Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Most red meat contains about 20% protein, so 500 g of meat contains roughly 100 g of protein. Since 1 Da=1.66×10-24 g, an amino acid with a mass of about 100 Da has a mass of 1.66×10-22 g. Dividing 100 g by 1.66×10-22 g gives approximately 6×1023 amino acids.
Why do humans eat beef but do not become a cow, eat fish but do not become fish? Because during digestion the proteins we eat are broken down into their amino acids. Then, using these amino acids, our bodies synthesize proteins that match our own genetic information, which makes us who we are.
Why are there only 20 natural amino acids? First of all, this shows that the evolutionary process has progressed using these 20 amino acids. Among infinitely many possibilities, these 20 amino acids with diverse chemical properties were selected. Using fewer amino acids may be advantageous for living organisms, both in terms of metabolic efficiency and reducing the error rate during protein synthesis.
Can you make other non-natural amino acids? Design some new amino acids. Yes, it is possible to create non-natural amino acids. Synthetic amino acids can be produced by modifying the side chain of an amino acid and adding functional groups with desired properties, such as increased hydrophobicity or metal binding ability.
Where did amino acids come from before enzymes that make them, and before life started? Especially from the Miller-Urey experiment, we know that amino acids can form spontaneously under suitable conditions. In the early Earth, which was a high energy environment, they could form in numbers much greater than the amino acids currently used by living organisms.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? L-amino acids form a right-handed helix, D-amino acids would be expected to form a left handed helix
Can you discover additional helices in proteins? Depending on the amino acid structure, additional helices can form
Why are most molecular helices right-handed? Most molecular helices are right-handed because proteins almost always contain L-amino acids
Why do β-sheets tend to aggregate? β-sheets tend to aggregate because their functional groups are exposed and they have a flat structure, allowing β-strands from different proteins to form intermolecular hydrogen bonds.
Part B: Protein Analysis and Visualization
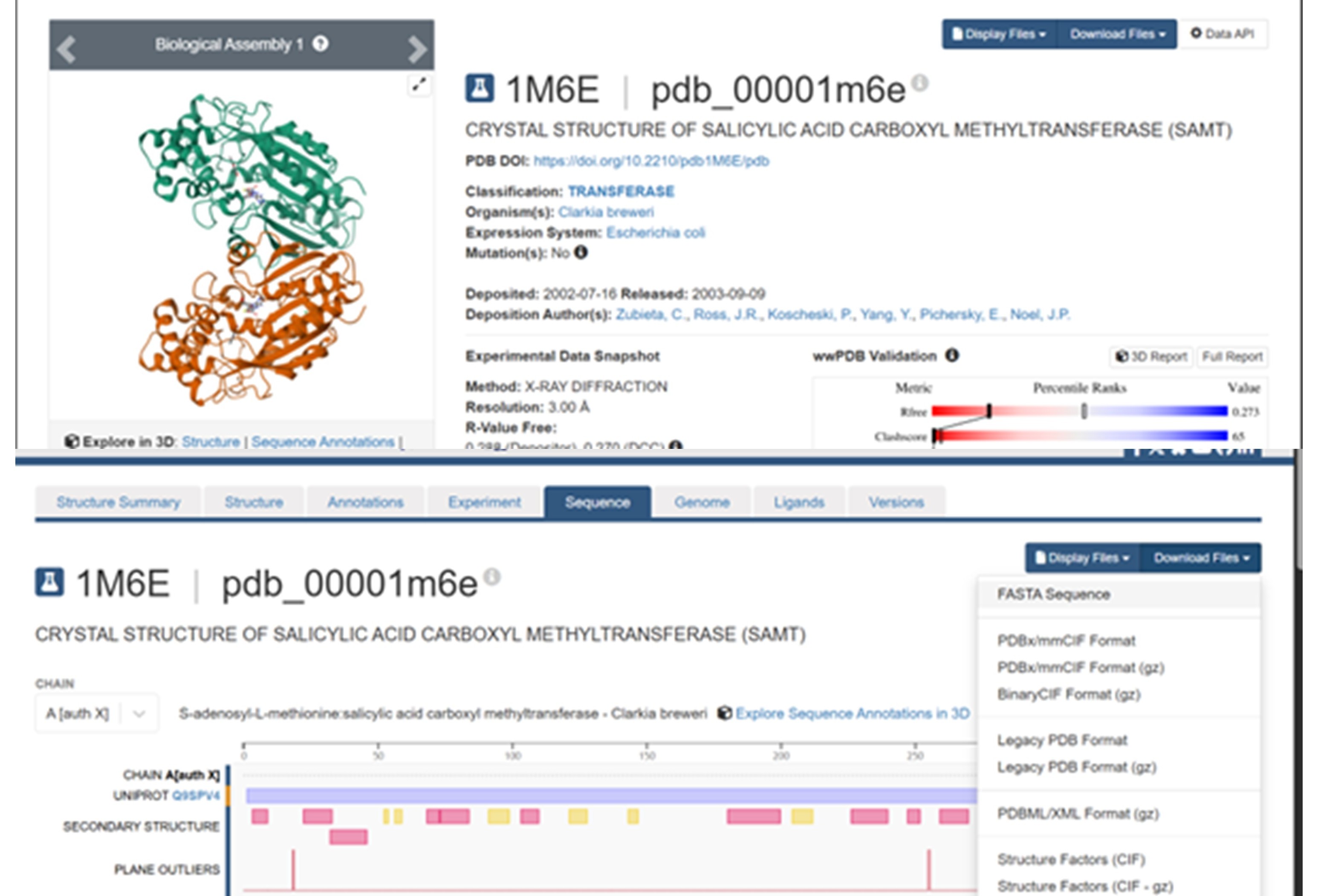
The protein I selected is salicylic acid carboxyl methyltransferase (SAMT) from Clarkia breweri. This enzyme converts salicylic acid into methyl salicylate, which is a volatile compound involved in plant signaling. I chose this protein because methyl salicylate can act as a detectable signal. Since my previous assignments focused on the NiMap-S project, where a microbial nickel biosensor produces a volatile output, SAMT could be a useful protein to generate that signal.
The amino acid sequence of this protein is: MDVRQVLHMKGGAGENSYAMNSFIQRQVISITKPITEAAITALYSGDTVTTRLAIADLGCSSGPNALFAVTELIKTVEELRKKMGRENSPEYQIFLNDLPGNDFNAIFRSLPIENDVDGVCFINGVPGSFYGRLFPRNTLHFIHSSYSLMWLSQVPIGIESNKGNIYMANTCPQSVLNAYYKQFQEDHALFLRCRAQEVVPGGRMVLTILGRRSEDRASTECCLIWQLLAMALNQMVSEGLIEEEKMDKFNIPQYTPSPTEVEAEILKEGSFLIDHIEASEIYWSSCTKDGDGGGSVEEEGYNVARCMRAVAEPLLLDHFGEAIIEDVFHRYKLLIIERMSKEKTKFINVIVSLIRKSD
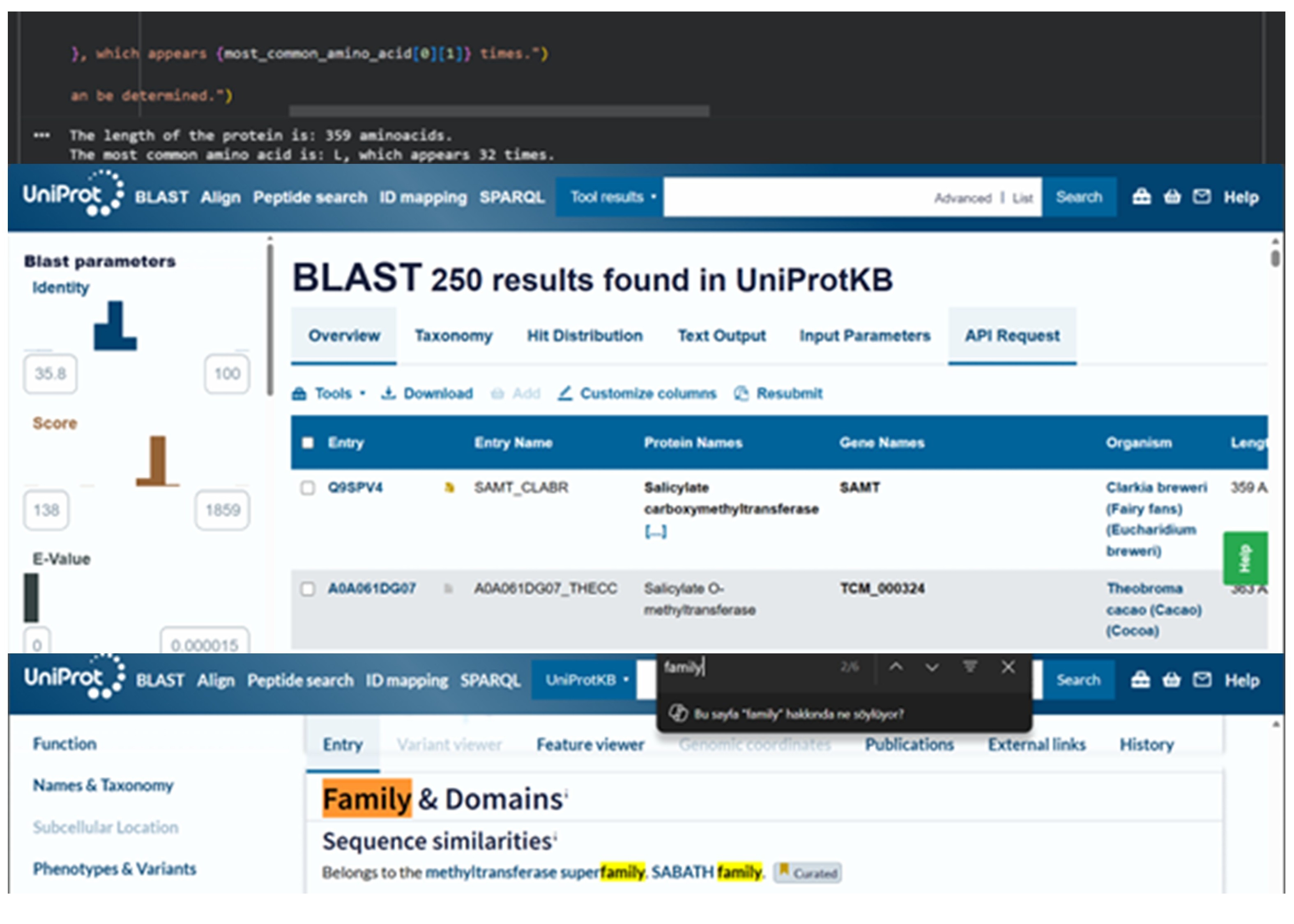
The SAMT protein contains 359 amino acids, and the most common amino acid in the sequence is leucine (L). BLAST search found 250 homologous sequences, and the protein belongs to the SABATH methyltransferase family.
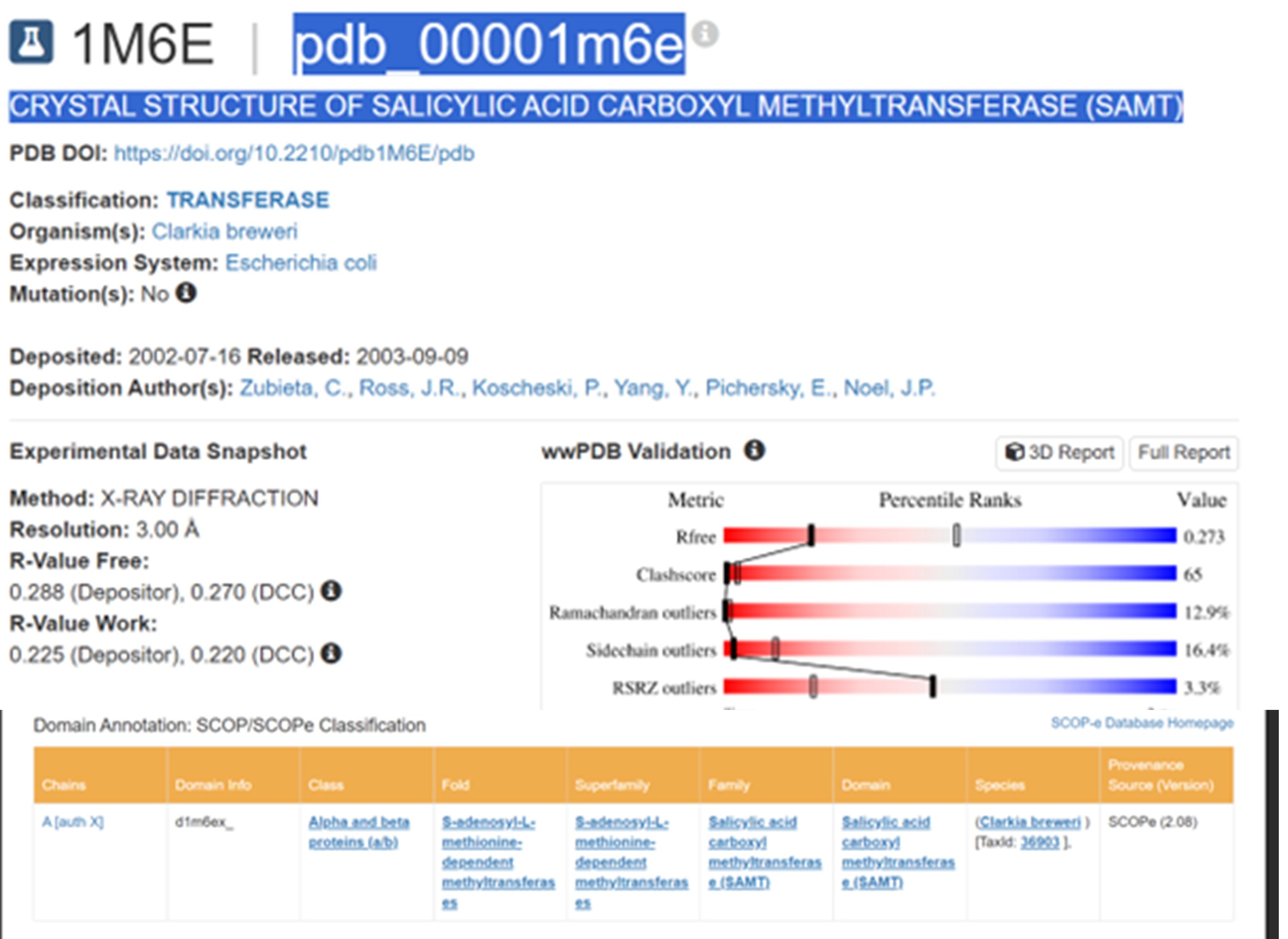
RCSB link for SAMT is: https://www.rcsb.org/structure/1M6E PDB ID: 1M6E
The structure was released on 2003. It was solved using X-ray diffraction with a resolution of 3.00 Å. Lower resolution values indicate better structural quality, and structures below about 2.7 Å are considered high quality, this structure can be considered to have a moderate resolution. According to PDB the class of SAMT is alpha and beta proteins (a/b).

According to the model generated in PyMOL, (a) cartoon, (b) ribbon, and (c) sticks and balls representations of the 1M6E SAMT protein are shown.
PyMOL visualization of the secondary structure of the 1M6E SAMT protein. Helices are shown in blue, β-sheets in magenta, and loops in salmon. The structure is mainly composed of helices, with some β-sheets located between them.
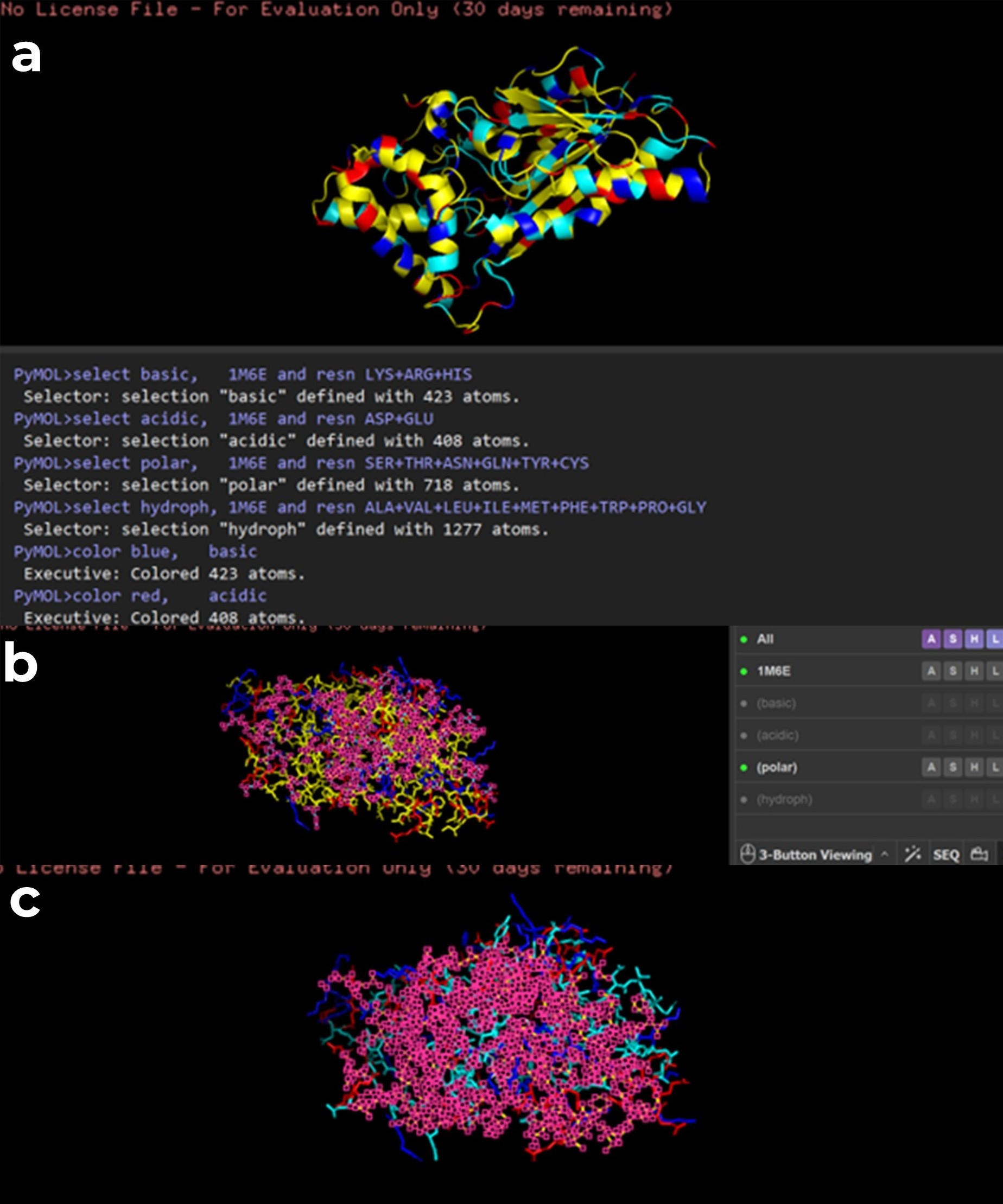
While trying to visualize hydrophobic and hydrophilic residues in PyMOL, I had difficulty. Therefore I used a script generated with ChatGPT 5.2 to color amino acids according to their chemical properties. Hydrophobic residues are shown in yellow, polar residues in cyan, basic residues in blue, and acidic residues in red. (a) all amino acids are displayed, (b) polar residues are highlighted, (c) hydrophobic residues are highlighted.
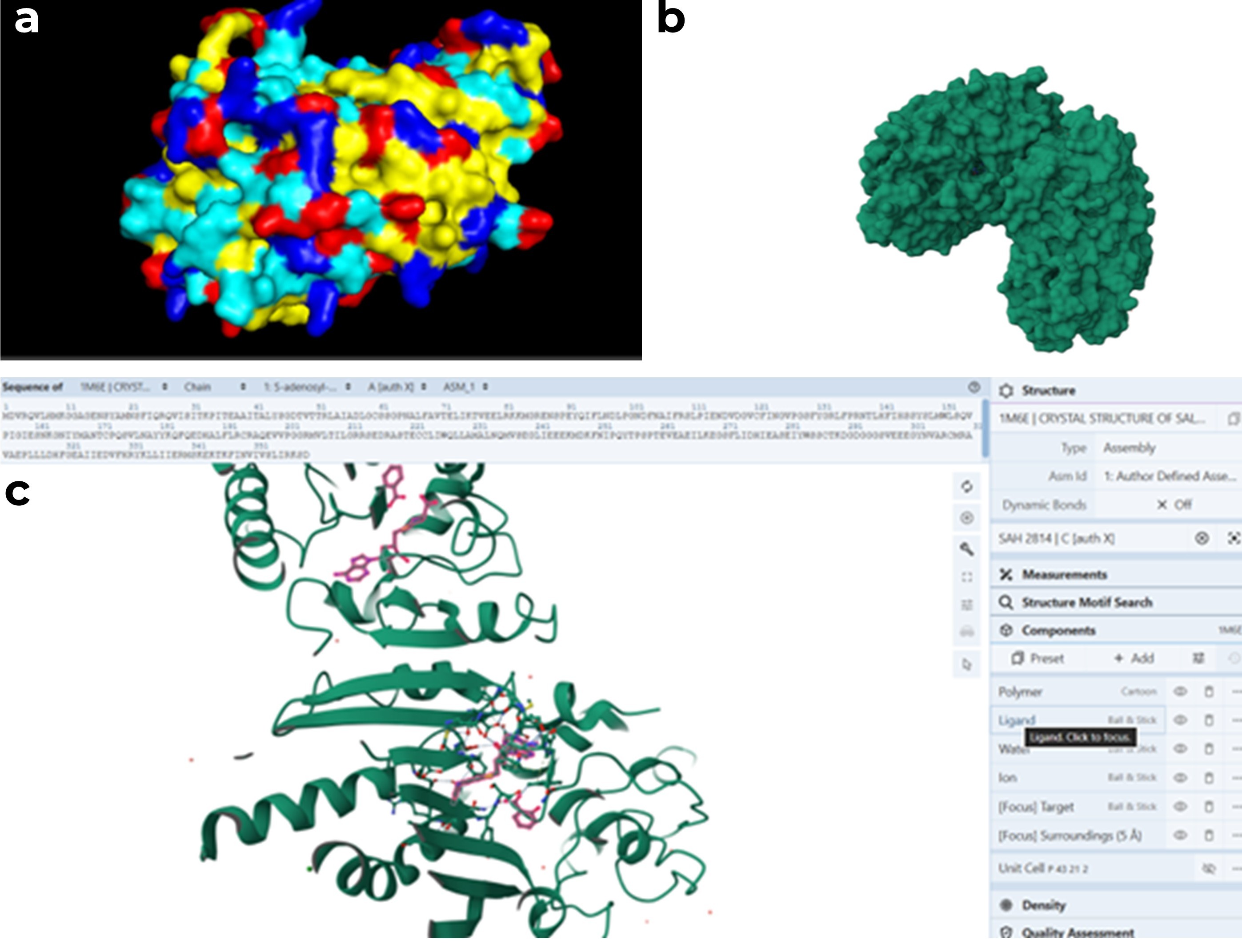
(a) Surface representation generated in PyMOL, (b) surface representation from RCSB PDB, and (c) cartoon representation of the protein with the ligand shown as balls and sticks. When examining the structure, there appears to be a pocket which is especially visible in the RCSB surface view. This region seemed likely to be the ligand binding site. To better understand this, I also visualized the ligand in RCSB, and it appears to be located in this pocket.
Part C. Using ML-Based Protein Design Tools
For this part, I selected the protein with PDB ID 4UIG, which is the copper sensitive operon repressor from Streptomyces lividans
C1. Protein Language Modeling
Deep Mutational Scans
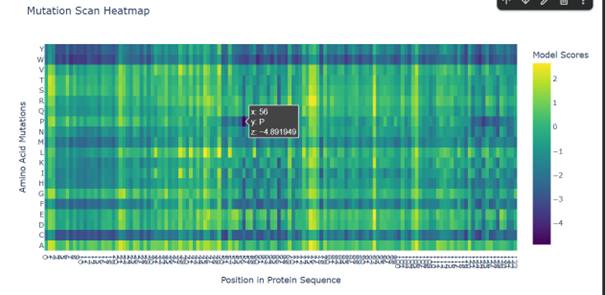
Mutations to W (tryptophan) and C (cysteine) seem to affect the protein in many cases. This may be because tryptophan is a large amino acid. Its size may make it difficult to fit into the protein structure, which can disturb the folding, while cysteine is a reactive residue that can form disulfide bonds, potentially altering the folding of the protein.
Mutations to P (proline) appear to affect especially certain positions. For example, replacing K (lysine) at position 56 seems to have one of the strongest effects. This may be related to the importance of the secondary structure, since proline can break α-helices and make the protein backbone less flexible.
Latent Space Analysis
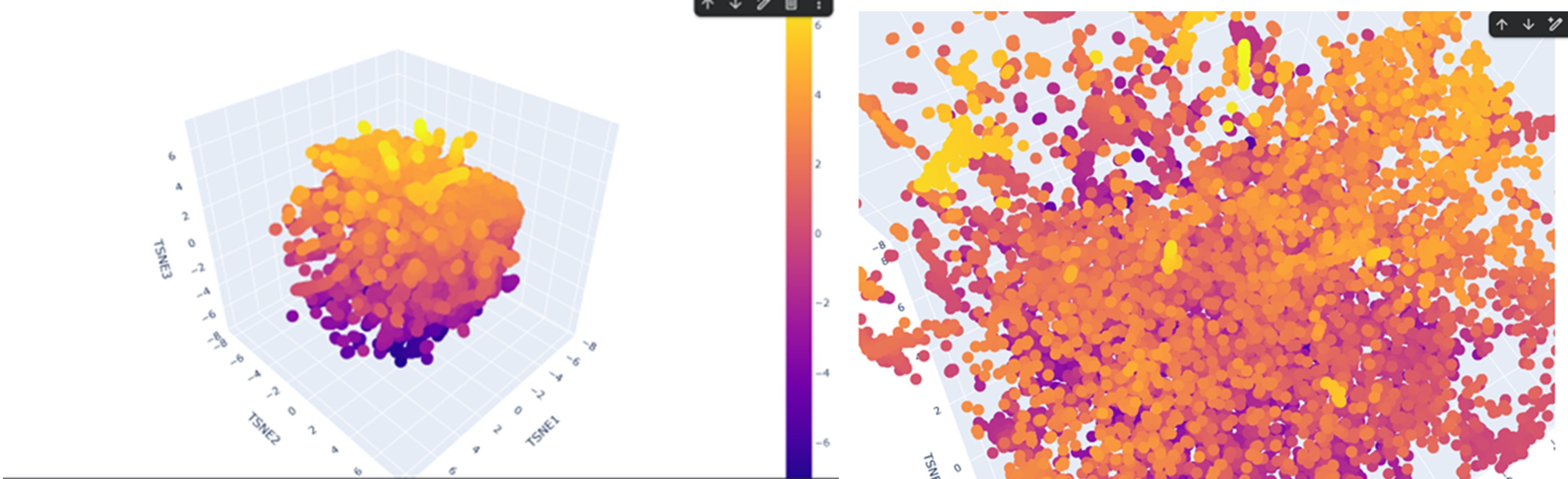
According to the latent space visualization, proteins do not form very large and clearly separated clusters. They show a more continuous distribution, sometimes forming smaller and tighter local groups. Considering the continuity and mechanisms of evolution this pattern is expected.
For example, when I examined one of the yellow clusters I observed that the proteins had SCOP classifications as d.17.4.x, where only the last number (x) sometimes changed while the earlier levels remained the same. This suggests that the grouped proteins share the same class, fold, and superfamily, indicating a possible evolutionary relationship.
If my protein were located in this cluster, proteins placed near it would likely share a similar SCOP classification to my protein.
C2. Protein Folding
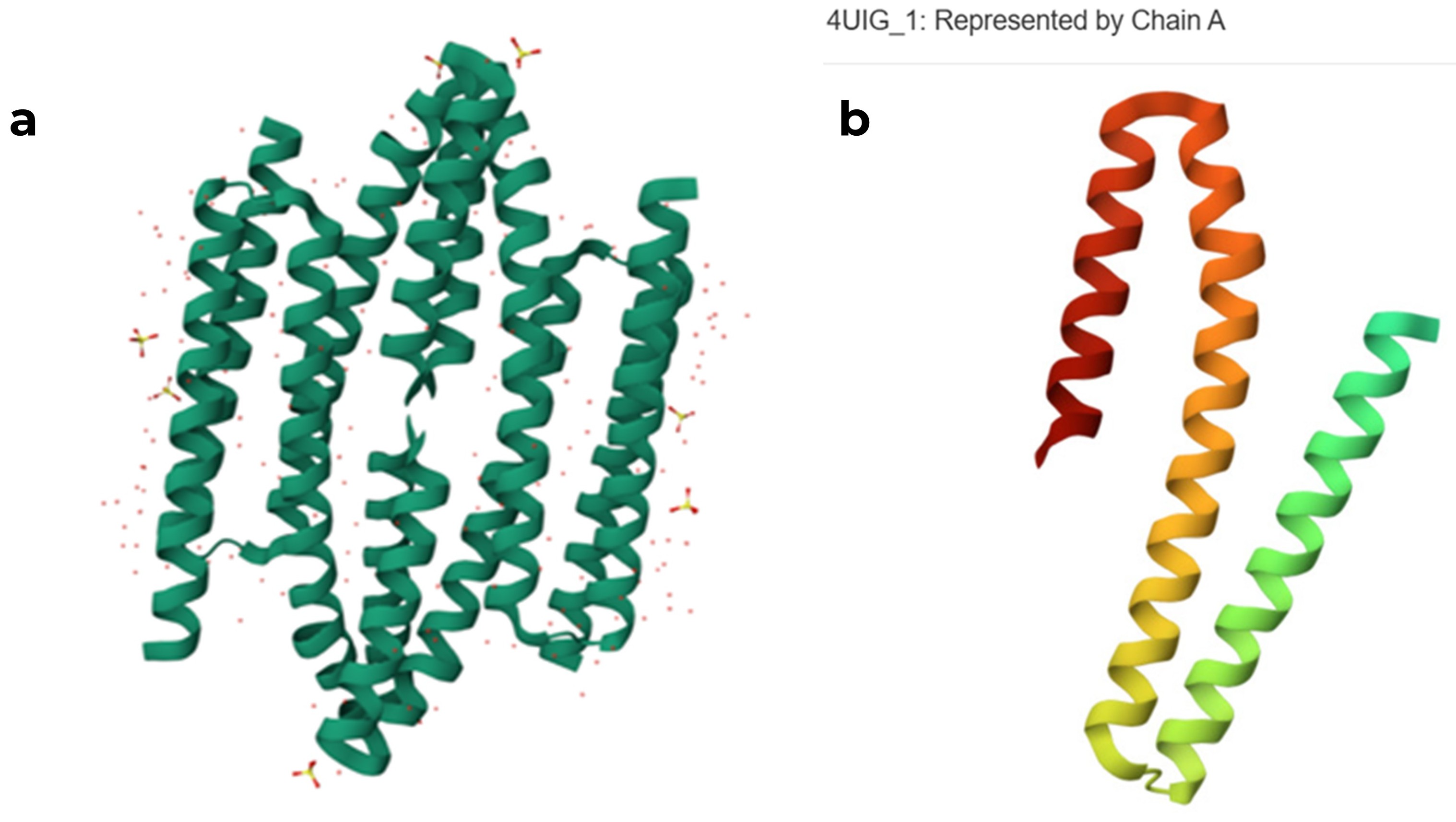
original RCSB PDB structure of the 4UIG protein (a) tetrameric assembly, (b) monomeric structure of chain A
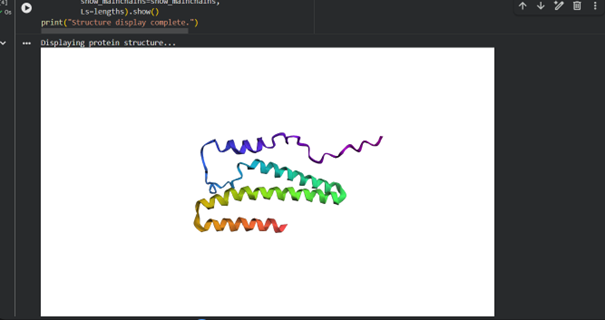
When I used ESMFold with the full sequence “GSHMTTTEAGASAPSPAVDGAVNQTARQAEADGTDIVTDHDRGVHGYHKQKAEHLKRLRRIEGQIRGLQRMVDEDVYCIDILTQVSASTKALQSFALQLLEEHLRHCVADAALKGGTEIDAKVEEATKAIGRLLRT” from RCSB, I noticed that the predicted structure in ESMFold is larger and different from the chain A structure in the RCSB model.
When examining the sequence on the PDB page to understand tge problem, I noticed that some residues are shown in grey. These residues are present in the sequence but were not resolved in the experimental structure. This is probably why they are not included in the PDB structural model, while ESMFold predicts their possible conformation. The new sequence is “HGYHKQKAEHLKRLRRIEGQIRGLQRMVDEDVYCIDILTQVSASTKALQSFALQLLEEHLRHCVADAALKGGTEIDAKVEEATKAIGRLLRT”
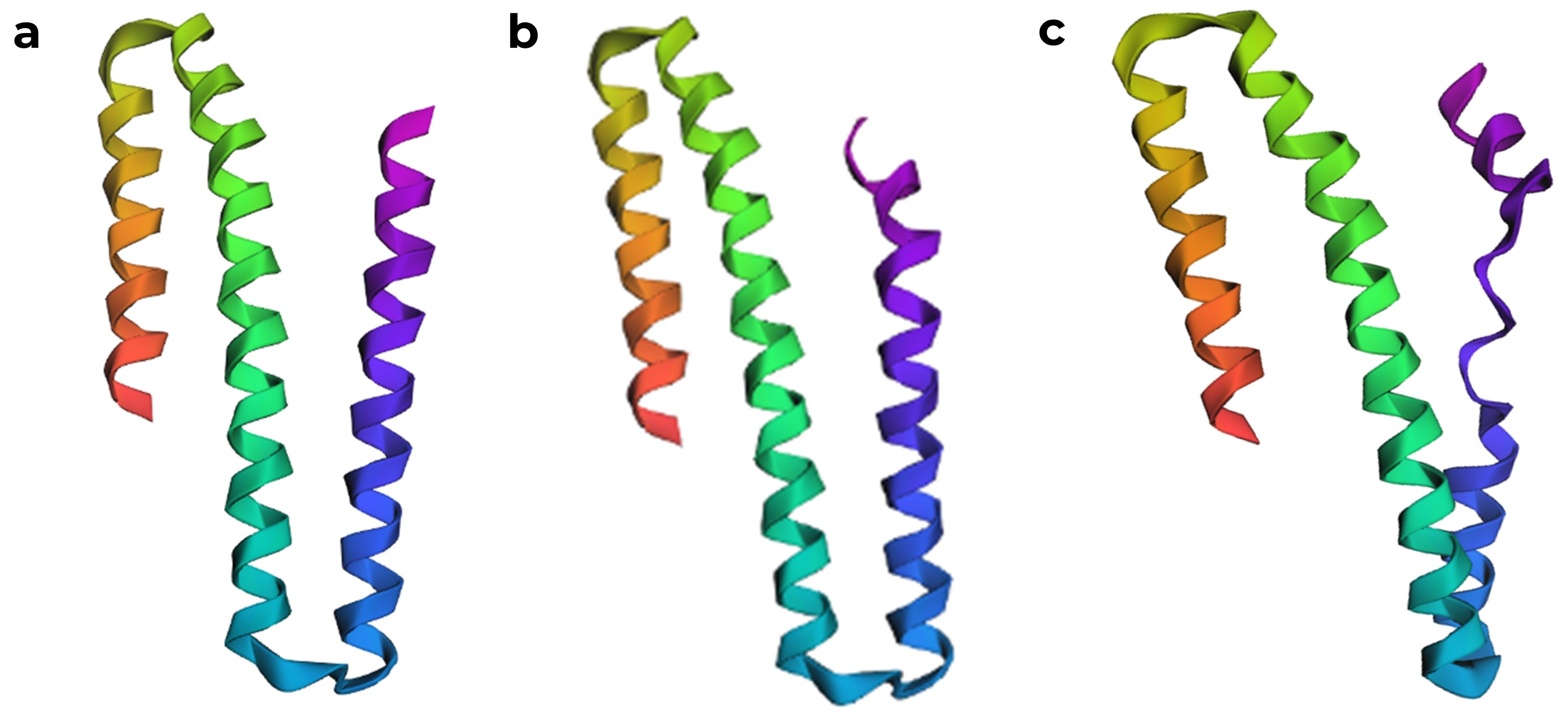
After correcting the sequence, ESMFold produced a model that is quite similar to the chain A structure in the RCSB PDB. ESMFold predicted structures, (a) model generated from the original sequence with missing residues removed, (b) structure with the one K to P mutation, which was predicted to have a high affect from the mutation scan, showing a slight disruption in the α helix, and (c) structure where seven amino acids were mutated to proline.
C3. Protein Generation
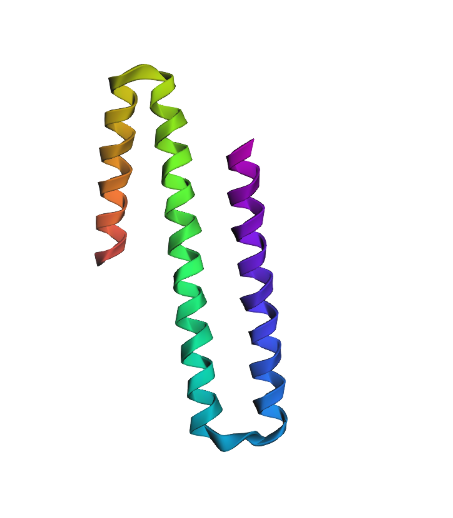
4UIG, score=2.4416, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 (original sequence) HGYHKQKAEHLKRLRRIEGQIRGLQRMVDEDVYCIDILTQVSASTKALQSFALQLLEEHLRHCVADAALKGGTEIDAKVEEATKAIGRLLR
T=0.2, sample=0, score=1.0736, seq_recovery=0.1758 (generated sequence from the structure) GAAAAAAAALAARRAEIAAGERELAAMRAAGAPPAEIAAREAALAAARAALAAAVLARLRATALAAARALGAAAAAAAAAALAAAEAEAAA
When modeled with ESMFold, the generated sequence produces a structure that looks quite similar to the original one, even though it shares only about 17% amino acid identity with the native sequence. This suggests that the same helix backbone can be supported by different amino acid compositions. Also when we look at the scores, the designed sequence even appears more compatible with the backbone than the native sequence. But protein function depends not only on the overall fold but also on the specific chemical properties and diversity of amino acids, and having a similar structure does not necessarily mean that the protein would perform the same biological function.