P. A. H. M., Ran, L., & Mercado-Blanco, J. (2014). Rhizobacterial salicylate production provokes headaches! Plant and Soil
Kerbarh, O., Ciulli, A., Howard, N. I., & Abell, C. (2005). Salicylate biosynthesis: Overexpression, purification, and characterization of Irp9, a bifunctional salicylate synthase from Yersinia enterocolitica. Journal of Bacteriology
Kim, H. M., Ahn, B.-E., Lee, J.-H., & Roe, J.-H. (2015). Regulation of a nickel/cobalt efflux system and nickel homeostasis in Streptomyces coelicolor. Metallomics.
Busenlehner, L. S., Pennella, M. A., & Giedroc, D. P. (2003). The SmtB/ArsR family of metalloregulatory transcriptional repressors: structural insights into prokaryotic metal resistance. FEMS Microbiology Reviews
Bai, C., Zhang, Y., Zhao, X., Hu, Y., Xiang, S., Miao, J., Lou, C., & Zhang, L. (2015). Exploiting a precise design of universal synthetic modular regulatory elements to unlock microbial natural products in Streptomyces. Proceedings of the National Academy of Sciences (PNAS)
Week 3 HW: Lab Automation
Part 1: Python Script for Opentrons Artwork
First, I created my design on the website and drew a bacteriophage
Then, I copied the coordinates of my design
I also published my design to the gallery.
After that, I went to colab
I sent my coordinates to ChatGPT 5.2 and asked it to generate code that would run my desired design
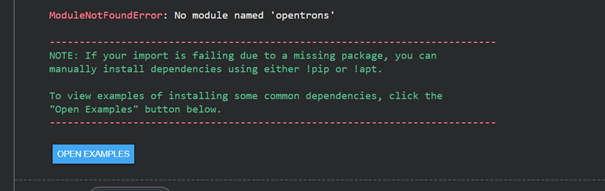
When I first tried to run it, I received an error
Then I executed the “Run this block once per runtime to set up your environment” section
Rice’s whale (Balaenoptera ricei) is an endangered whale species living in the Gulf of Mexico. Because it is very rare and difficult to observe directly researchers in this article used environmental DNA (eDNA) from seawater samples to detect its presence. Since whales could occasionally be visually observed during surveys, these encounters were used to validate the eDNA detection method by collecting seawater samples shortly after a whale surfaced. Seawater samples were first filtered using different filter pore sizes to capture DNA fragments released by the whale. DNA was then extracted from the filters and analyzed using qPCR assay targeting a mitochondrial DNA region.
The assay was validated through in silico, in vitro, and in situ testing: computational analyses were used to ensure primer specificity, laboratory experiments tested the assay against whale DNA and non-target species, and environmental seawater samples collected in the field were used to confirm that the method could successfully detect Rice’s whale eDNA. An Opentrons OT-2 was used to automate the preparation of qPCR plates by pipetting reagents, standards etc. before the analysis was performed with a qPCR device.
2. How would I utilize opetrons for the final project
To test the feasibility of NiMap-S from the week 1 HW, I would first engineer E. coli with my nickel-responsive promoter before moving into Streptomyces. The promoter from cassette 2 (from week 2 HW) would drive GFP expression instead of the methyl salicylate (MeSA) pathway enzymes. Cassette 1 from week 2 HW would remain unchanged, expressing the regulatory protein nmrT to regulate the expression of cassette 2 depending on different Ni levels.
By measuring GFP fluorescence, I could evaluate two things: whether the nickel-responsive promoter is functioning at all (GFP vs. no GFP), and whether GFP expression changes quantitatively in response to different nickel concentrations. These preliminary experiments are important before transitioning into a more complex organism and a more complex genetic circuit. This allows me to verify that the promoter and sensing module (which uses nmrT as the regulatory protein for gradual Ni response) behave as expected before adding metabolic burden from the MeSA biosynthetic pathway. This strategy helps isolate potential problems early. If something fails, by implementing this preliminary research I can determine whether the issue lies in the promoter system or in the metabolic pathway.
So, I would use Opentrons robot in this experiment to prepare different nickel concentrations in a 96-well plate. The robot would create a nickel gradient using serial dilutions, add the engineered E. coli to each well, and make sure that all volumes are consistent. I would measure GFP fluorescence to see how the promoter responds to increasing nickel levels. This would allow me to generate a curve and understand whether the promoter response is gradual based on increasing nickel levels or switch like.
If the desired GFP expression patterns are observed in E. coli, I would then replace GFP with the methyl salicylate biosynthesis enzymes and test whether volatile production behaves as expected (I do not know if opentrons can be used to detect volatile signals). If that stage is successful, the validated circuit could then be transferred into Streptomyces for further development, since Streptomyces is a more complex organism to work with compared to E. coli. This gradual progression ensures that each layer of the system is validated before moving to the next level of biological complexity.
Final Project Ideas
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Most red meat contains about 20% protein, so 500 g of meat contains roughly 100 g of protein. Since 1 Da=1.66×10-24 g, an amino acid with a mass of about 100 Da has a mass of 1.66×10-22 g. Dividing 100 g by 1.66×10-22 g gives approximately 6×1023 amino acids.
Why do humans eat beef but do not become a cow, eat fish but do not become fish? Because during digestion the proteins we eat are broken down into their amino acids. Then, using these amino acids, our bodies synthesize proteins that match our own genetic information, which makes us who we are.
Why are there only 20 natural amino acids? First of all, this shows that the evolutionary process has progressed using these 20 amino acids. Among infinitely many possibilities, these 20 amino acids with diverse chemical properties were selected. Using fewer amino acids may be advantageous for living organisms, both in terms of metabolic efficiency and reducing the error rate during protein synthesis.
Can you make other non-natural amino acids? Design some new amino acids. Yes, it is possible to create non-natural amino acids. Synthetic amino acids can be produced by modifying the side chain of an amino acid and adding functional groups with desired properties, such as increased hydrophobicity or metal binding ability.
Where did amino acids come from before enzymes that make them, and before life started? Especially from the Miller-Urey experiment, we know that amino acids can form spontaneously under suitable conditions. In the early Earth, which was a high energy environment, they could form in numbers much greater than the amino acids currently used by living organisms.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? L-amino acids form a right-handed helix, D-amino acids would be expected to form a left handed helix
Can you discover additional helices in proteins? Depending on the amino acid structure, additional helices can form
Why are most molecular helices right-handed? Most molecular helices are right-handed because proteins almost always contain L-amino acids
Why do β-sheets tend to aggregate? β-sheets tend to aggregate because their functional groups are exposed and they have a flat structure, allowing β-strands from different proteins to form intermolecular hydrogen bonds.
Part B: Protein Analysis and Visualization
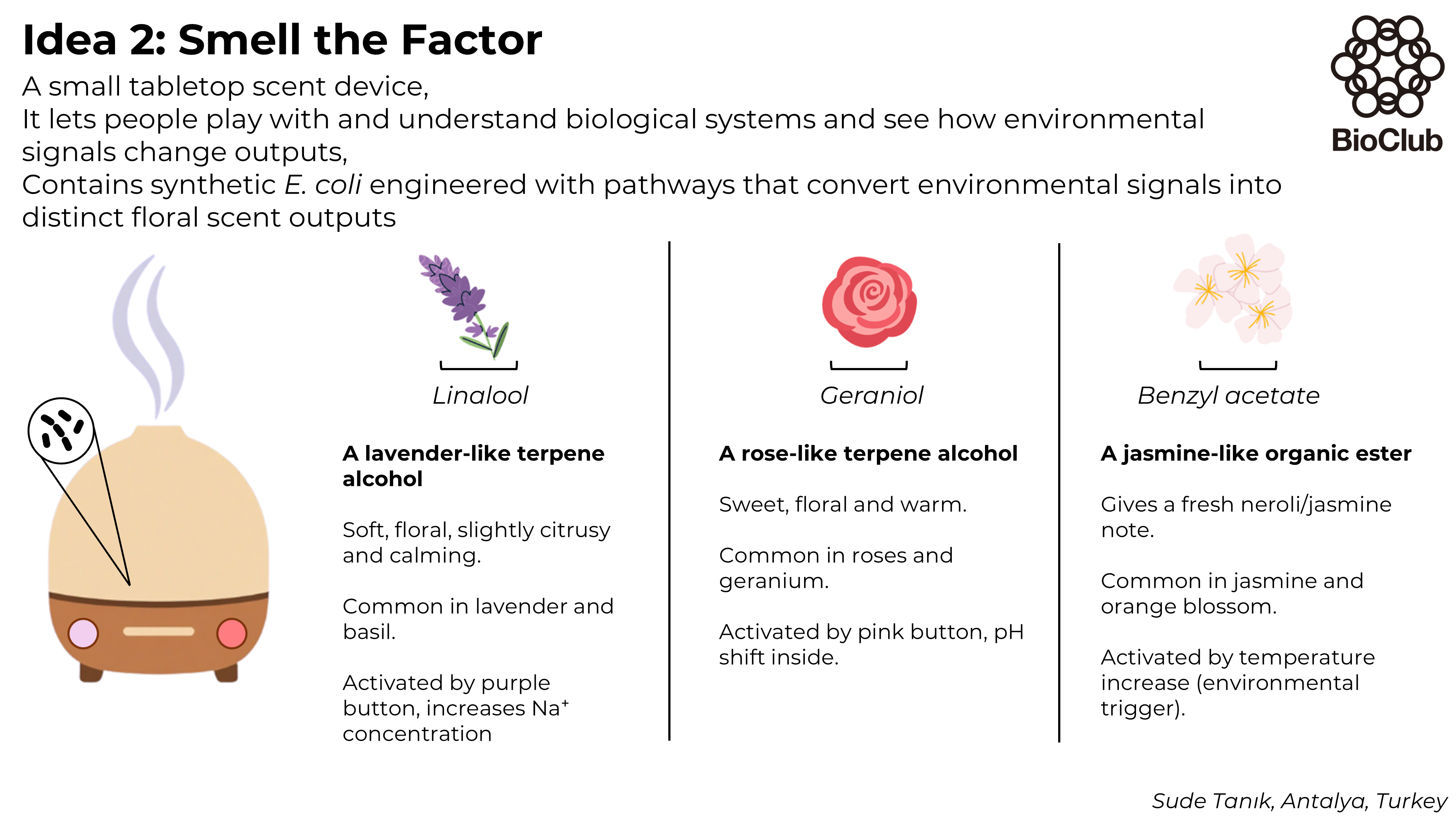
The protein I selected is salicylic acid carboxyl methyltransferase (SAMT) from Clarkia breweri. This enzyme converts salicylic acid into methyl salicylate, which is a volatile compound involved in plant signaling. I chose this protein because methyl salicylate can act as a detectable signal. Since my previous assignments focused on the NiMap-S project, where a microbial nickel biosensor produces a volatile output, SAMT could be a useful protein to generate that signal.
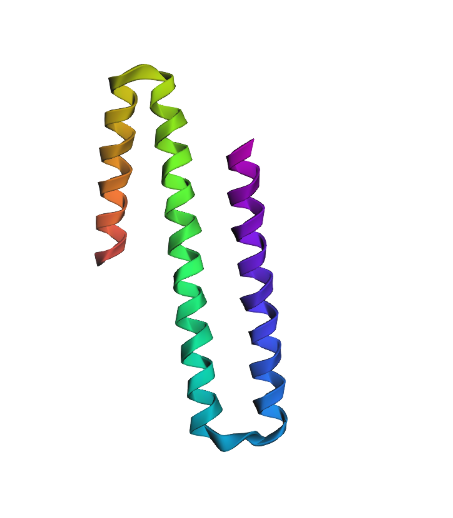
The amino acid sequence of this protein is: MDVRQVLHMKGGAGENSYAMNSFIQRQVISITKPITEAAITALYSGDTVTTRLAIADLGCSSGPNALFAVTELIKTVEELRKKMGRENSPEYQIFLNDLPGNDFNAIFRSLPIENDVDGVCFINGVPGSFYGRLFPRNTLHFIHSSYSLMWLSQVPIGIESNKGNIYMANTCPQSVLNAYYKQFQEDHALFLRCRAQEVVPGGRMVLTILGRRSEDRASTECCLIWQLLAMALNQMVSEGLIEEEKMDKFNIPQYTPSPTEVEAEILKEGSFLIDHIEASEIYWSSCTKDGDGGGSVEEEGYNVARCMRAVAEPLLLDHFGEAIIEDVFHRYKLLIIERMSKEKTKFINVIVSLIRKSD
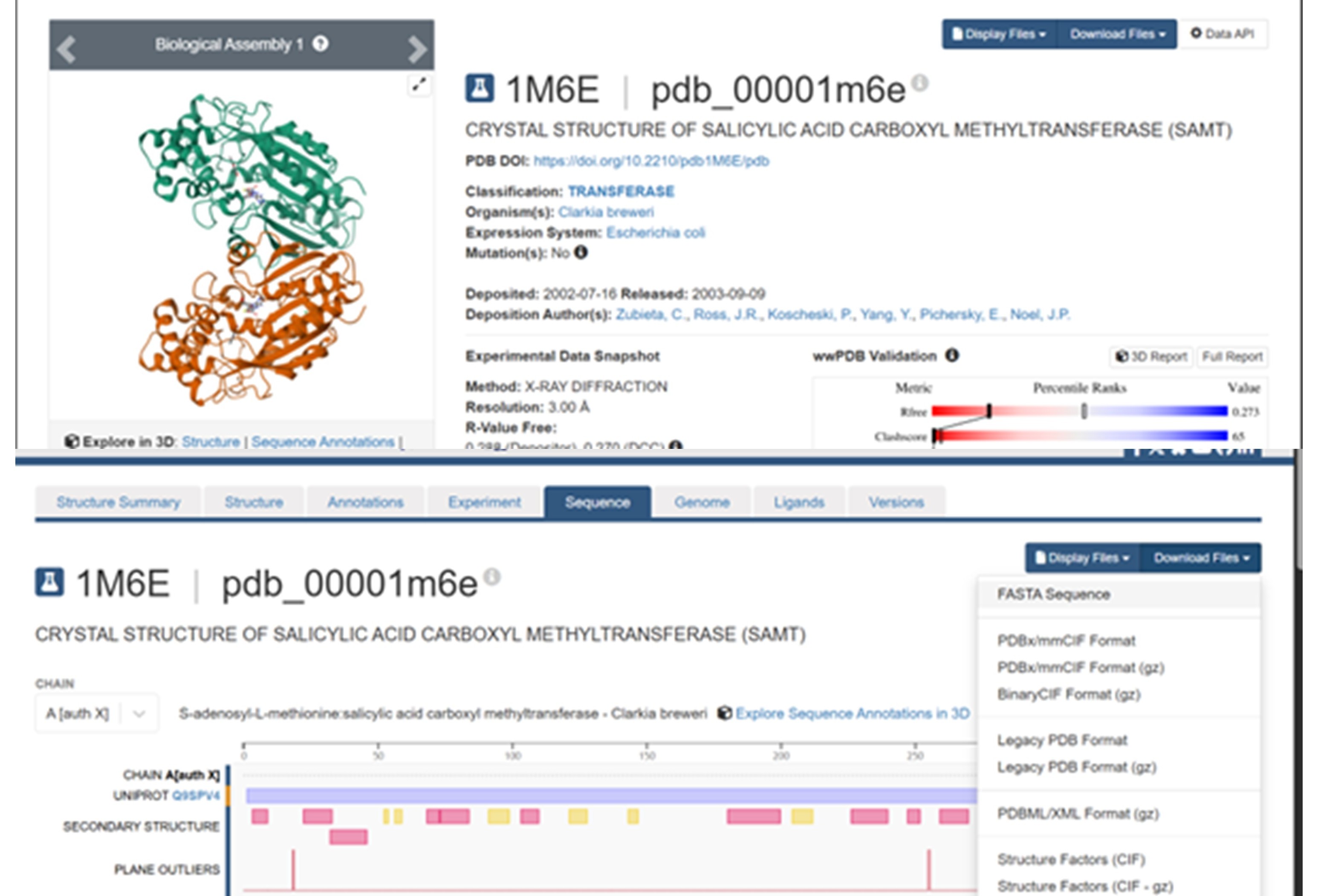
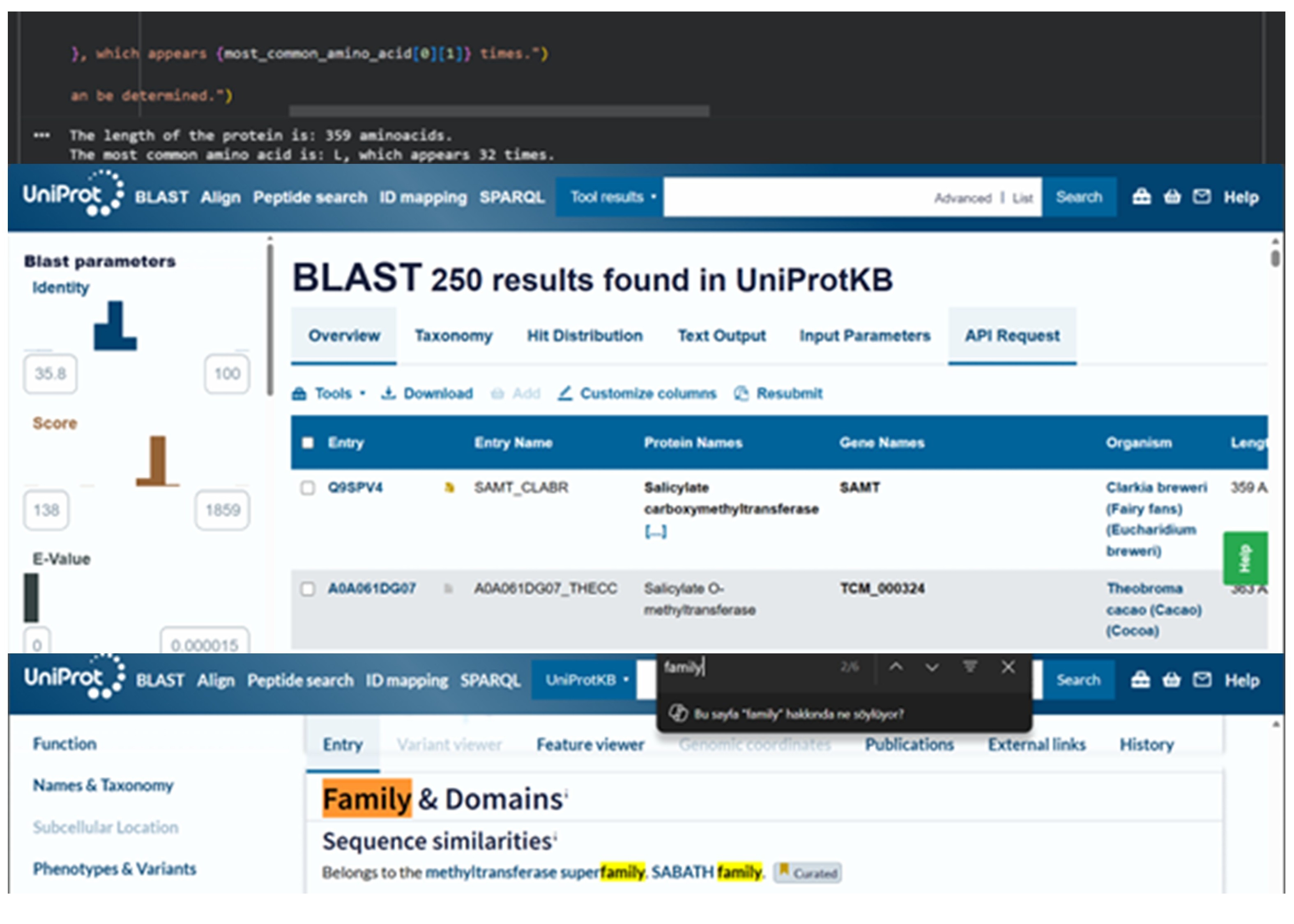
The SAMT protein contains 359 amino acids, and the most common amino acid in the sequence is leucine (L). BLAST search found 250 homologous sequences, and the protein belongs to the SABATH methyltransferase family.
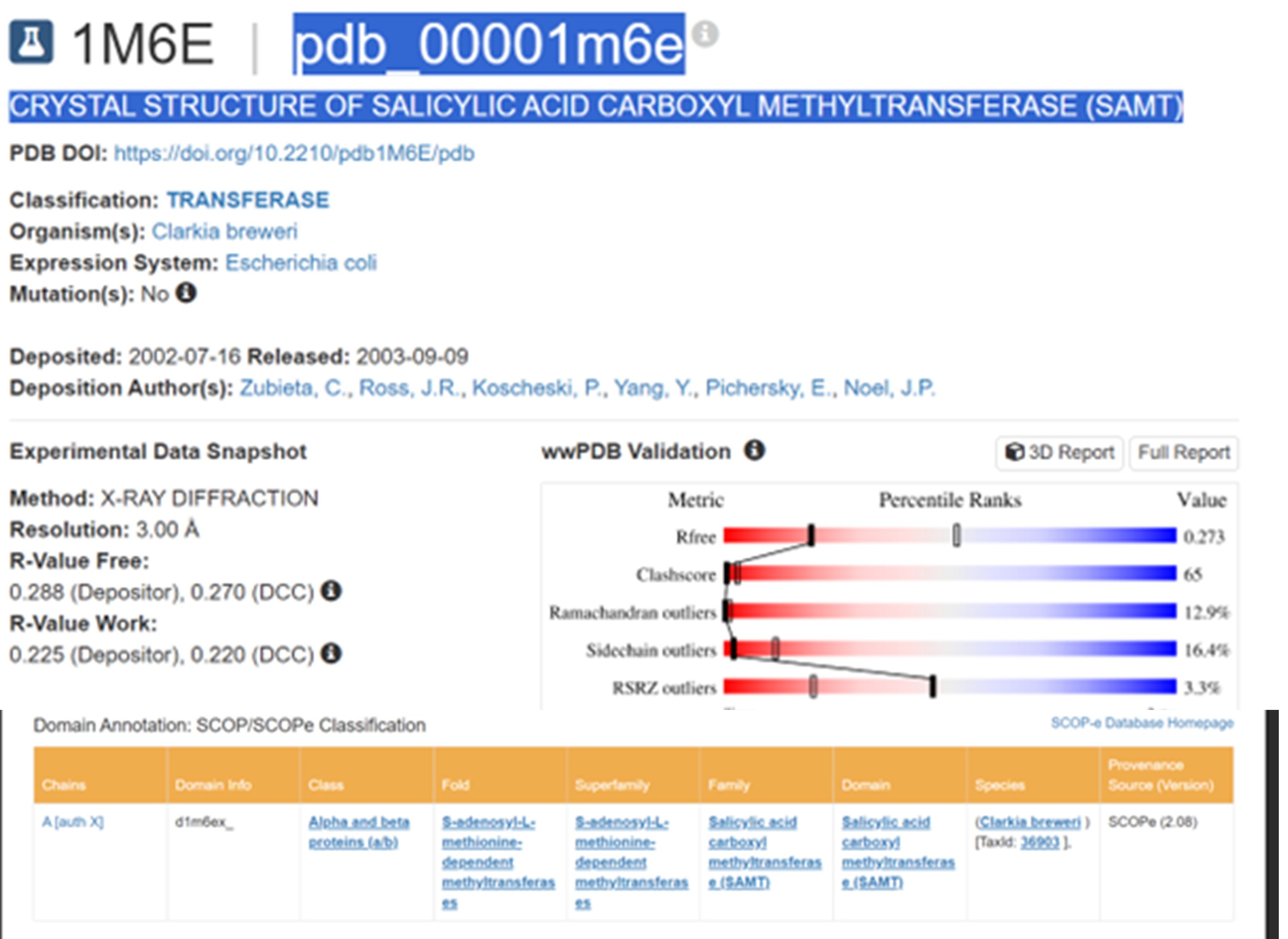
The structure was released on 2003. It was solved using X-ray diffraction with a resolution of 3.00 Å. Lower resolution values indicate better structural quality, and structures below about 2.7 Å are considered high quality, this structure can be considered to have a moderate resolution. According to PDB the class of SAMT is alpha and beta proteins (a/b).
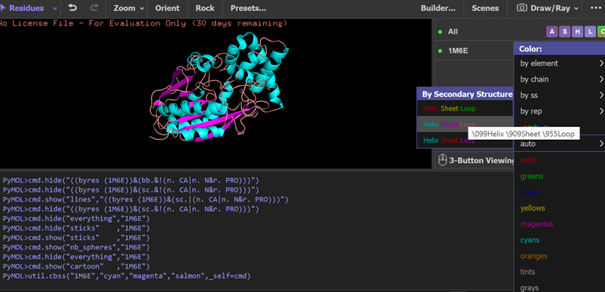
According to the model generated in PyMOL, (a) cartoon, (b) ribbon, and (c) sticks and balls representations of the 1M6E SAMT protein are shown.
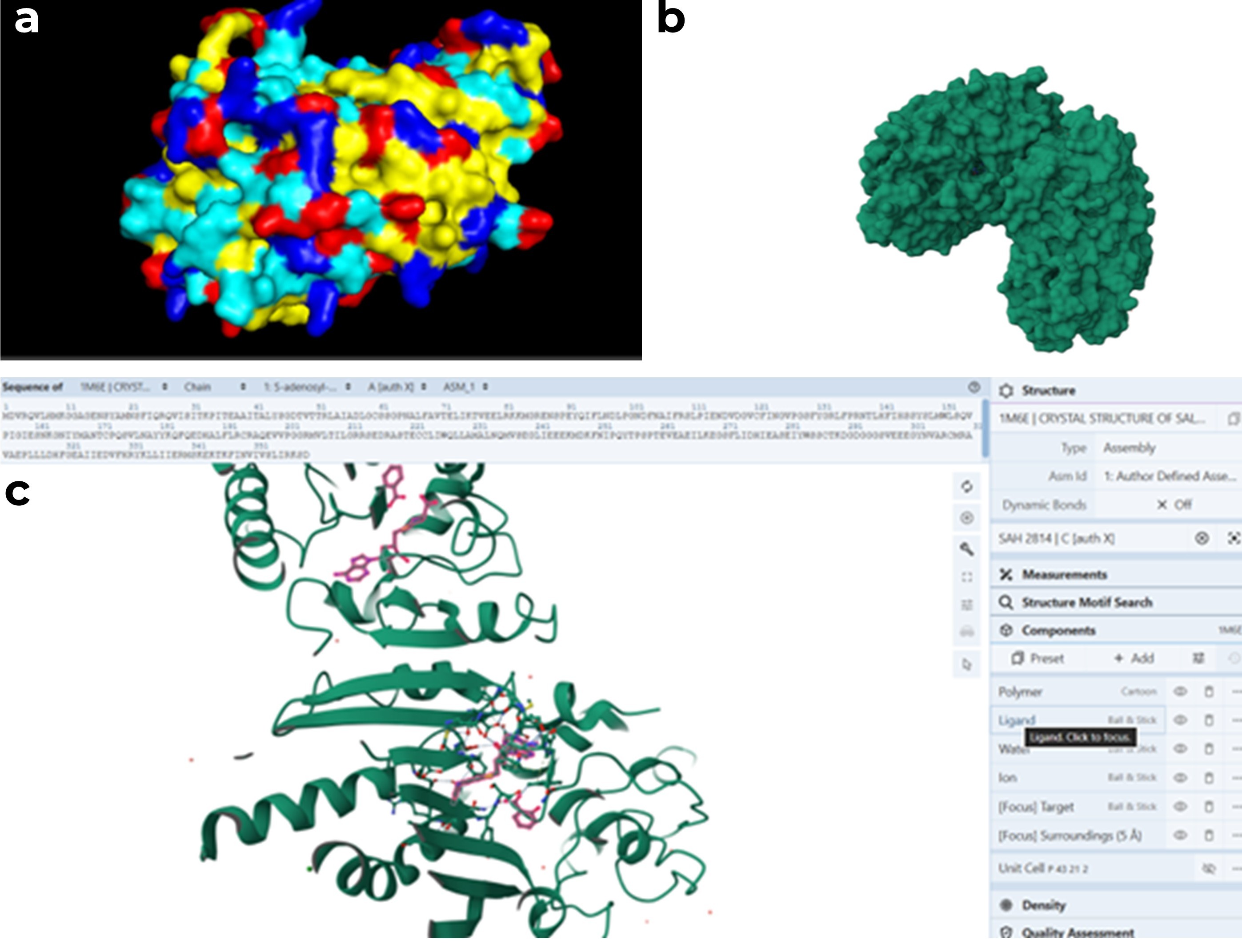
PyMOL visualization of the secondary structure of the 1M6E SAMT protein. Helices are shown in blue, β-sheets in magenta, and loops in salmon. The structure is mainly composed of helices, with some β-sheets located between them.
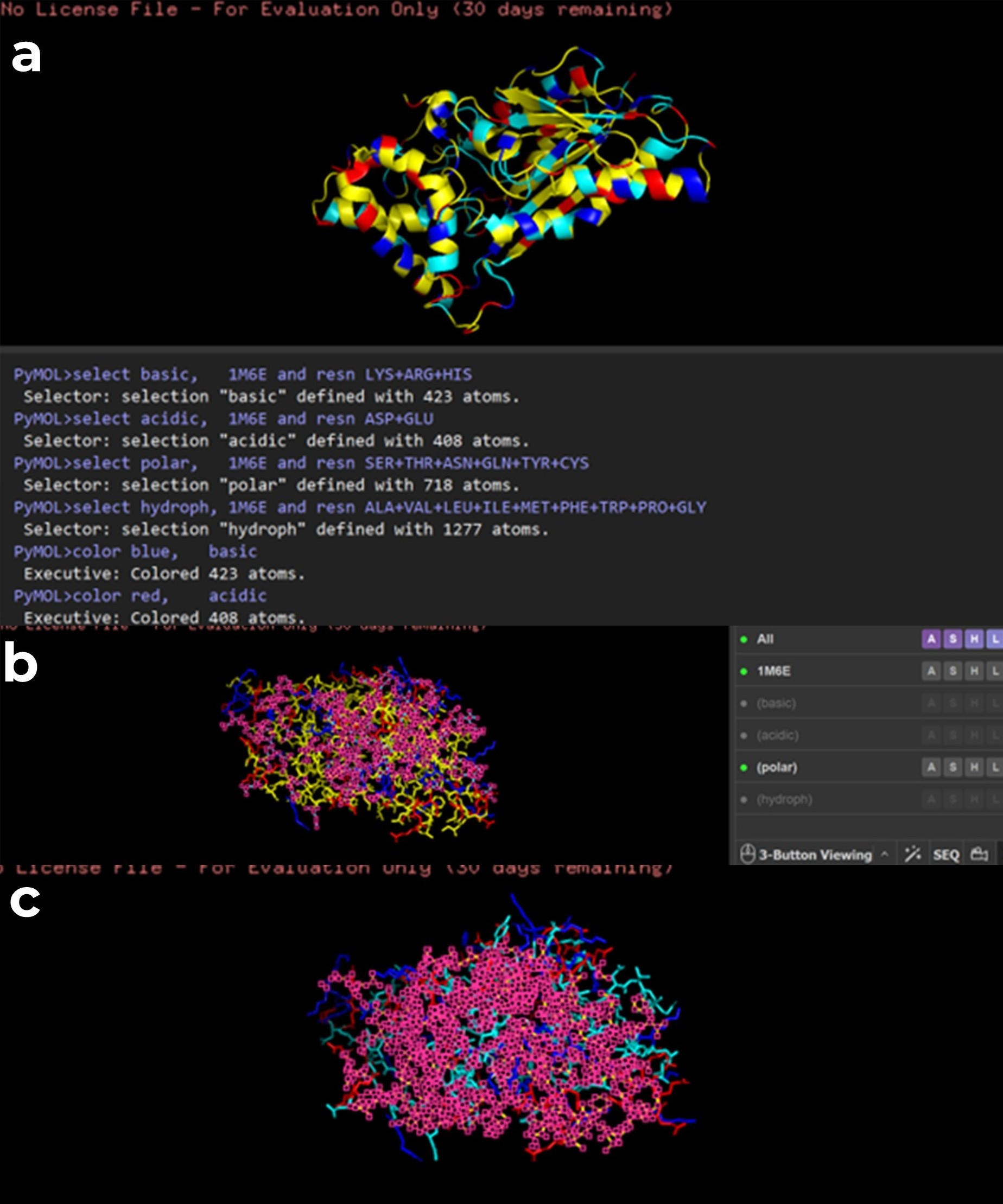
While trying to visualize hydrophobic and hydrophilic residues in PyMOL, I had difficulty. Therefore I used a script generated with ChatGPT 5.2 to color amino acids according to their chemical properties. Hydrophobic residues are shown in yellow, polar residues in cyan, basic residues in blue, and acidic residues in red. (a) all amino acids are displayed, (b) polar residues are highlighted, (c) hydrophobic residues are highlighted.
(a) Surface representation generated in PyMOL, (b) surface representation from RCSB PDB, and (c) cartoon representation of the protein with the ligand shown as balls and sticks. When examining the structure, there appears to be a pocket which is especially visible in the RCSB surface view. This region seemed likely to be the ligand binding site. To better understand this, I also visualized the ligand in RCSB, and it appears to be located in this pocket.
Part C. Using ML-Based Protein Design Tools
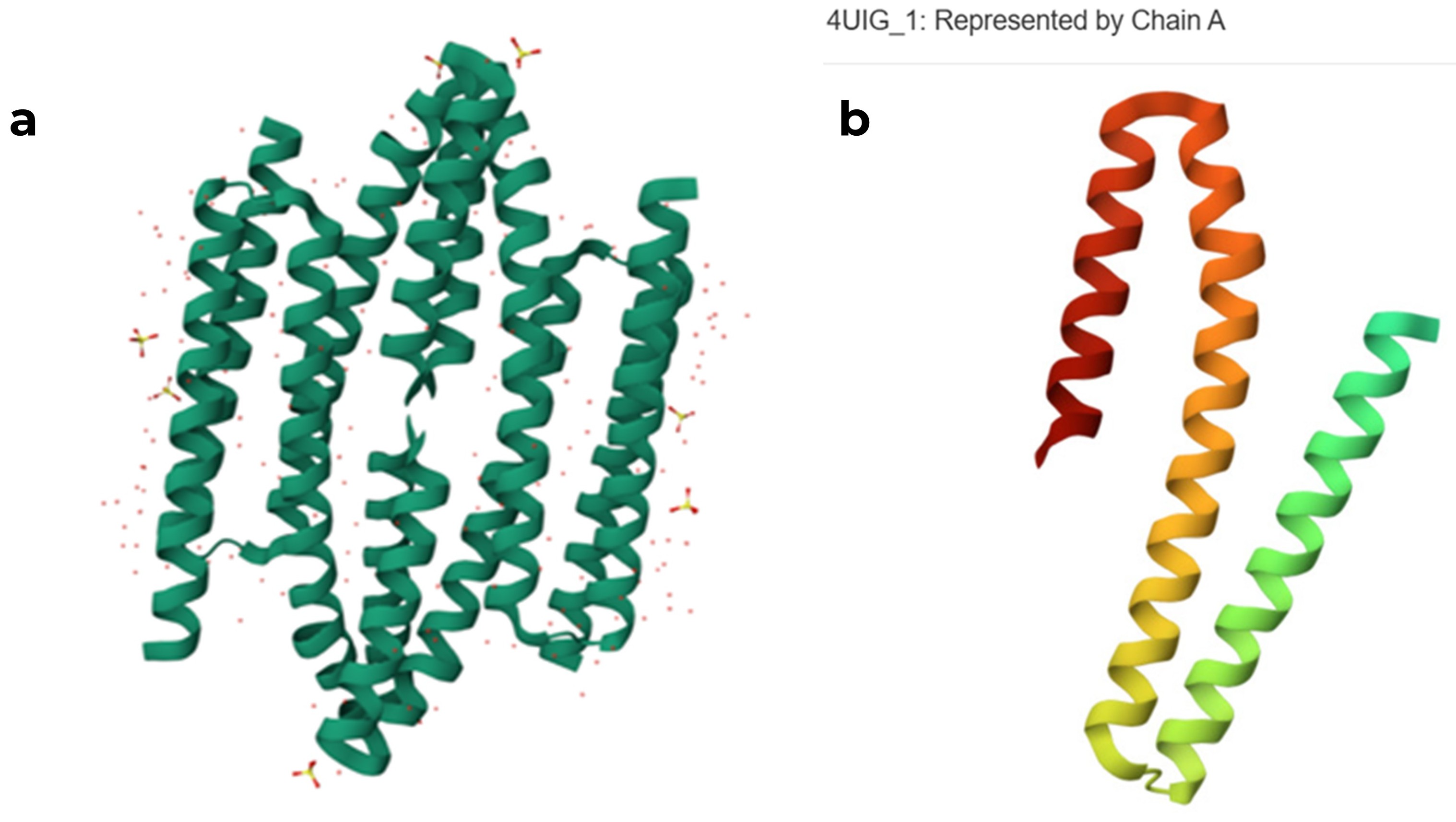
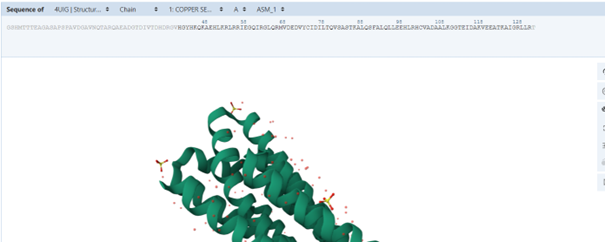
For this part, I selected the protein with PDB ID 4UIG, which is the copper sensitive operon repressor from Streptomyces lividans
C1. Protein Language Modeling
Deep Mutational Scans
Mutations to W (tryptophan) and C (cysteine) seem to affect the protein in many cases. This may be because tryptophan is a large amino acid. Its size may make it difficult to fit into the protein structure, which can disturb the folding, while cysteine is a reactive residue that can form disulfide bonds, potentially altering the folding of the protein.
Mutations to P (proline) appear to affect especially certain positions. For example, replacing K (lysine) at position 56 seems to have one of the strongest effects. This may be related to the importance of the secondary structure, since proline can break α-helices and make the protein backbone less flexible.
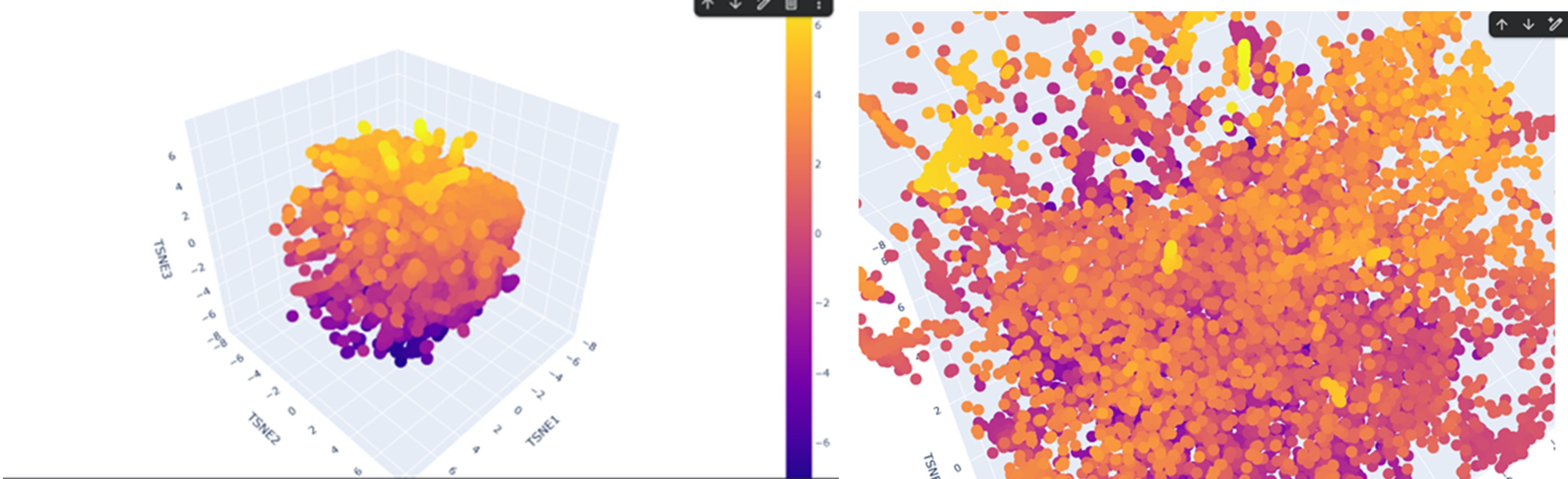
Latent Space Analysis
According to the latent space visualization, proteins do not form very large and clearly separated clusters. They show a more continuous distribution, sometimes forming smaller and tighter local groups. Considering the continuity and mechanisms of evolution this pattern is expected.
For example, when I examined one of the yellow clusters I observed that the proteins had SCOP classifications as d.17.4.x, where only the last number (x) sometimes changed while the earlier levels remained the same. This suggests that the grouped proteins share the same class, fold, and superfamily, indicating a possible evolutionary relationship.
If my protein were located in this cluster, proteins placed near it would likely share a similar SCOP classification to my protein.
C2. Protein Folding
original RCSB PDB structure of the 4UIG protein (a) tetrameric assembly, (b) monomeric structure of chain A
When I used ESMFold with the full sequence “GSHMTTTEAGASAPSPAVDGAVNQTARQAEADGTDIVTDHDRGVHGYHKQKAEHLKRLRRIEGQIRGLQRMVDEDVYCIDILTQVSASTKALQSFALQLLEEHLRHCVADAALKGGTEIDAKVEEATKAIGRLLRT” from RCSB, I noticed that the predicted structure in ESMFold is larger and different from the chain A structure in the RCSB model.
When examining the sequence on the PDB page to understand tge problem, I noticed that some residues are shown in grey. These residues are present in the sequence but were not resolved in the experimental structure. This is probably why they are not included in the PDB structural model, while ESMFold predicts their possible conformation. The new sequence is “HGYHKQKAEHLKRLRRIEGQIRGLQRMVDEDVYCIDILTQVSASTKALQSFALQLLEEHLRHCVADAALKGGTEIDAKVEEATKAIGRLLRT”
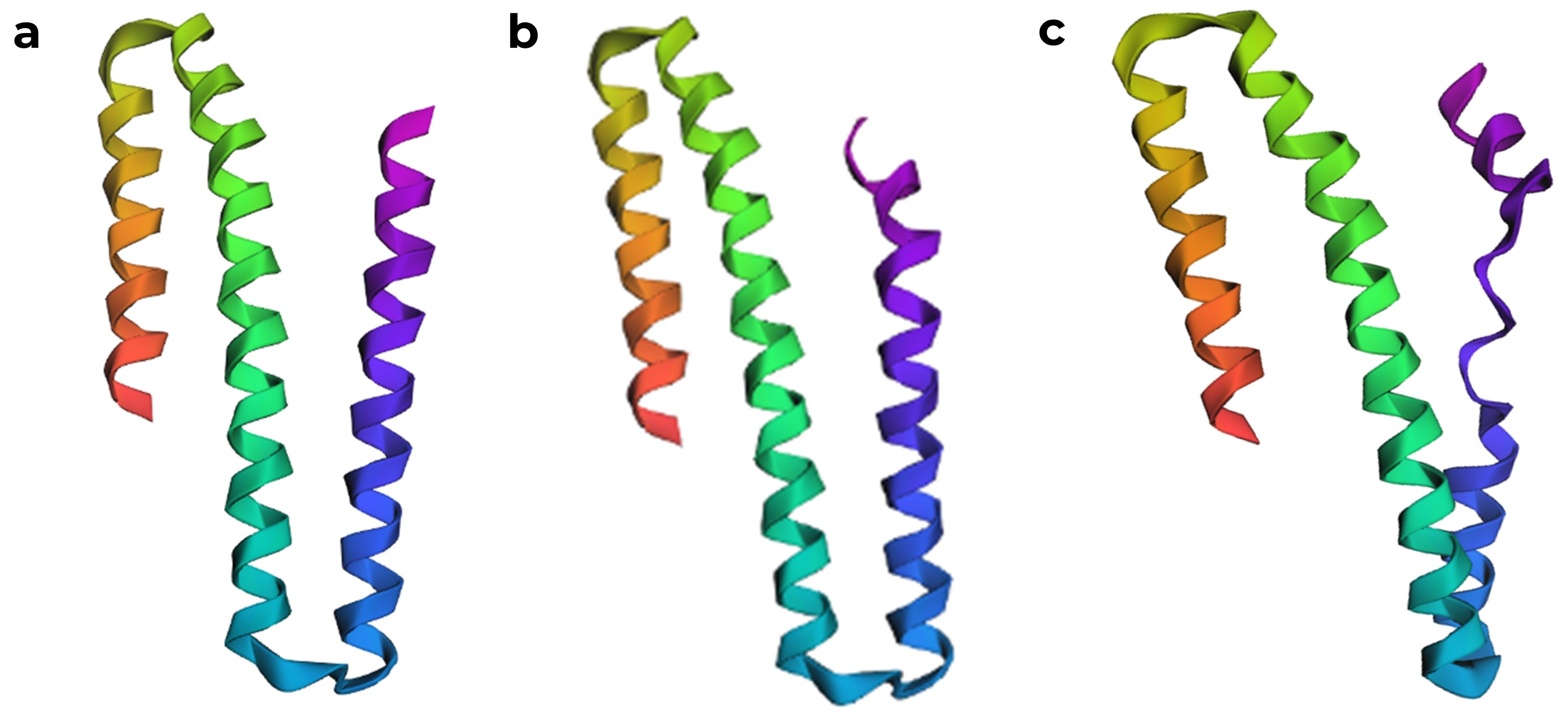
After correcting the sequence, ESMFold produced a model that is quite similar to the chain A structure in the RCSB PDB. ESMFold predicted structures, (a) model generated from the original sequence with missing residues removed, (b) structure with the one K to P mutation, which was predicted to have a high affect from the mutation scan, showing a slight disruption in the α helix, and (c) structure where seven amino acids were mutated to proline.
T=0.2, sample=0, score=1.0736, seq_recovery=0.1758 (generated sequence from the structure)
GAAAAAAAALAARRAEIAAGERELAAMRAAGAPPAEIAAREAALAAARAALAAAVLARLRATALAAARALGAAAAAAAAAALAAAEAEAAA
When modeled with ESMFold, the generated sequence produces a structure that looks quite similar to the original one, even though it shares only about 17% amino acid identity with the native sequence. This suggests that the same helix backbone can be supported by different amino acid compositions. Also when we look at the scores, the designed sequence even appears more compatible with the backbone than the native sequence.
But protein function depends not only on the overall fold but also on the specific chemical properties and diversity of amino acids, and having a similar structure does not necessarily mean that the protein would perform the same biological function.
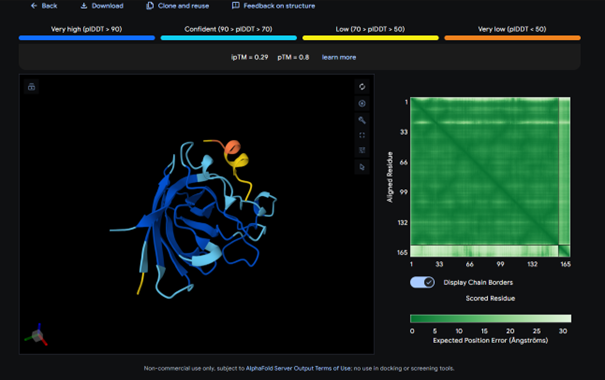
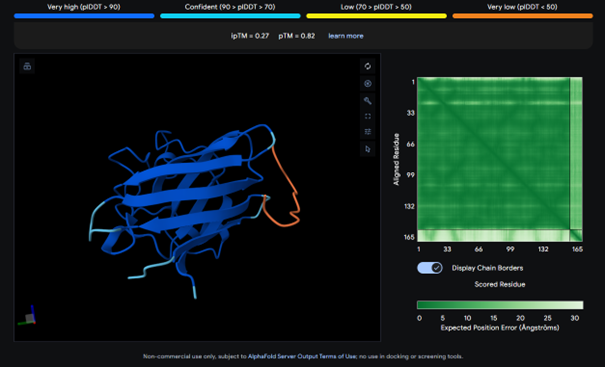
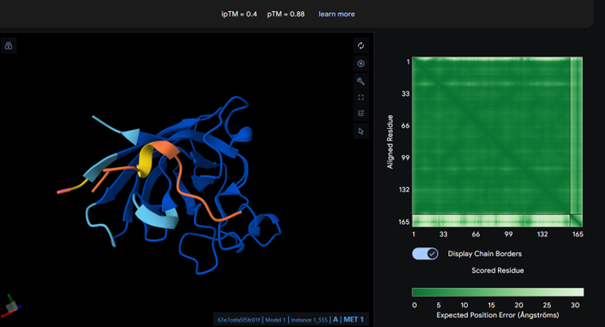
The complex has a low ipTM score (0.29), indicating weak binding. The peptide does not localize near the N-terminus where A4V sits. It appears to bind to a surface loop region and is surface bound rather than partially buried. It does not engage the β-barrel region or approach the dimer interface.
AlphaFold results for peptide 2:
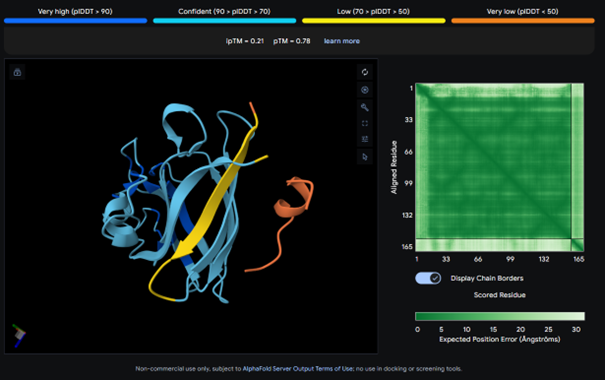
This complex also has a low ipTM score (ipTM 0.21). It is surface bound and is not binded near terminus. It seems to bind on the outside of the β-barrel.
AlphaFold results for peptide 3:
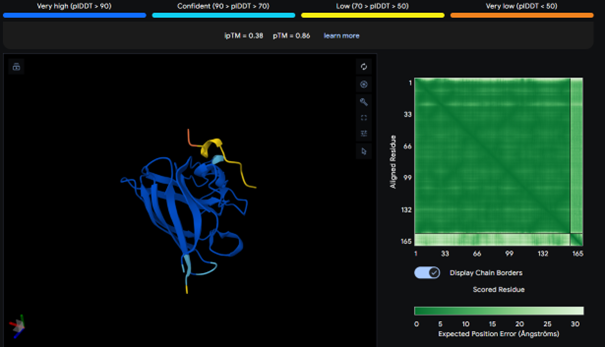
It has the highest binding score (ipTM = 0.38) among the tested models, but this still indicates relatively weak binding. The peptide does not bind to the N-terminus and appears surface-bound. There is no strong interaction with the β-barrel or the dimer interface.
AlphaFold results for peptide 4:
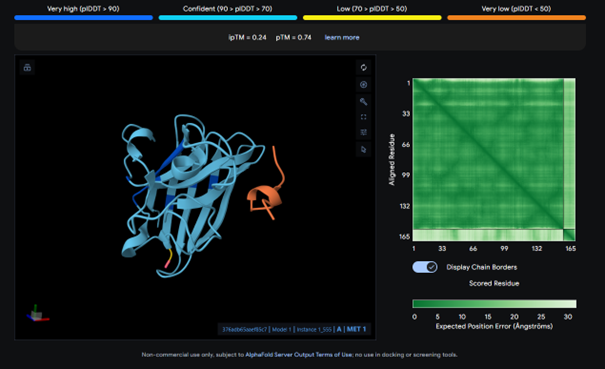
This complex has a low binding score (ipTM = 0.24), meaning weak interaction. The peptide does not bind to the N-terminus and bounds to surface. There is no strong interaction with the β-barrel or the dimer interface.
AlphaFold results for the known binder:
I expected the known binder to have a higher score, but it also shows a low binding score (ipTM = 0.27). Considering this, the result for peptide 3 is actually higher than the known binder, even though it is still relatively low.
All ipTM values indicate weak binding confidence. The known binder has an ipTM of about 0.27, which is also relatively low. Among the PepMLM-generated peptides, peptide 3 shows the highest ipTM (0.38) and therefore exceeds the score of the known binder. In addition, none of the peptides appear to bind near the N-terminus and they all appear surface-bound.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
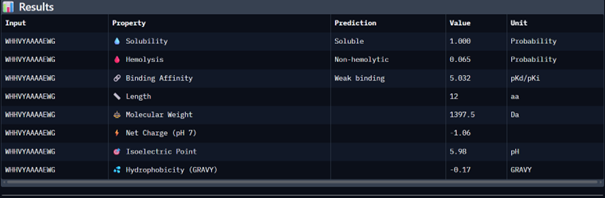
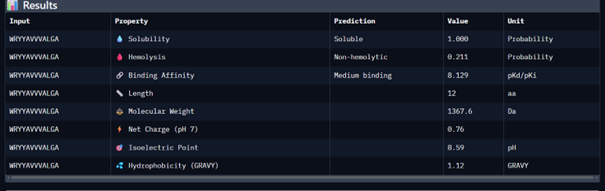
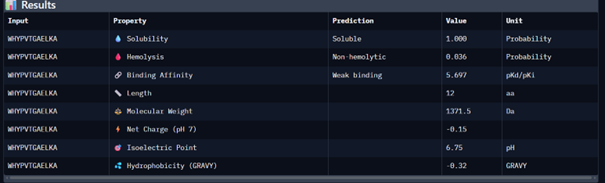
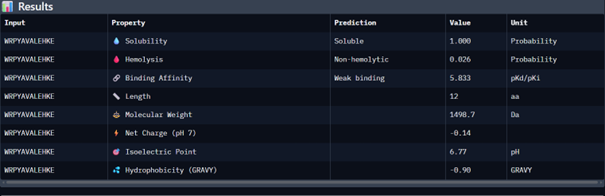
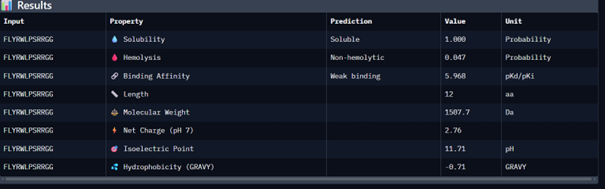
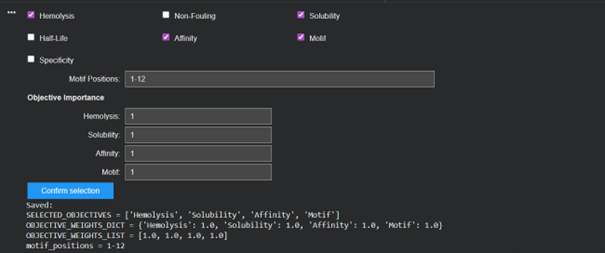
Then I moved to Peptiverse and evaluated the predicted binding affinity, solubility, hemolysis probability, net charge (pH 7), and molecular weight for all peptides.
Peptide 1 appeared slightly hydrophilic, peptide 2 more hydrophobic, and peptides 3 and 4 strongly hydrophilic. All peptides were predicted to be soluble. None showed a significant hemolytic effect, although peptide 2 had the highest hemolysis probability, while peptides 3 and 4 had the lowest. Predicted binding affinity was generally low for all peptides, including the known binder, but unlike the AlphaFold results, Peptiverse predicted the highest binding potential for peptide 2.
The peptide binds near the N-terminal region, which is where the A4V mutation site is located, as intended in the design. The peptide still appears mostly surface-bound rather than deeply buried, but it is positioned closer to the targeted N-terminal region compared to the other peptides, which was the motif I chose for the generated peptides to bind.
The main reason for the difference between the peptides suggested by the two models is likely that PepMLM generates peptides that could bind anywhere on the protein, whereas moPPIt works by specifying the region we want to target and then designing sequences optimized according to multiple criteria. Because of this, moPPIt provides peptides that are not only target-guided but also optimized during the design process, while PepMLM searches more broadly for generally compatible binders.
Before moving to clinical studies, I would first evaluate whether the peptide is safe, starting by checking that it does not cause hemolysis. Then I would examine whether the peptide is soluble. Among the peptides that meet these criteria, I would select those with the highest predicted binding affinity. From these candidates, I would choose the peptide that is biochemically easier to produce and has suitable biological properties (such as pH-related charge and molecular weight) before advancing it toward clinical studies.